
Intensification of Gas Hydrate Formation Processes by Renewal of Interfacial Area between Phases



Abstract
:1. Introduction
2. Research Background
2.1. Intensification of GH Formation Processes by Renewing the Phase Boundary
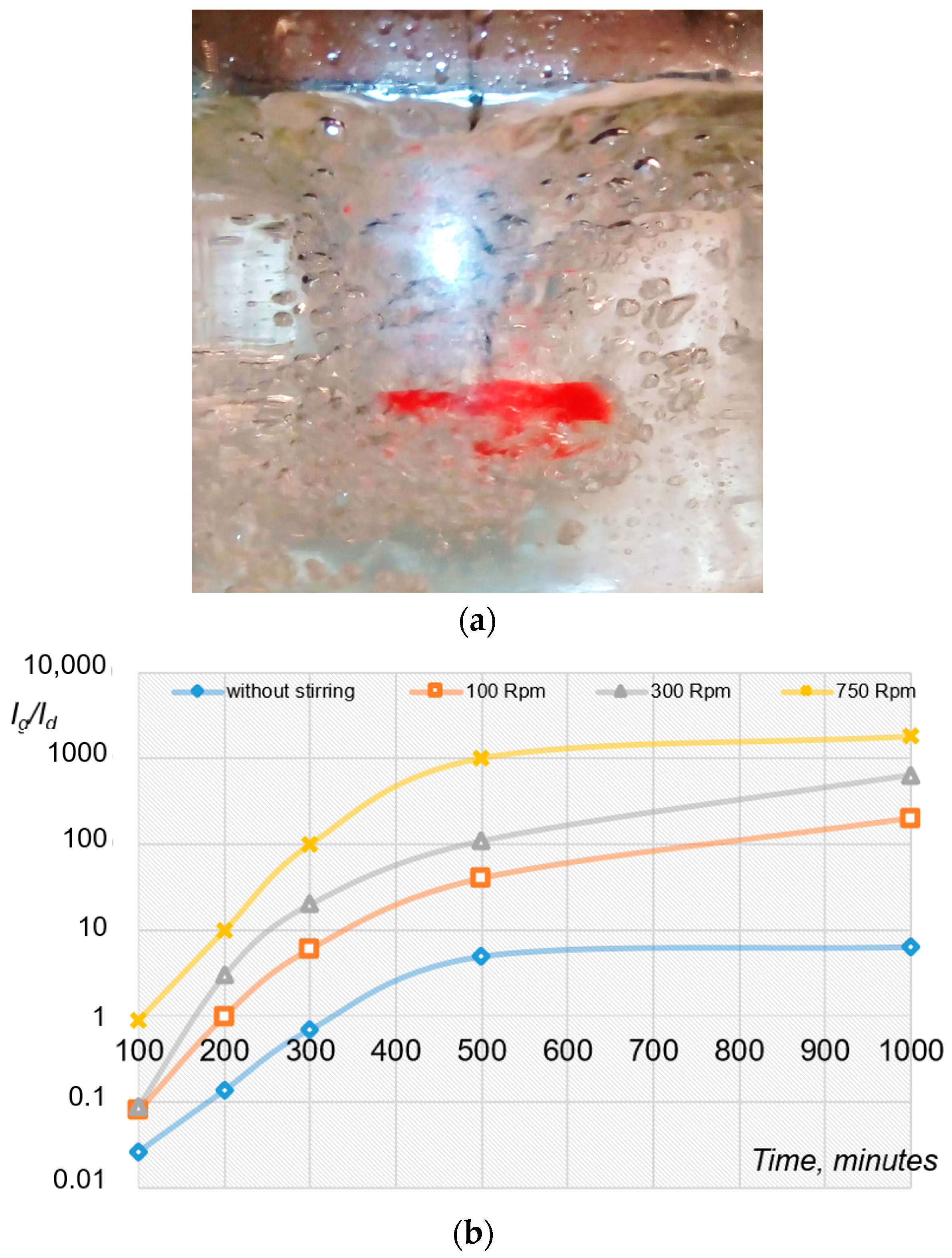
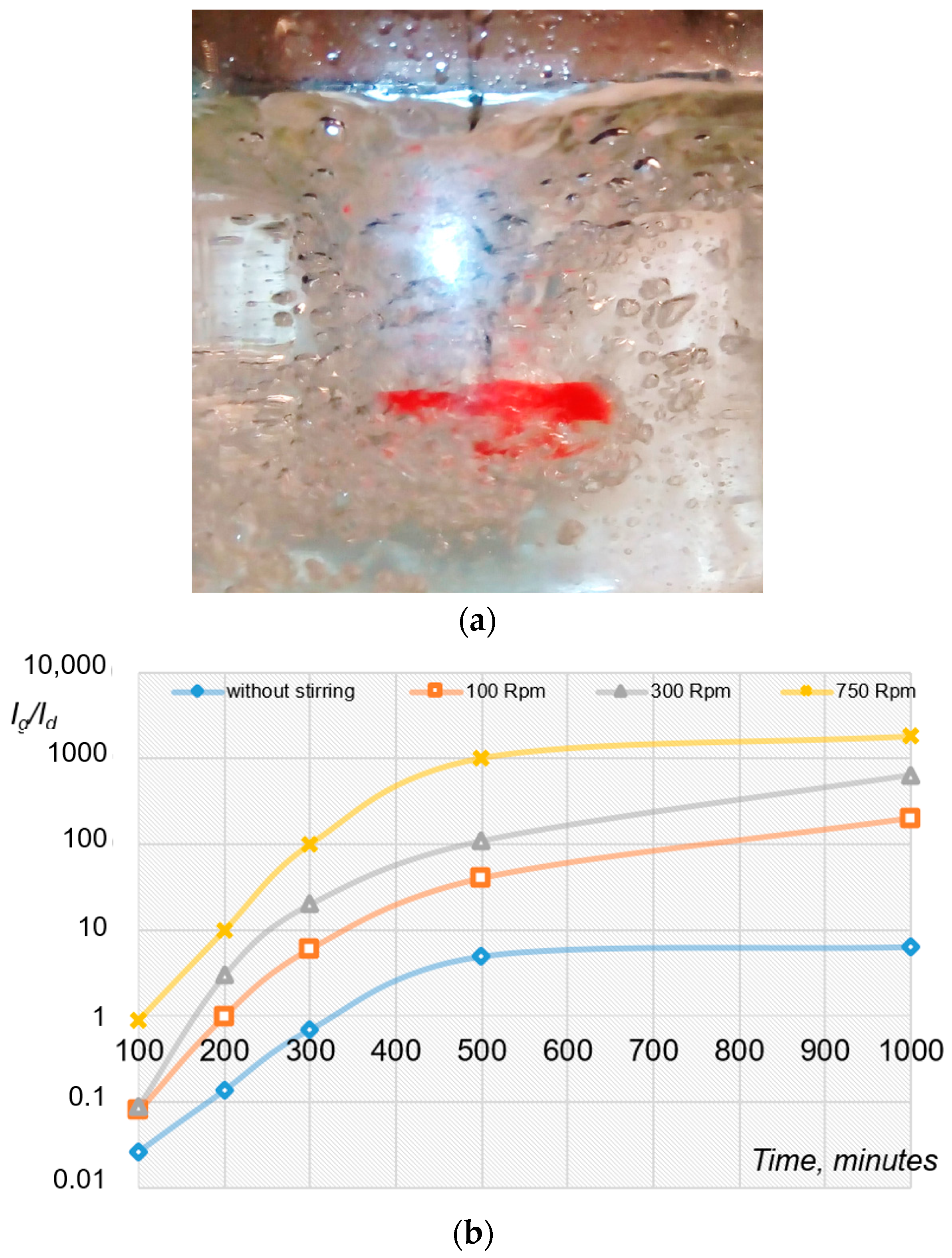
2.2. Mechanical Initiation of Hydrate Formation Processes Using Mixing Devices
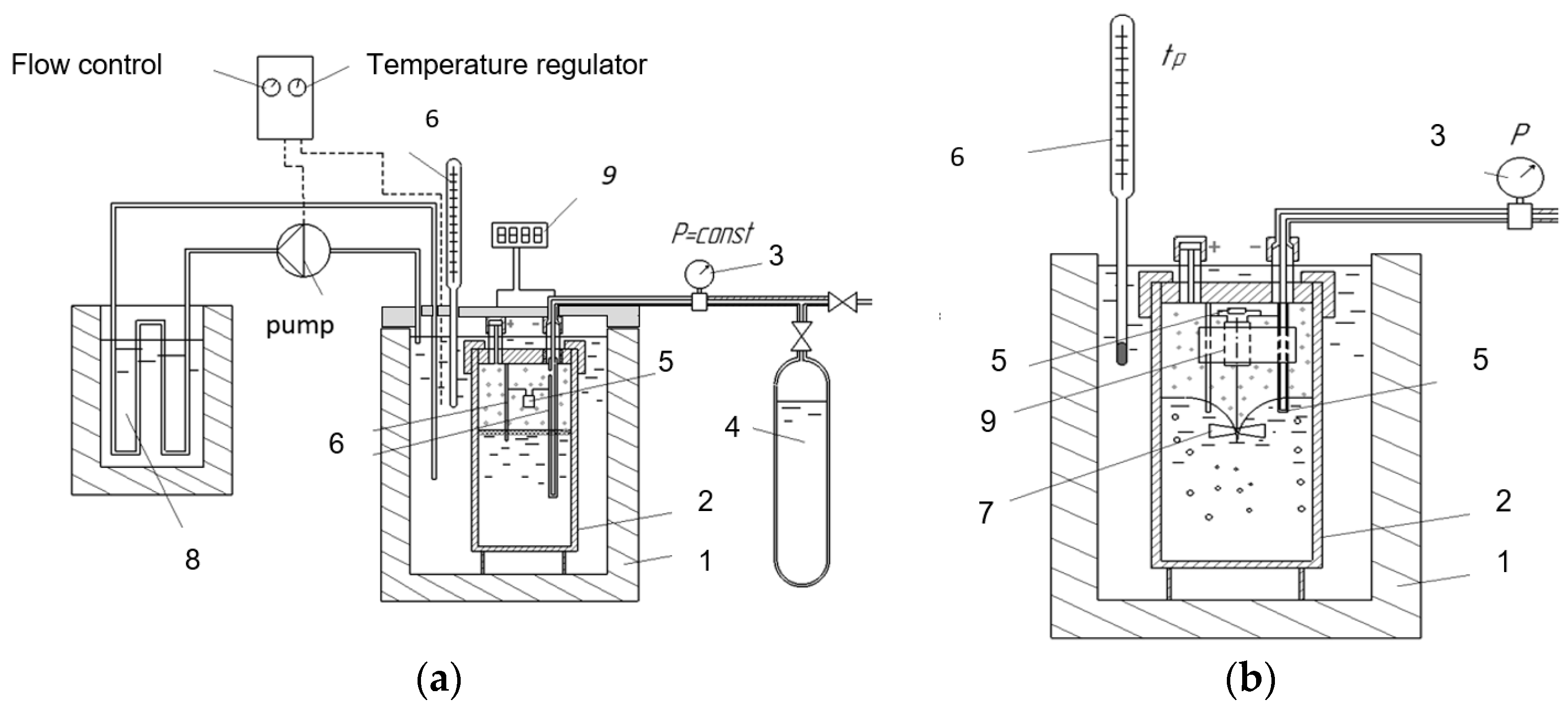
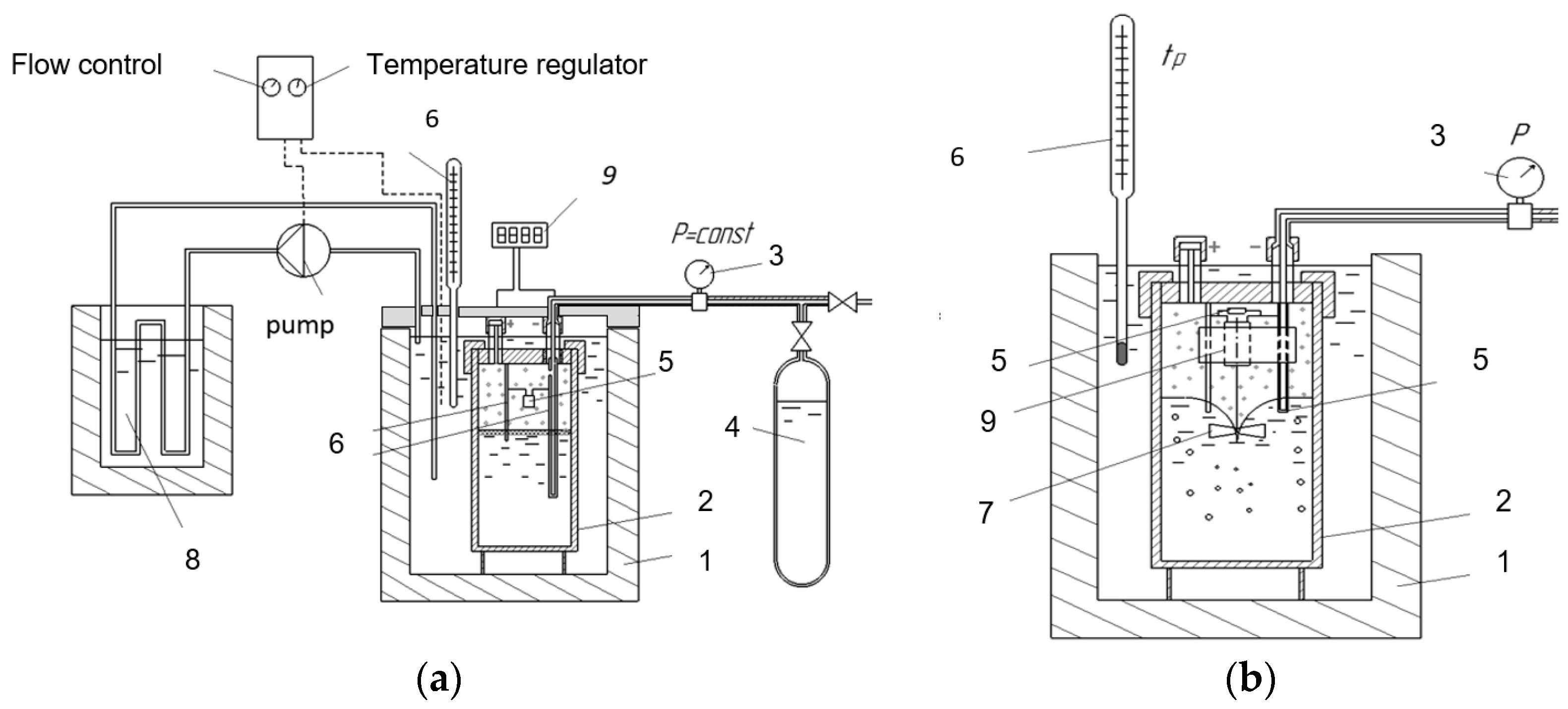
3. Materials and Methods
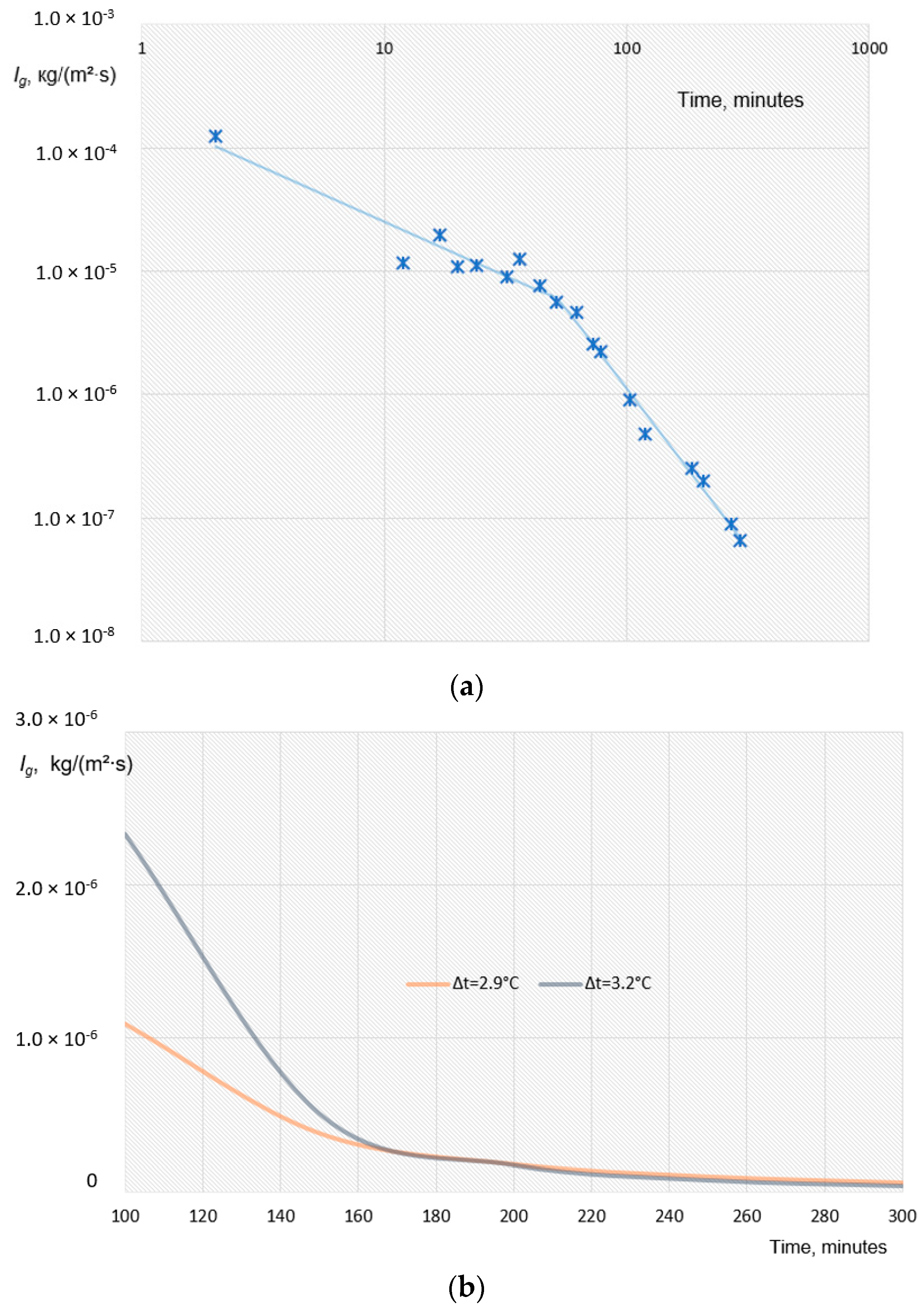
4. Results
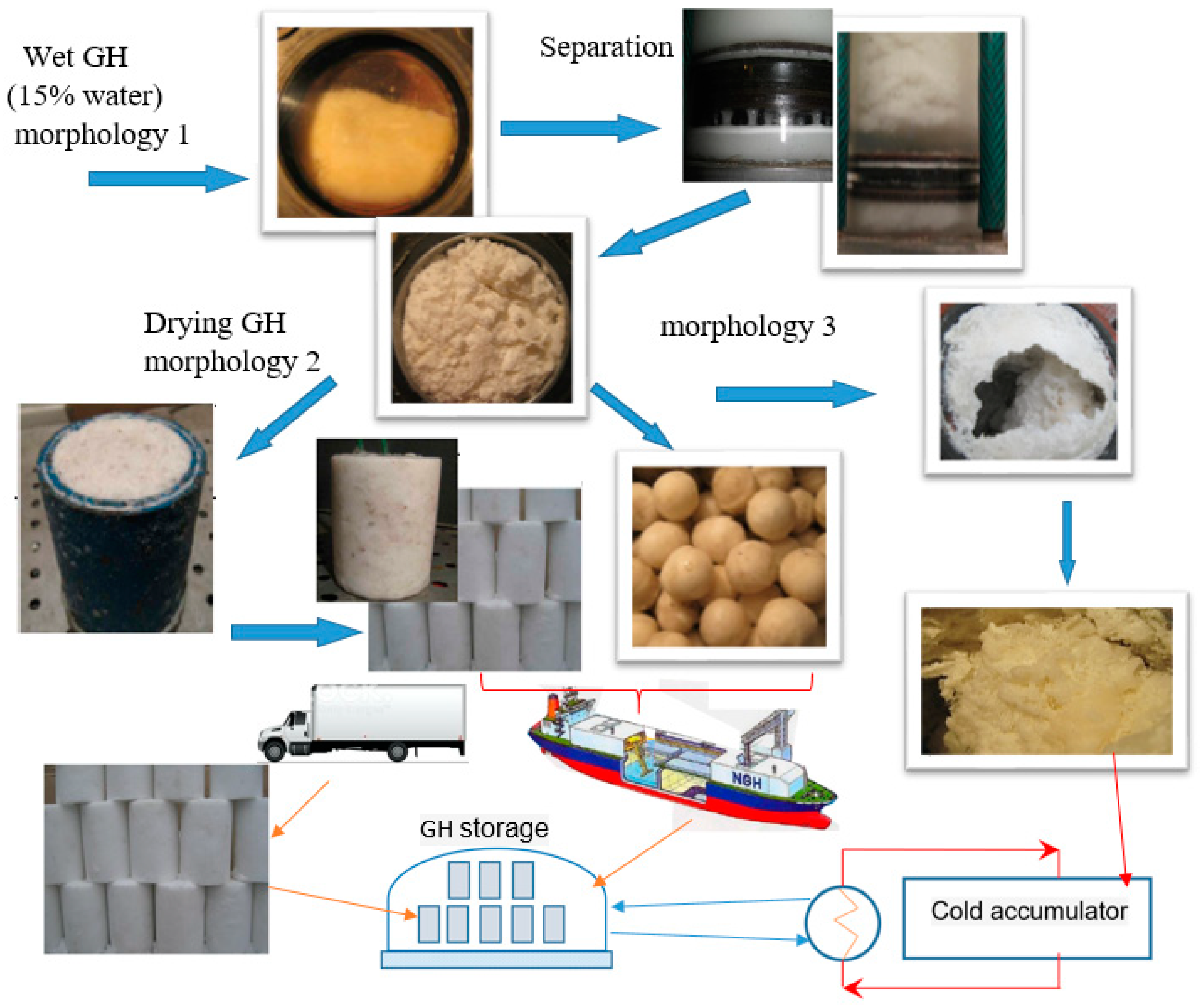
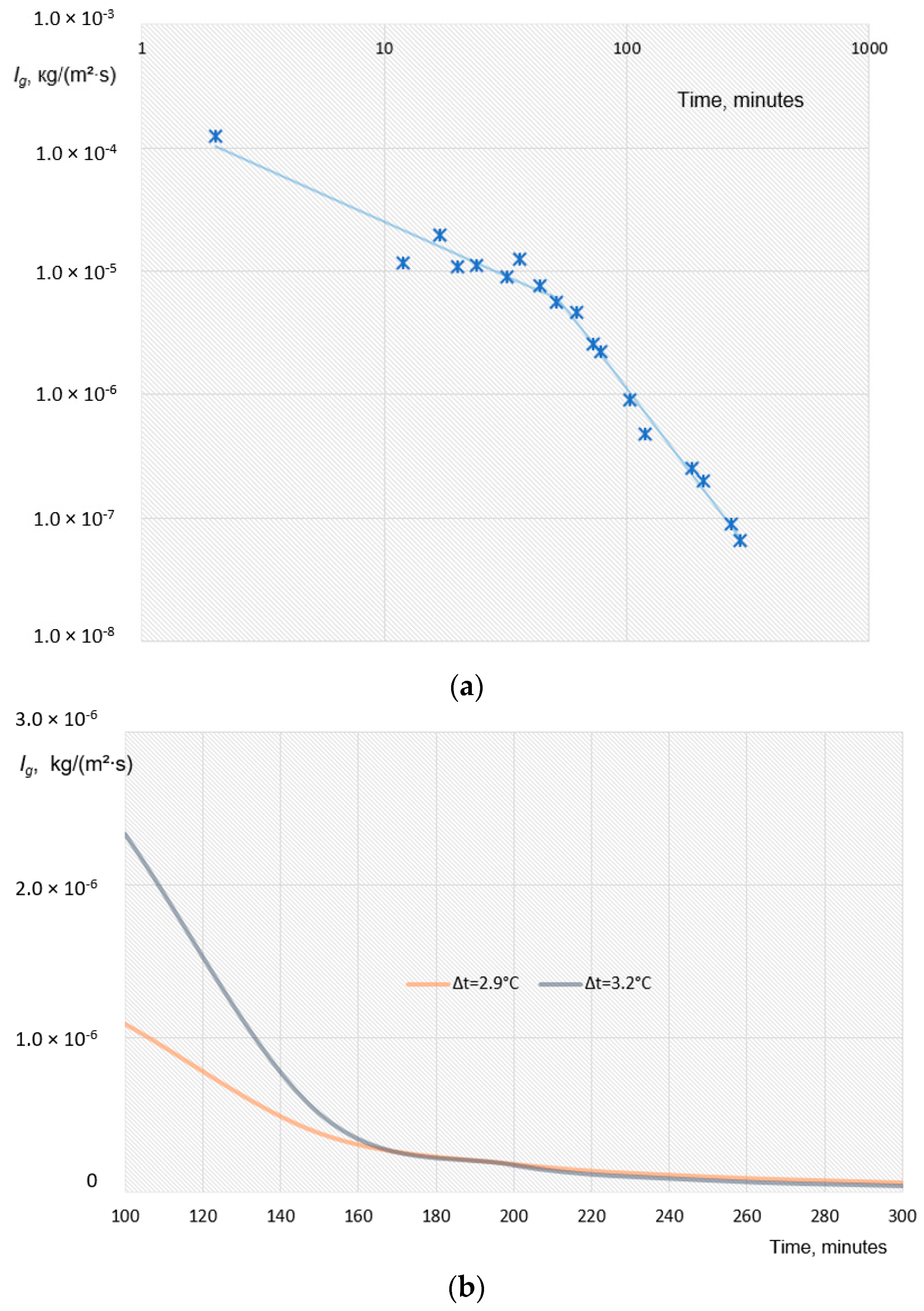
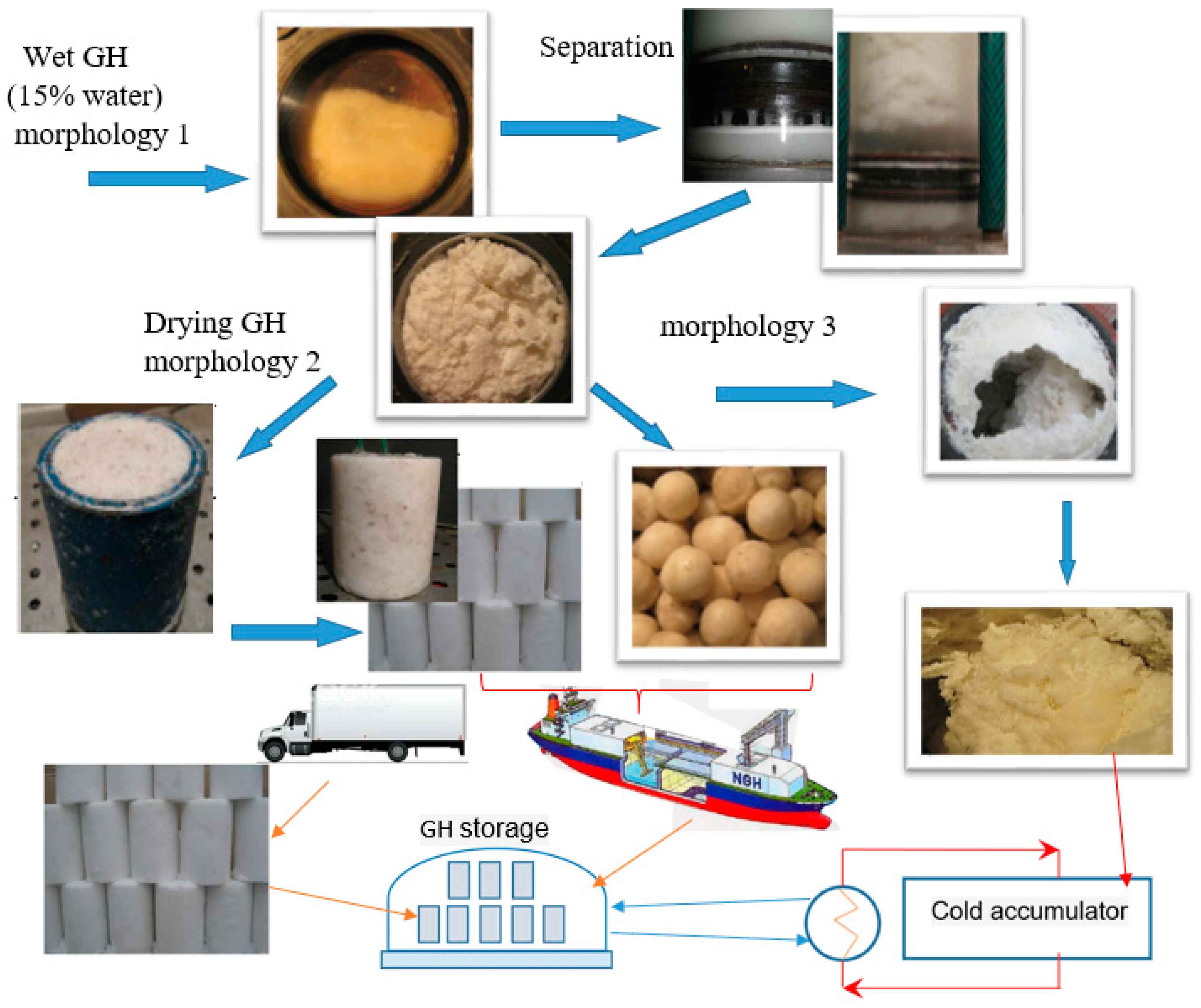
4.1. Water Removal from Gas Hydrate Mass
4.2. Hydrate Mass Drying
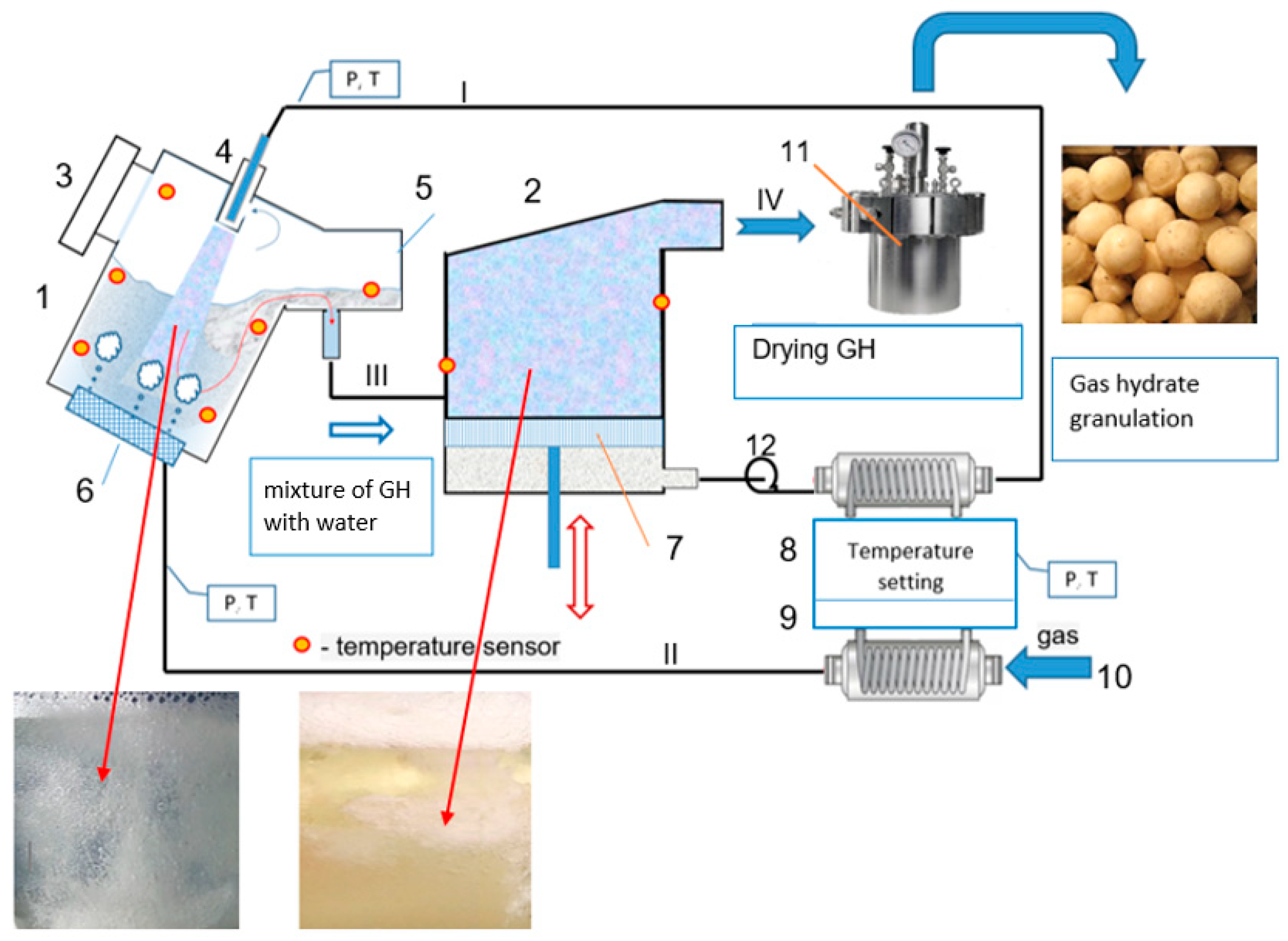
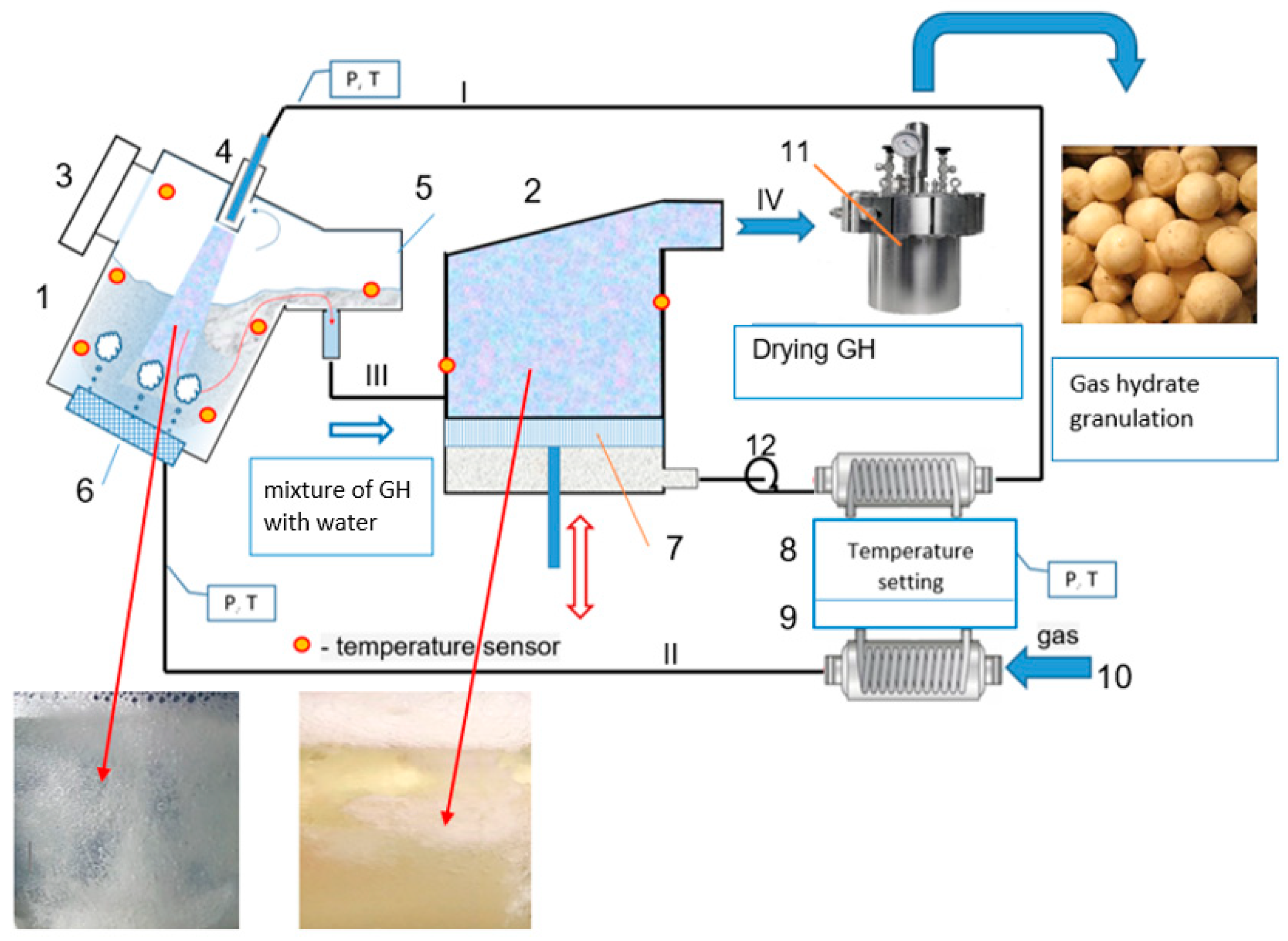
4.3. General Process Flow Diagram of GH Synthesis
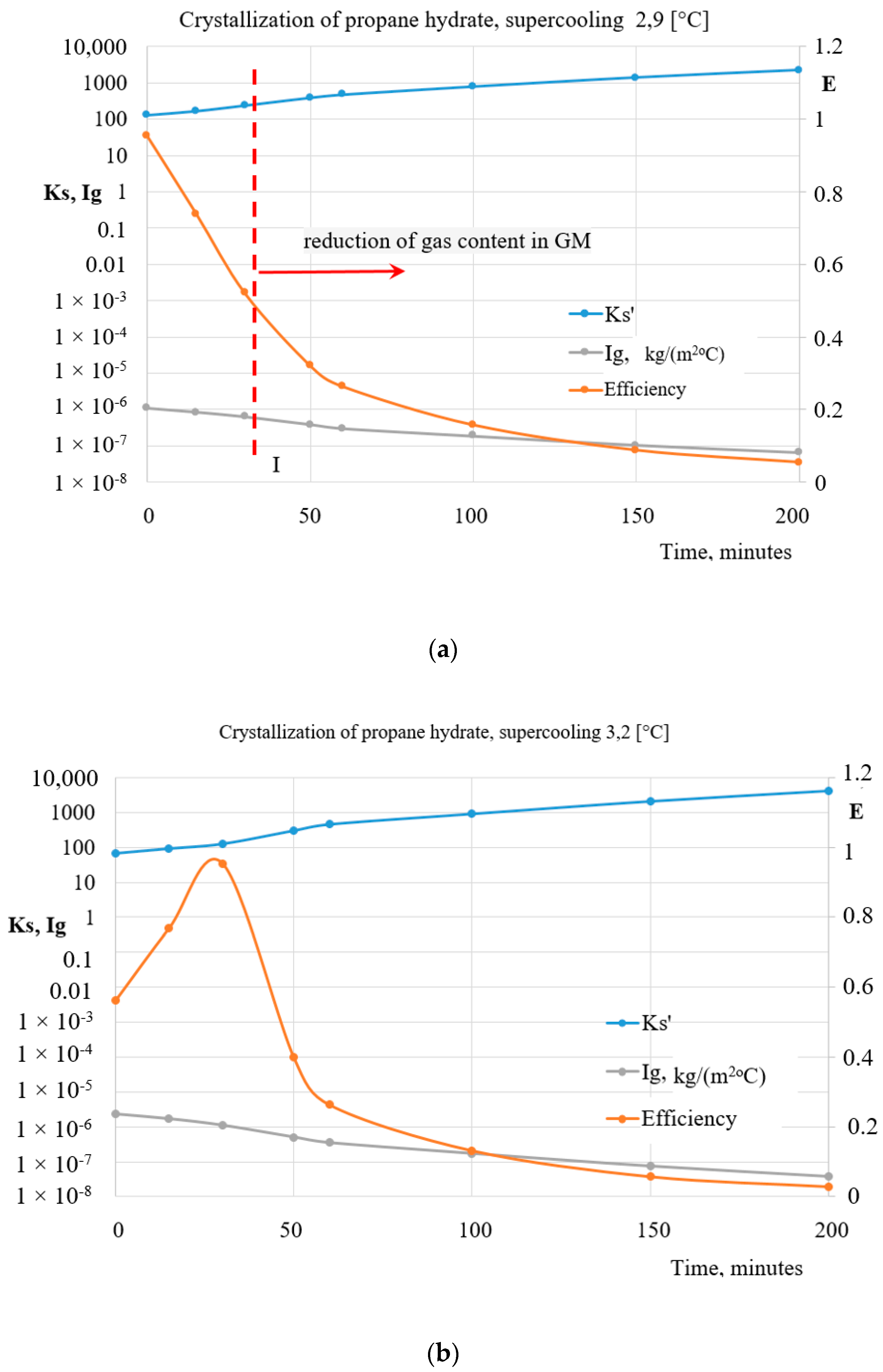
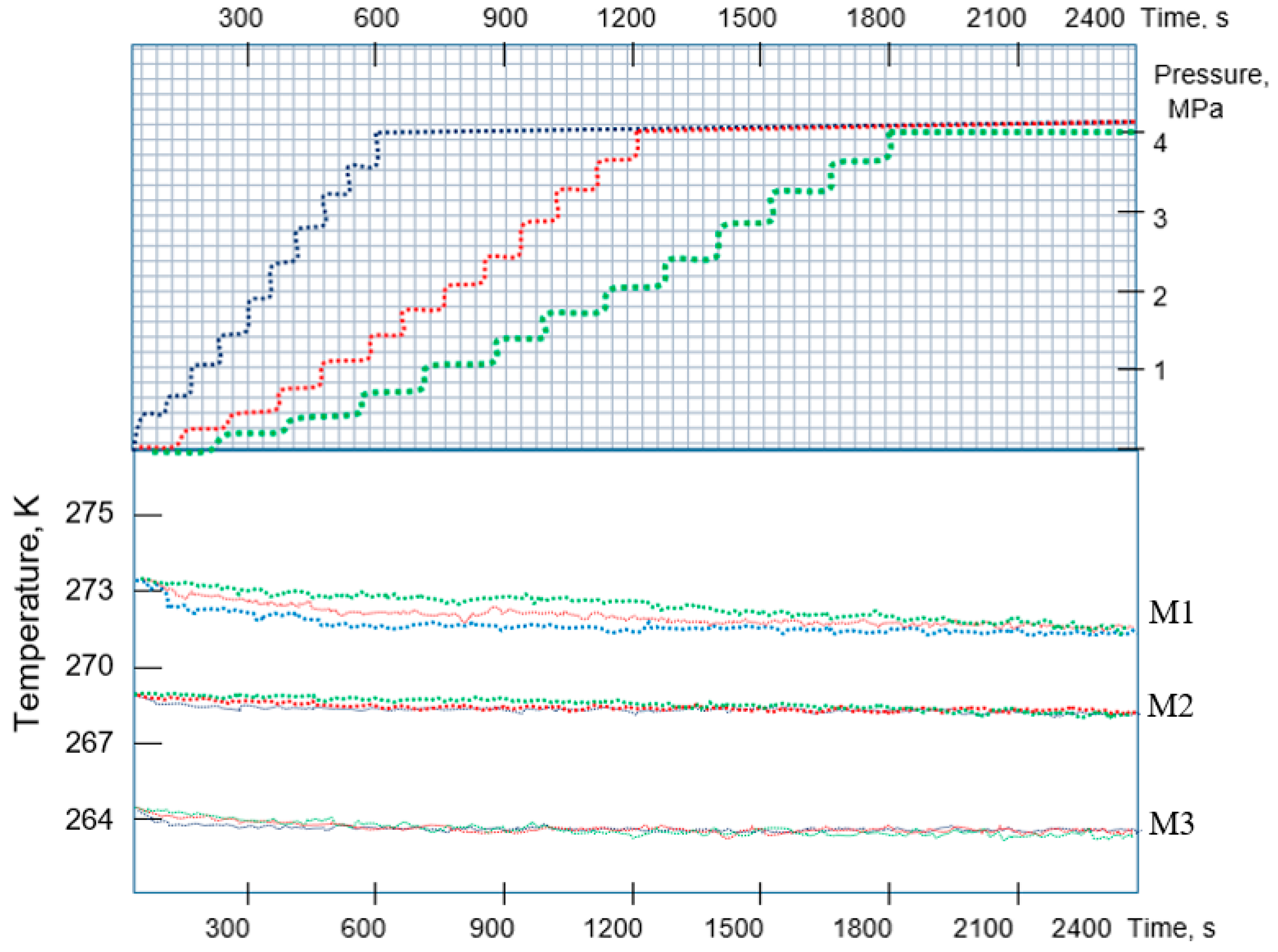
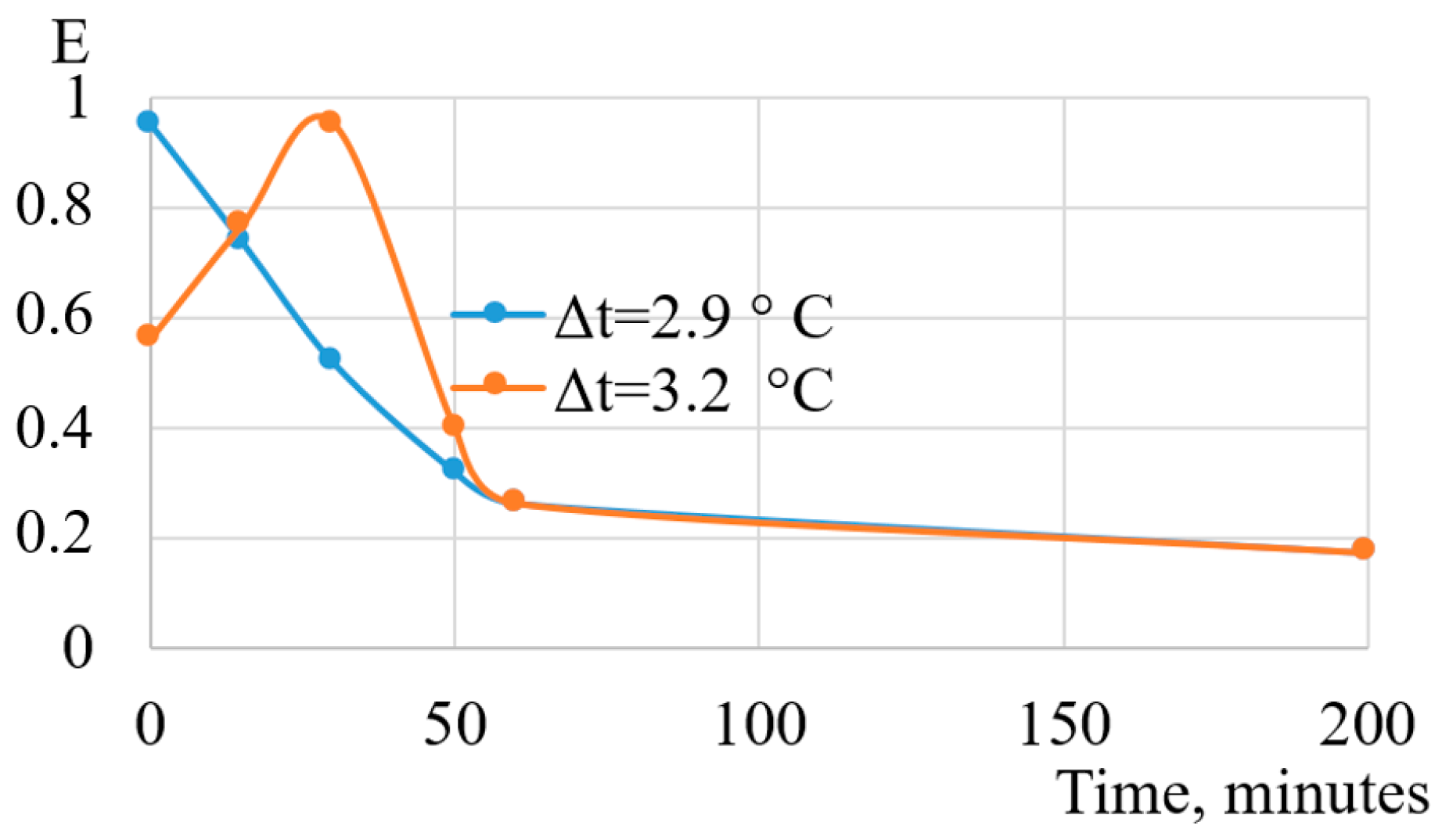
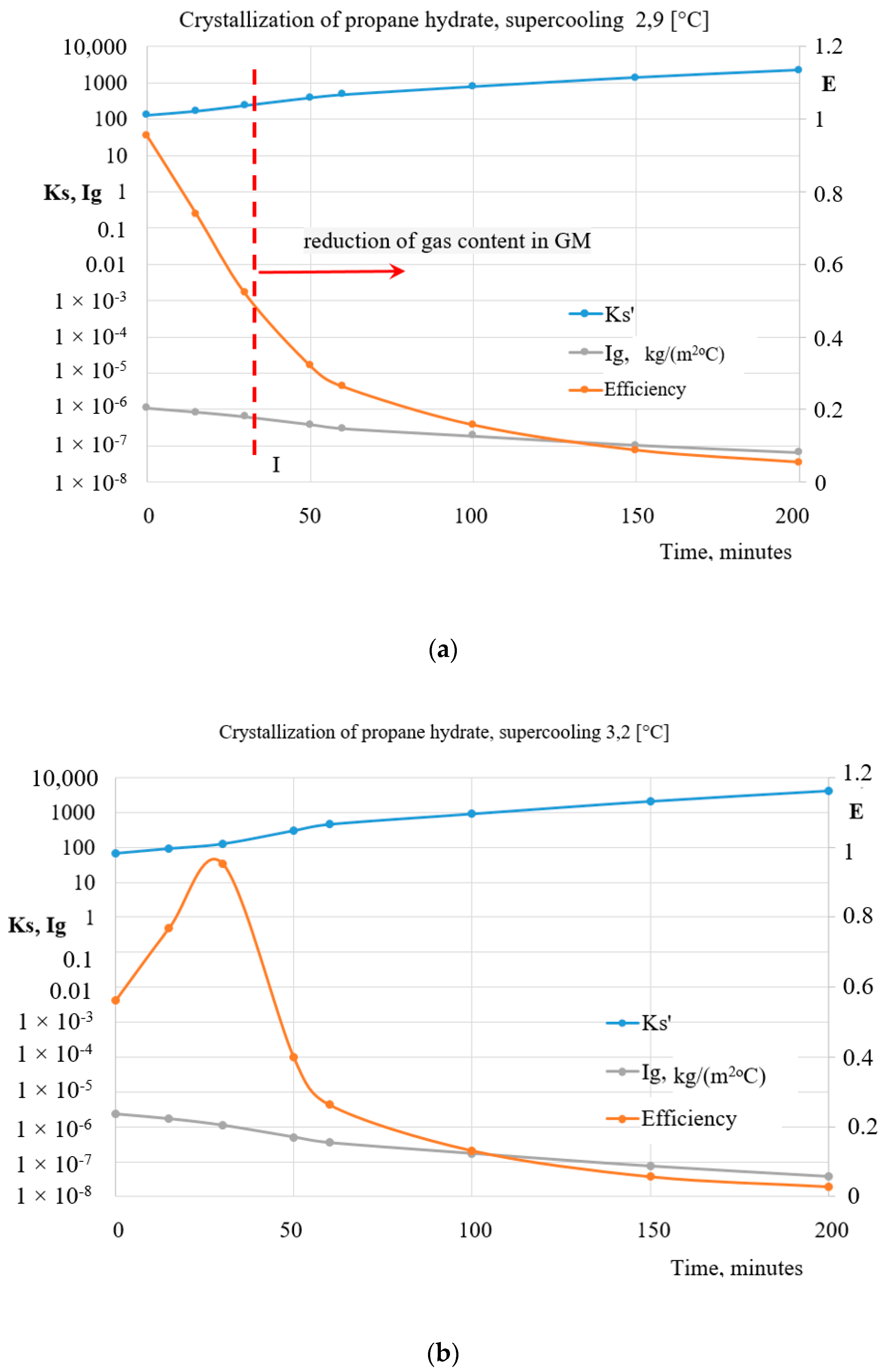
5. Heat Exchange Processes on the Interfacial Surface
6. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Koh, C.A.; Sum, A.K.; Sloan, E.D. State of the art: Natural gas hydrates as a natural resource. J. Nat. Gas Sci. Eng. 2012, 8, 132–138. [Google Scholar] [CrossRef]
- Boswell, R.; Hancock, S.; Yamamoto, K.; Collett, T.; Pratap, M.; Lee, S. Natural Gas Hydrates: Status of Potential as an Energy Resource. Future Energy, 3rd ed.; Improved Sustainable and Clean Options for our Planet; Elsevier: Amsterdam, The Netherlands, 2020; pp. 111–131. [Google Scholar] [CrossRef]
- Xu, H.; Kong, W.; Yang, F. Decomposition characteristics of natural gas hydrates in hydraulic lifting pipelines. Nat. Gas Ind. B 2019, 6, 159–167. [Google Scholar] [CrossRef]
- Alexei, V. Milkov Global estimates of hydrate-bound gas in marine sediments: How much is really out there? Earth-Sci. Rev. 2004, 66, 183–197. [Google Scholar]
