
Current and Future Therapeutic Strategies for Limb Girdle Muscular Dystrophy Type R1: Clinical and Experimental Approaches



Abstract
1. Introduction
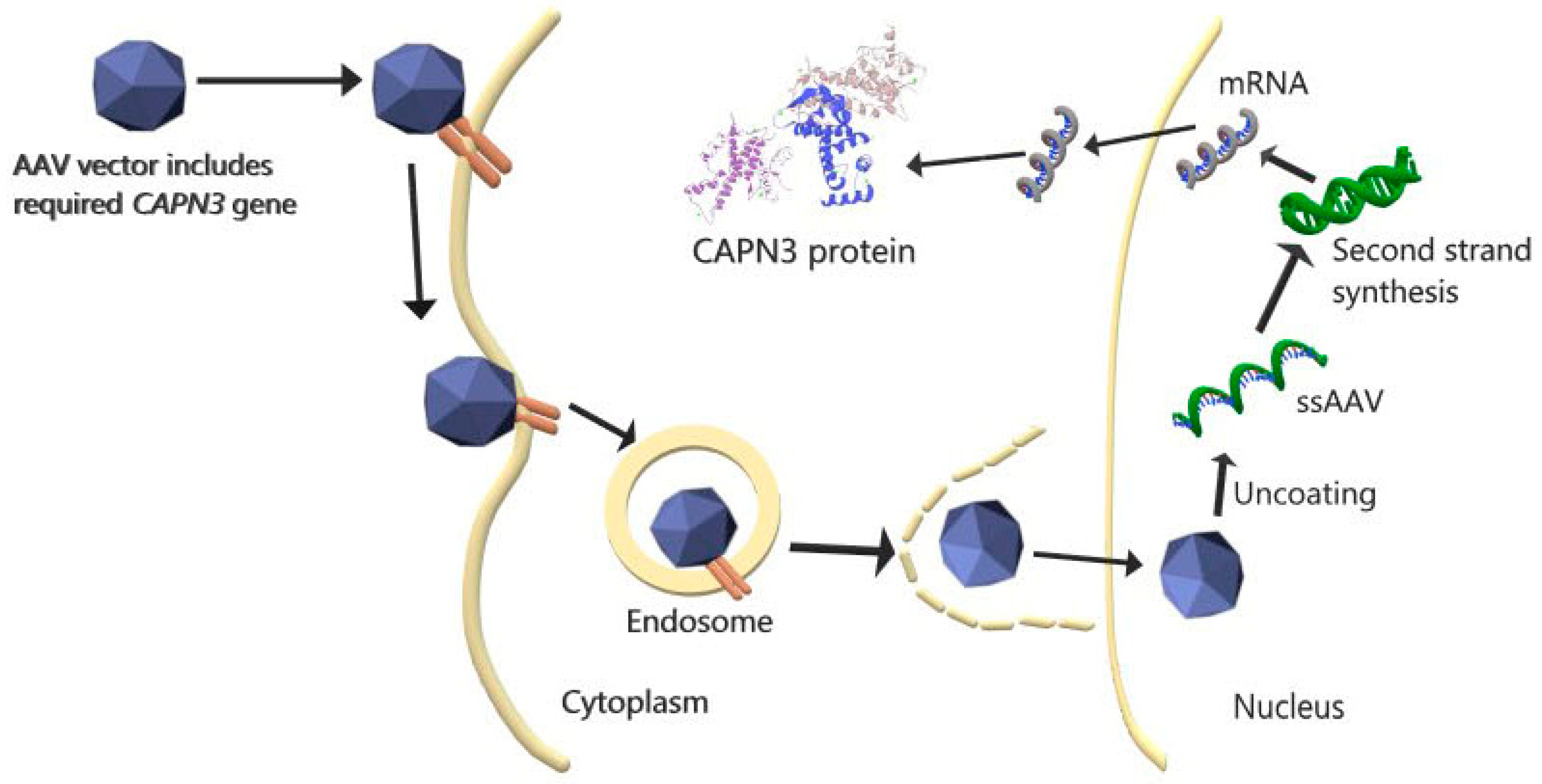
2. Current Clinical and Experimental Studies
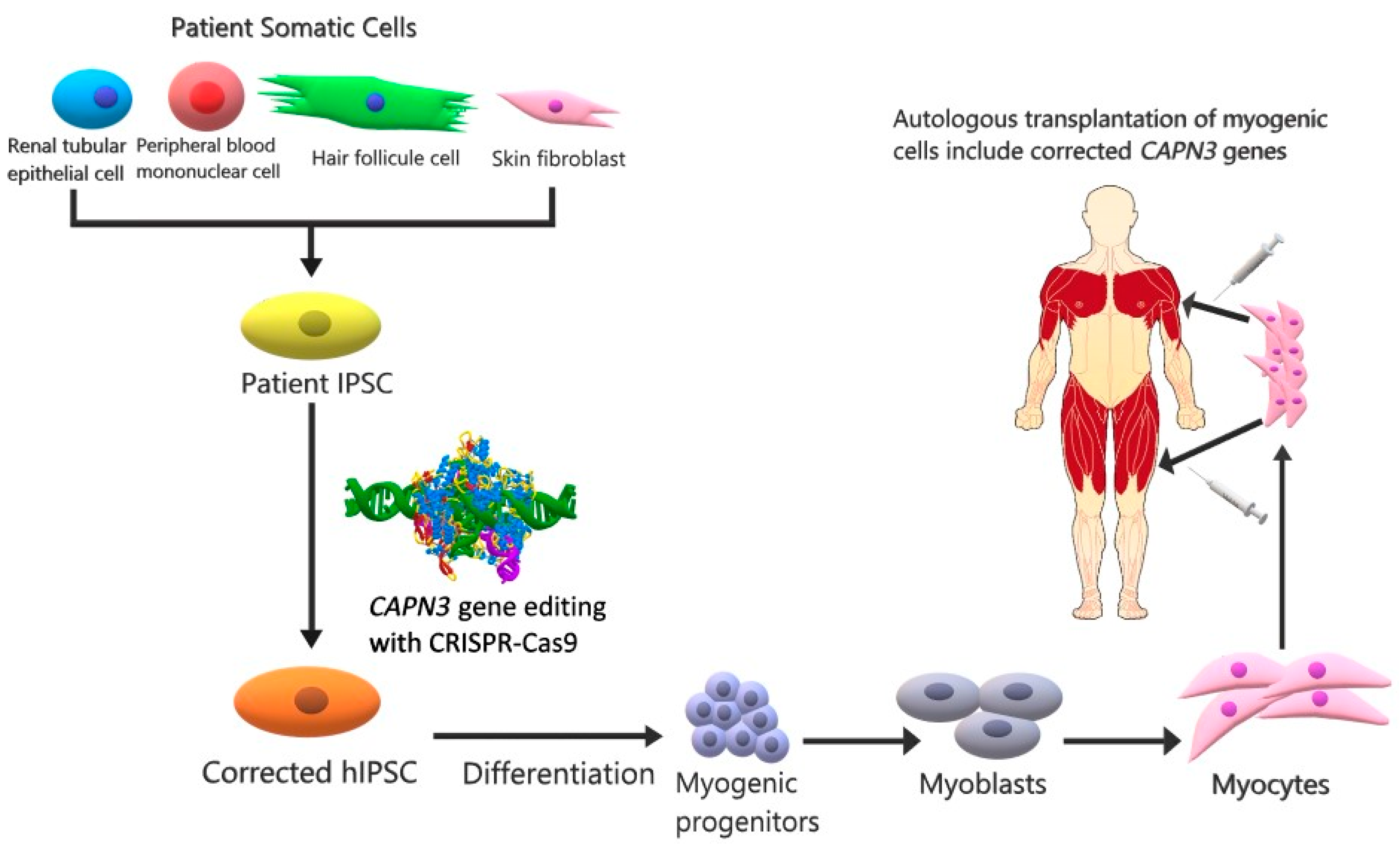
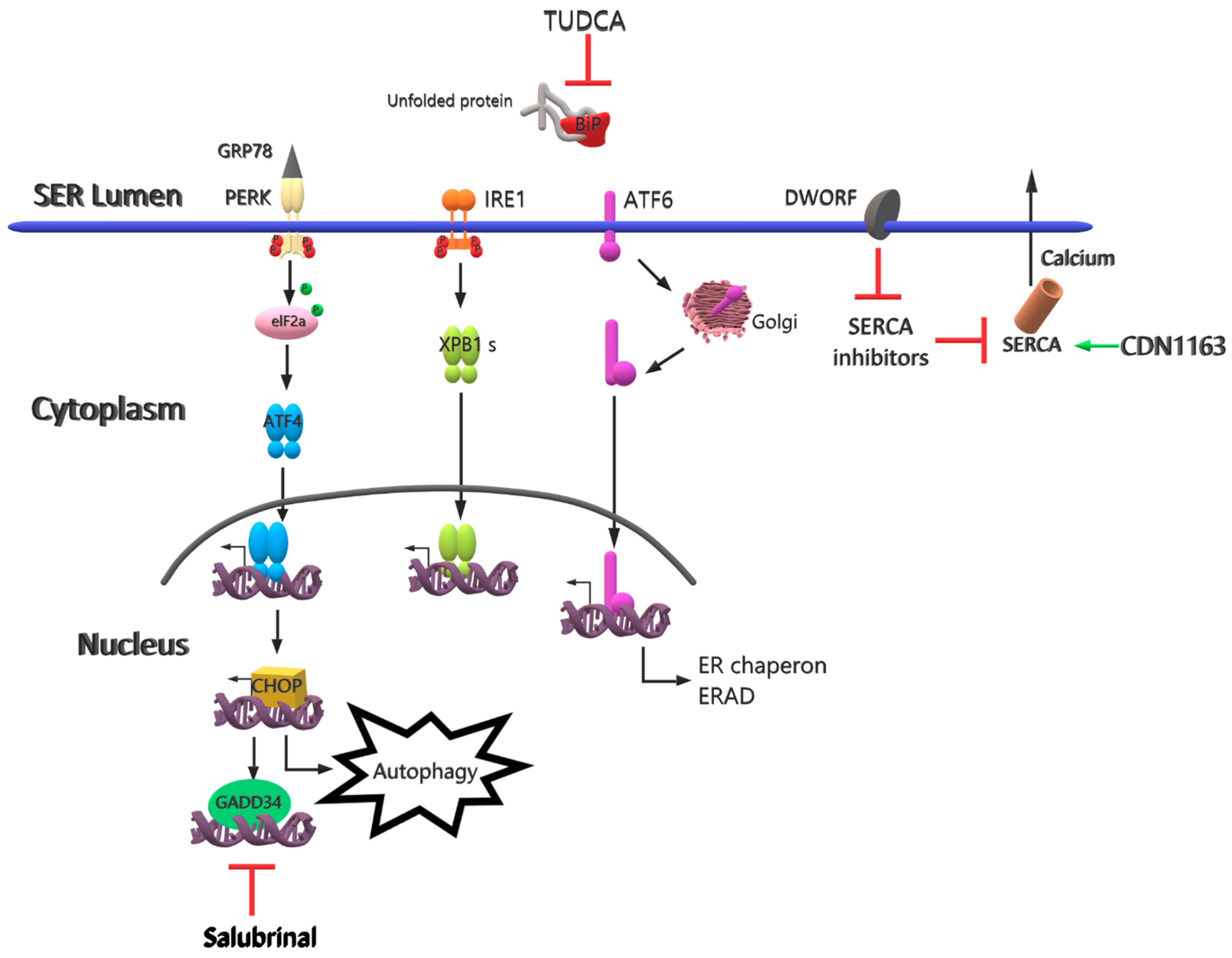
3. Future Therapy Strategies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AAV | Adeno-associated virus |
| AAVrh74.tMCK.hCAPN3 | AAV rhesus 74 truncated muscle creatine kinase human CAPN3 |
| AMBMP | 2-amino-4-(3,4-(methylenedioxy)benzylamino)-6-(3-methoxyphenyl) pyrimidine |
| ATF6 | Activating transcription factor 6 |
| C3KO | Calpain 3 knock out |
| CaMKII | Ca2+/calmodulin-dependent protein kinase II |
| CaMKII | Ca2+/calmodulin (CaM)-dependent protein kinase II |
| CAPN3 | Calpain 3 |
| Cas9 | Native Cas9 nuclease |
| CHOP | C/-EBP homologous protein |