- Boswell, R.; Collett, T.S. Collett Current perspectives on gas hydrate resources. Energy Environ. Sci. 2011, 4, 1045–1528. [Google Scholar] [CrossRef]
- Zhao, J.; Zhu, Z.; Song, Y.; Liu, W.; Zhang, Y.; Wang, D. Analysing the process of gas production for natural gas hydrate using depressurization. Appl. Energy 2015, 142, 125–134. [Google Scholar] [CrossRef]
- Taheri, Z.; Shabani, M.R.; Nazari, K.; Mehdizaheh, A. Natural gas transportation and storage by hydrate technology: Iran case study. J. Nat. Gas Sci. Eng. 2014, 21, 846–849. [Google Scholar] [CrossRef]
- Hassanpouryouzband, A.; Joonaki, E.; Farahani, M.V.; Takeya, S.; Ruppel, C.; Yang, J.; English, N.J.; Schicks, J.M.; Edlmann, K.; Mehrabian, H.; et al. Gas hydrates in sustainable chemistry. Chem. Soc. Rev. 2020, 49, 5225–5309. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Wang, F.; Tian, Y.; Wu, X.; Zheng, J.; Zhang, J.; Li, L.; Yang, P.; Zhao, J. Review and prospects of hydrate cold storage technology. Renew. Sustain. Energy Rev. 2020, 117, 109492. [Google Scholar] [CrossRef]
- Siažik, J.; Malcho, M. Accumulation of primary energy into natural gas hydrates. Procedia Eng. 2017, 192, 782–787. [Google Scholar] [CrossRef]
- Zhou, S.; Chen, W.; Li, Q.; Zhou, J.; Shi, H. Research on the solid fluidization well testing and production for shallow non-diagenetic natural gas hydrate in deep water area China Offshore. Oil Gas 2017, 29, 1–8. [Google Scholar]
- Bahadori, A. Chapter Liquefied Natural Gas (LNG). In Natural Gas Processing; Bahadori, A., Ed.; Gulf Professional Publishing: Boston, MA, USA, 2014; pp. 591–632. [Google Scholar]
- Kiran, B.S.; Sowjanya, K.; Prasad, P.S.; Yoon, J.-H. Experimental investigations on tetrahydrofuran-methanewater system: Rapid methane gas storage in hydrates. Oil Gas Sci. Technol.-Rev. D’IFP Energ. Nouv. 2019, 74, 12. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Sun, H.; Li, K.; Wang, D.; Yang, M. Experimental Investigation of Natural Gas Hydrate Production Characteristics via Novel Combination Modes of Depressurization With Water Flow Erosion. Fuel 2019, 252, 295–303. [Google Scholar] [CrossRef]
- Zhao, J.; Lv, Q.; Li, Y.; Yang, M.; Liu, W.; Yao, L.; Wang, S.; Zhang, Y.; Song, Y. In-situ visual observation for the formation and dissociation of methane hydrates in porous media by magnetic resonance imaging. Magn. Reson. Imaging 2015, 33, 485–490. [Google Scholar] [CrossRef] [PubMed]
- ZareNezhad, B.; Mottahedin, M.; Varaminian, F. Experimental and Theoretical Investigations on the Enhancement of Methane Gas Hydrate Formation Rate by Using the Kinetic Additives. Pet. Sci. Technol. 2015, 33, 857–864. [Google Scholar] [CrossRef]
- Koh, D.Y.; Kang, H.; Lee, J.W.; Park, Y.; Kim, S.J.; Lee, J.; Lee, J.Y.; Lee, H. Energy–efficient natural gas hydrate production using gas exchange. Appl. Energy 2016, 162, 114–130. [Google Scholar] [CrossRef]
- Kumar, K.V.; Preuss, K.; Titirici, M.-M.; Rodríguez-Reinoso, F. Nanoporous materials for the onboard storage of natural gas. Chem. Rev. 2017, 117, 1796–1825. [Google Scholar] [CrossRef]
- Song, Y.M.; Wang, F.; Guo, G.; Luo, S.-J.; Guo, R.B. Energy-efficient storage of methane in the formed hydrates with metal nanoparticles-grafted carbon nan- otubes as promoter. Appl. Energy 2018, 224, 175–183. [Google Scholar] [CrossRef]
- Chong, Z.R.; Yang, S.H.B.; Babu, P.; Linga, P.; Li, X.S. Review of natural gas hydrates as an energy resource: Prospects and challenges. Appl. Energy 2016, 162, 1633–1652. [Google Scholar] [CrossRef]
- Baek, S.; Ahn, Y.H.; Zhang, J.S.; Min, J.; Lee, H.; Lee, J.W. Enhanced methane hydrate formation with cyclopentane hydrate seeds. Appl. Energy 2017, 202, 32–41. [Google Scholar] [CrossRef]
- Veluswamy, H.P.; Kumar, A.; Kumar, R.; Linga, P. An innovative approach to enhance methane hydrate formation kinetics with leucine for energy storage application. Appl. Energy 2017, 188, 190–199. [Google Scholar] [CrossRef]