| CRISPR-Cas9 | Clustered regularly interspaced short palindromic repeats CRISPR-associated proteins 9 |
| CSQ | Calsequestrin |
| DMD | Duchenne muscular dystrophy |
| elF2α | Eukaryotic initiation factor 2α |
| ER | Endoplasmic reticulum |
| ERAD | ER-associated protein degradation |
| FRZB | Frizzled-related protein |
| GRP78 | Glucose-regulated protein |
| IPSC | Induced pluripotent stem cell |
| IRE1α | Inositol-requiring enzyme 1α |
| LGMDR1 | Limb girdle muscular dystrophy R1 |
| LIM | Lin-11 Isl-1 Mec-3 |
| Mss51 | Mitochondrial translational activator |
| MuRF1 | Muscle RING-finger protein-1 |
| MYO-029 | Stamulumab |
| MyoD | Myogenic differentiation antigen |
| NCX3 | Na+-Ca2+ exchanger 3 |
| pAAV-CMV-mSeAPpropmyoD76A | Plasmid AAV-cytomegalovirus- murine-secreted alkaline phosphatase myogenic differentiation antigen murine-secreted alkaline phosphatase |
| Pax3/Pax7 | Paired box gene 3/Paired box gene 7 |
| PERK | PKR-like ER kinase |
| RyR1 | Ryanodine receptor 1 |
| SERCA | Sarcoplasmic/endoplasmic reticulum Ca2+ ATPase |
| TCF/LEF | T-cell factor/lymphoid enhancer factor |
| TRIM32 | Tripartite motif-containing protein 32 |
| TUDCA | Tauroursodeoxycholic acid |
| UPR | Unfolded protein response |
| WNT | Wingless-related integration site |
| XBP1 | X-box binding protein 1 |
References
- Richard, I.; Hogrel, J.Y.; Stockholm, D.; Payan, C.A.M.; Fougerousse, F.; Eymard, B.; Mignard, C.; Lopez de Munain, A.; Fardeau, M.; Urtizberea, J.A. Natural history of LGMD2A for delineating outcome measures in clinical trials. Ann. Clin. Transl. Neurol. 2016, 3, 248–265. [Google Scholar] [CrossRef] [PubMed]
- OMIM—Online Mendelian Inheritance in Man. Available online: https://www.omim.org/ (accessed on 20 October 2020).