- Veluswamy, H.P.; Kumar, A.; Seo, Y.; Lee, J.D.; Linga, P. A review of solidified natural gas (SNG) technology for gas storage via clathrate hydrates. Appl. Energy 2018, 216, 262–285. [Google Scholar] [CrossRef]
- Zhang, P.; Chen, X.; Li, S.; Wu, Q.; Xu, Z. Heat transfer and water migration rules during formation/dissociation of methane hydrate under temperature fields with gradient. Int. J. Heat Mass Transf. 2021, 169, 120929. [Google Scholar] [CrossRef]
- He, Y.; Sun, M.-T.; Chen, C.; Zhang, G.-D.; Chao, K.; Lin, Y.; Wang, F. Surfactan—T-based promotion to gas hydrate formation for energy storage. J. Mater. Chem. 2019, A7, 21634–21661. [Google Scholar] [CrossRef]
- Kumari, A.; Balomajumder, C.; Arora, A.; Dixit, G.; Gomari, S.R. Physio-Chemical and Mineralogical Characteristics of Gas Hydrate-Bearing Sediments of the Kerala-Konkan, Krishna-Godavari, and Mahanadi Basins. J. Mar. Sci. Eng. 2021, 9, 808. [Google Scholar] [CrossRef]
- Thoutam, P.; Gomari, S.R.; Chapoy, A.; Ahmad, F.; Islam, M. Study on CO2 Hydrate Formation Kinetics in Saline Water in the Presence of Low Concentrations of CH4. ACS Omega 2019, 4, 18210–18218. [Google Scholar] [CrossRef] [Green Version]
- Arora, A.; Cameotra, S.S.; Kumar, R.; Balomajumder, C.; Singh, A.K.; Santhakumari, B.; Kumar, P.; Laik, S. Biosurfactant as a promoter of methane hydrate formation: Thermodynamic and kinetic studies. Sci. Rep. 2016, 6, 20893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Bhattacharjee, G.; Kulkarni, B.D.; Kumar, R. Role of surfactants in promoting gas hydrate formation. Ind. Eng. Chem. Res. 2015, 54, 12217–12232. [Google Scholar] [CrossRef]
- Veluswamy, H.P.; Hong, Q.W.; Linga, P. Morphology study of methane hydrate formation and dissociation in the presence of amino acid. Cryst. Growth Des. 2016, 16, 5932–5945. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, B.; Chen, Y.; Zhang, S.; Guo, W.; Cai, Y.; Tan, B.; Wang, W. Methane storage in a hydrated form as promoted by leucines for possible application to natural gas transportation and storage. Energy Technol. 2015, 3, 815–819. [Google Scholar] [CrossRef]
- Veluswamy, H.P.; Lee, P.Y.; Premasinghe, K.; Linga, P. Effect of biofriendly amino acids on the kinetics of methane hydrate formation and dissociation. Ind. Eng. Chem. Res. 2017, 56, 6145–6154. [Google Scholar] [CrossRef]
- Mohammad-Taheri, M.; Moghaddam, A.Z.; Nazari, K.; Zanjani, N.G. Methane hydrate stability in the presence of water-soluble hydroxyalkyl cellulose. J. Nat. Gas. Chem. 2012, 21, 119–125. [Google Scholar] [CrossRef]
- Gnanendran, N.; Amin, R. Modelling hydrate formation kinetics of a hydrate promoter–water–natural gas system in a semi-batch spray reactor. Chem. Eng. Sci. 2004, 59, 3849–3863. [Google Scholar] [CrossRef]
- Sowjanya, Y.; Prasad, P.S.R. Formation kinetics & phase stability of double hydrates of C4H8O and CO2/CH4: A comparison with pure systems. J. Nat. Gas Sci. Eng. 2014, 18, 58–63. [Google Scholar]
- Veluswamy, H.P.; Kumar, S.; Kumar, R.; Rangsunvigit, P.; Linga, P. Enhanced clathrate hydrate formation kinetics at near ambient temperatures and moderate pressures: Application to natural gas storage. Fuel 2016, 182, 907–919. [Google Scholar] [CrossRef]
- Delahaye, A.; Fournaison, L.; Marinhas, S.; Chatti, I.; Petitet, J.P.; Dalmazzone, D.; Fürst, W. Effect of THF on equilibrium pressure and dissociation enthalpy of CO2 hydrates applied to secondary refrigeration. Ind. Eng. Chem. Res. 2006, 45, 391–397. [Google Scholar] [CrossRef]
- Sharma, D.; Sowjanya, Y.; Chari, V.D.; Prasad, P.S.R. Methane storage in mixed hydrates with tetrahydrofuran. Indian J. Chem. Technol. 2014, 21, 114–119. [Google Scholar]
- Chernov, A.A.; Pil’Nik, A.A.; Elistratov, D.S.; Mezentsev, I.V.; Meleshkin, A.V.; Bartashevich, M.V.; Vlasenko, M.G. New hydrate formation methods in a liquid-gas medium. Sci. Rep. 2017, 7, 40809. [Google Scholar] [CrossRef] [Green Version]