- Richard, I.; Broux, O.; Allamand, V.; Fougerousse, F.; Chiannilkulchai, N.; Bourg, N.; Brenguier, L.; Devaud, C.; Pasturaud, P.; Roudaut, C.; et al. Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell 1995, 81, 27–40. [Google Scholar] [CrossRef]
- Vissing, J.; Barresi, R.; Witting, N.; Van Ghelue, M.; Gammelgaard, L.; Bindoff, L.A.; Straub, V.; Lochmüller, H.; Hudson, J.; Wahl, C.M.; et al. A heterozygous 21-bp deletion in CAPN3 causes dominantly inherited limb girdle muscular dystrophy. Brain 2016, 139, 2154–2163. [Google Scholar] [CrossRef]
- Lasa-Elgarresta, J.; Mosqueira-Martín, L.; Naldaiz-Gastesi, N.; Sáenz, A.; de Munain, A.L.; Vallejo-Illarramendi, A. Calcium Mechanisms in Limb-Girdle Muscular Dystrophy with CAPN3 Mutations. Int. J. Mol. Sci. 2019, 20, 4548. [Google Scholar] [CrossRef]
- Calpainopathy-GeneReviews®-NCBI Bookshelf. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1313/#lgmd2a.Molecular_Genetics (accessed on 4 May 2021).