- Lucia, B.; Castellani, B.; Rossi, F.; Cotana, F.; Morini, E.; Nicolini, A.; Filipponi, M. Experimental investigations on scaled-up methane hydrate production with surfactant promotion: Energy considerations. J. Pet. Sci. Eng. 2014, 120, 187–193. [Google Scholar] [CrossRef]
- Rossi, F.; Filipponi, M.; Castellani, B. Investigation on a novel reactor for gas hydrate production. Appl. Energy 2012, 99, 167–172. [Google Scholar] [CrossRef]
- Thoutam, P.; Gomari, S.; Ahmad, F.; Islam, M. Comparative analysis of hydrate nucleation for methane and carbon dioxide. Molecules 2019, 24, 1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filarsky, F.; Schmuck, C.; Schultz, H.J. Impact of Modified Silica Beads on Methane Hydrate Formation in a Fixed-Bed Reactor. Ind. Eng. Chem. Res. 2019, 58, 16687–16695. [Google Scholar] [CrossRef]
- Lang, X.; Fan, S.; Wang, Y. Intensification of methane and hydrogen storage in clathrate hydrate and future prospect. J. Nat. Gas Chem. 2010, 19, 203–209. [Google Scholar] [CrossRef]
- Murakami, T.; Kuritsuka, H.; Fujii, H.; Mori, Y.H. Forming a Structure-H Hydrate Using Water and Methylcyclohexane Jets Impinging on Each Other in a Methane Atmosphere. Energy Fuels 2009, 23, 1619–1625. [Google Scholar] [CrossRef]
- Anatoliy, M. Pavlenko Thermodynamic Features of the Intensive Formation of Hydrocarbon Hydrates. Energies 2020, 13, 3396. [Google Scholar] [CrossRef]
- Veluswamy, H.P.; Wong, A.J.H.; Babu, P.; Kumar, R.; Kulprathipanja, S.; Rangsunvigit, P.; Linga, P. Rapid methane hydrate formation to develop a cost effective large scale energy storage system. Chem. Eng. J. 2016, 290, 161–173. [Google Scholar] [CrossRef]
- Filarsky, F.; Wieser, J.; Schultz, H.J. Rapid Gas Hydrate Formation—Evaluation of Three Reactor Concepts and Feasibility Study. Molecules 2021, 26, 3615. [Google Scholar] [CrossRef] [PubMed]
- Linga, P.; Daraboina, N.; Ripmeester, J.A.; Englezos, P. Enhanced rate of gas hydrate formation in a fixed bed column filled with sand compared to a stirred vessel. Chem. Eng. Sci. 2012, 68, 617–623. [Google Scholar] [CrossRef]
- Qureshi, M.F.; Atilhan, M.; Altamash, T.; Aparicio, S.; Aminnaji, M.; Tohidi, B. High-pressure gas hydrate auto-clave hydraulic experiments and scale-up modeling on the effect of stirring RPM effect. J. Nat. Gas Sci. Eng. 2017, 38, 50–58. [Google Scholar] [CrossRef]
- Filarsky, F.; Schmuck, C.; Schultz, H.J. Development of a Surface-Active Coating for Promoted Gas Hydrate Formation. Chem. Ing. Tech. 2019, 91, 85–91. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Khan, M.N.; Peters, C.J.; Sloan, E.D.; Koh, C.A. Hydrate-Based Desalination Using Cyclopentane Hy-drates at Atmospheric Pressure. J. Chem. Eng. Data 2018, 63, 1081–1087. [Google Scholar] [CrossRef]
- Brown, T.D.; Taylor, C.E.; Bernardo, M.P. Rapid Gas Hydrate Formation Processes: Will They Work? Energies 2010, 3, 1154–1175. Available online: https://doaj.org/article/de59e138d3bd48f392f152cd9a68b233 (accessed on 7 June 2010). [CrossRef] [Green Version]
- Mori, Y. On the Scale-up of Gas-Hydrate-Forming Reactors: The Case of Gas-Dispersion-Type Reactors. Energies 2015, 8, 1317–1335. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, T.; Imura, N.; Ohmura, R.; Mori, Y.H. Clathrate Hydrate Formation by Water Spraying in a Me-thane + Ethane + Propane Gas Mixture: Search for the Rate-Controlling Mechanism of Hydrate Formation in the Presence of Methylcyclohexane. Energy Fuels 2007, 21, 545–553. Available online: https://keio.pure.elsevier.com/en/publications (accessed on 7 March 2007). [CrossRef]
- Al-Otaibi, F.; Clarke, M.; Maini, B.; Bishnoi, P.R. Kinetics of structure II gas hydrate formation for propane and ethane using an in-situ particle size analyzer and a Raman spectrometer. Chem. Eng. Sci. 2011, 66, 2468–2474. [Google Scholar] [CrossRef]
- Al-Otaibi, F.; Clarke, M.; Maini, B.; Bishnoi, P.R. Formation Kinetics of Structure I Clathrates of Methane and Ethane Using an in Situ Particle Size Analyzer. Energy Fuels 2010, 24, 5012–5022. [Google Scholar] [CrossRef]
- Kumar, A.; Maini, B.; Bishnoi, P.R.; Clarke, M. Investigation of the Variation of the Surface Area of Gas Hydrates during Dissociation by Depressurization in Porous Media. Energy Fuels 2013, 27, 5757–5769. [Google Scholar] [CrossRef]
- Kalogerakis, N.; Jamaluddin, A.; Dholabhai, P.; Bishnoi, P. Effect of Surfactants on Hydrate Formation Kinetics. In Proceedings of the SPE International Symposium on Oilfield Chemistry, New Orleans, LA, USA, 2–5 March 1993. [Google Scholar] [CrossRef]
- Vlasov, V.A.; Nesterov, A.N.; Reshetnikov, A.M. Kinetics of Gas Hydrate Film Growth along the Water–Gas Interface. Russ. J. Phys. Chem. 2020, 94, 1949–1951. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
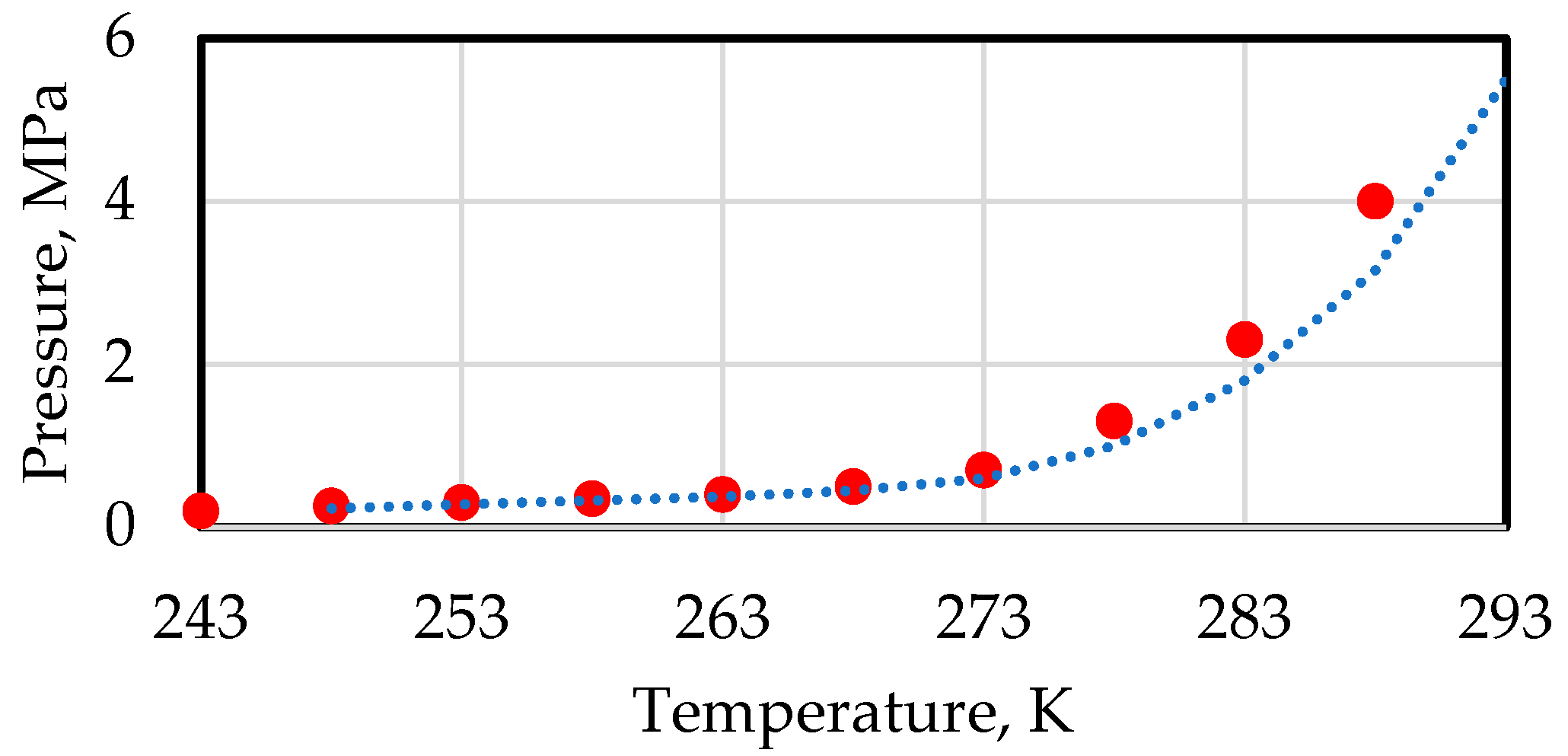
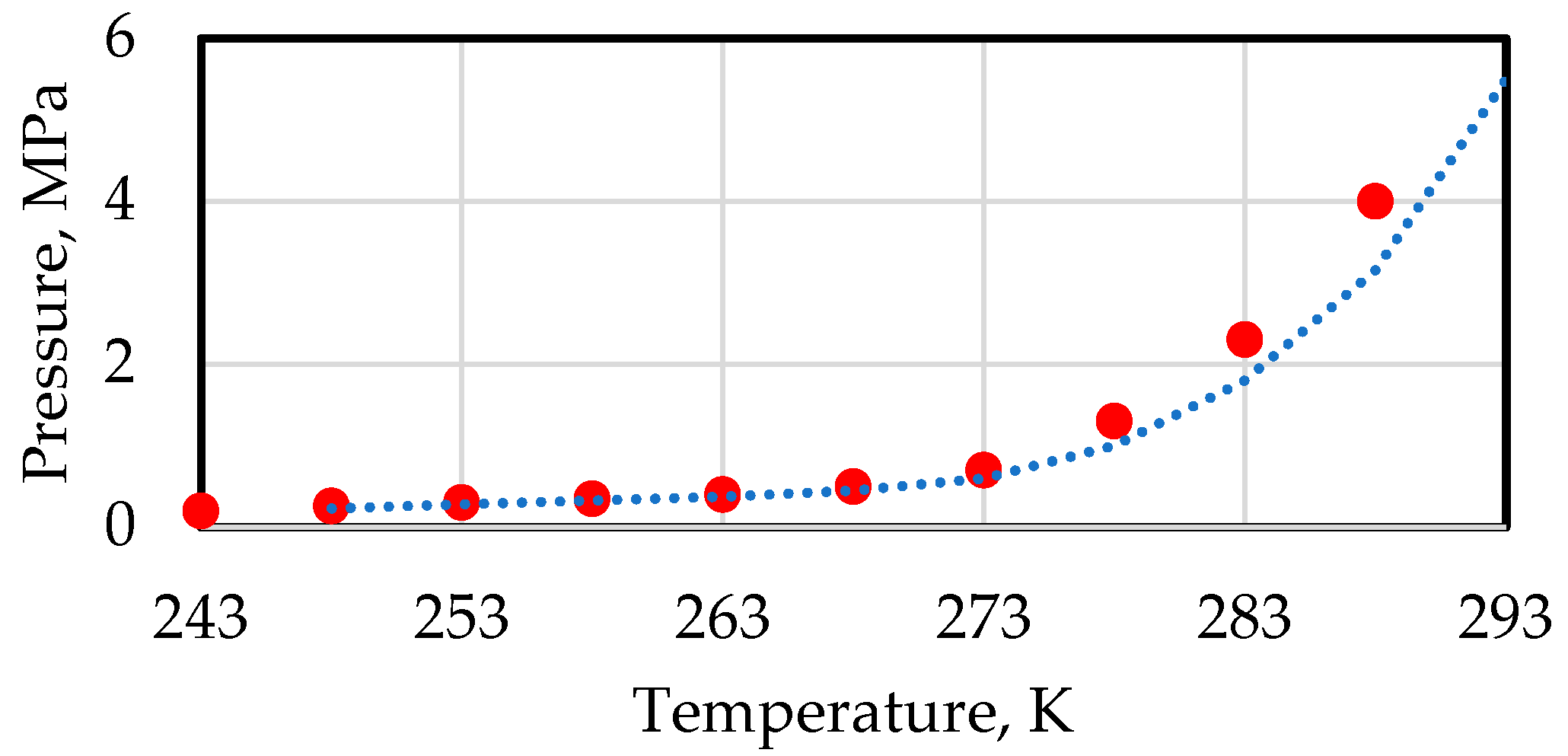
| Gas | Temperature Range, °C | Equilibrium Pressure, Pa (Absolute) |
|---|---|---|
| Methane | 0 < t < 22.0 | |
| t < 0 | ||
| Ethane | 0 < t < 14.5 | |
| t < 0 | ||
| Propane | 0 < t < 5.5 | |
| Isobutane | 0 < t < 2.5 |
| Gas | KS |
|---|---|
| Methane | 36–40 |
| Ethane | 55–57 |
| Propane | 115–124 |
| Isobutane | 173–186 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pavlenko, A.M.; Koshlak, H. Intensification of Gas Hydrate Formation Processes by Renewal of Interfacial Area between Phases. Energies 2021, 14, 5912. https://doi.org/10.3390/en14185912
Pavlenko AM, Koshlak H. Intensification of Gas Hydrate Formation Processes by Renewal of Interfacial Area between Phases. Energies. 2021; 14(18):5912. https://doi.org/10.3390/en14185912
Chicago/Turabian StylePavlenko, Anatoliy M., and Hanna Koshlak. 2021. "Intensification of Gas Hydrate Formation Processes by Renewal of Interfacial Area between Phases" Energies 14, no. 18: 5912. https://doi.org/10.3390/en14185912
APA StylePavlenko, A. M., & Koshlak, H. (2021). Intensification of Gas Hydrate Formation Processes by Renewal of Interfacial Area between Phases. Energies, 14(18), 5912. https://doi.org/10.3390/en14185912