- The CAPN3 gene homepage—Global Variome shared LOVD. Available online: https://databases.lovd.nl/shared/genes/CAPN3 (accessed on 10 October 2020).
- Sorimachi, H.; Imajoh-Ohmi, S.; Emori, Y.; Kawasaki, H.; Ohno, S.; Minami, Y.; Suzuki, K. Molecular cloning of a novel mammalian calcium-dependent protease distinct from both m- and μ-types. Specific expression of the mRNA in skeletal muscle. J. Biol. Chem. 1989, 264, 20106–20111. [Google Scholar] [CrossRef]
- Ojima, K.; Ono, Y.; Ottenheijm, C.; Hata, S.; Suzuki, H.; Granzier, H.; Sorimachi, H. Non-proteolytic functions of calpain-3 in sarcoplasmic reticulum in skeletal muscles. J. Mol. Biol. 2011, 407, 439–449. [Google Scholar] [CrossRef]
- Toral-Ojeda, I.; Aldanondo, G.; Lasa-Elgarresta, J.; Lasa-Fernandez, H.; Vesga-Castro, C.; Mouly, V.; de Munain, A.L.; Vallejo-Illarramendi, A. A Novel Functional In Vitro Model that Recapitulates Human Muscle Disorders. In Muscle Cell and Tissue-Current Status of Research Field; InTech: London, UK, 2018. [Google Scholar]
- Kramerova, I.; Kudryashova, E.; Ermolova, N.; Saenz, A.; Jaka, O.; López de munain, A.; Spencer, M.J. Impaired calcium calmodulin kinase signaling and muscle adaptation response in the absence of calpain 3. Hum. Mol. Genet. 2012, 21, 3193–3204. [Google Scholar] [CrossRef]
- Michel, L.Y.M.; Hoenderop, J.G.J.; Bindels, R.J.M. Calpain-3-mediated regulation of the Na+-Ca2+ exchanger isoform 3. Pflugers Arch. Eur. J. Physiol. 2016, 468, 243–255. [Google Scholar] [CrossRef]
- Ono, Y.; Ojima, K.; Torii, F.; Takaya, E.; Doi, N.; Nakagawa, K.; Hata, S.; Abe, K.; Sorimachi, H. Skeletal muscle-specific calpain is an intracellular Na+- dependent protease. J. Biol. Chem. 2010, 285, 22986–22998. [Google Scholar] [CrossRef]
- Ono, Y.; Ojima, K.; Shinkai-Ouchi, F.; Hata, S.; Sorimachi, H. An eccentric calpain, CAPN3/p94/calpain-3. Biochimie 2016, 122, 169–187. [Google Scholar] [CrossRef]
- Beckmann, J.S.; Spencer, M. Calpain 3, the “gatekeeper” of proper sarcomere assembly, turnover and maintenance. Neuromuscul. Disord. 2008, 18, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Sorimachi, H.; Toyama-Sorimachi, N.; Saido, T.C.; Kawasaki, H.; Sugita, H.; Miyasaka, M.; Arahata, K.I.; Ishiura, S.; Suzuki, K. Muscle-specific calpain, p94, is degraded by autolysis immediately after translation, resulting in disappearance from muscle. J. Biol. Chem. 1993, 268, 10593–10605. [Google Scholar] [CrossRef]
- de Paula, F.; Vainzof, M.; Passos-Bueno, M.R.; Pavanello, R.d.C.; Matioli, S.R.; Anderson, L.V.B.; Nigro, V.; Zatz, M. Clinical variability in calpainopathy: What makes the difference? Eur. J. Hum. Genet. 2002, 10, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Schessl, J.; Walter, M.C.; Schreiber, G.; Schara, U.; Müller, C.R.; Lochmüller, H.; Bönnemann, C.G.; Korinthenberg, R.; Kirschner, J. Phenotypic variability in siblings with calpainopathy (LGMD2A). Acta Myol. 2008, 27, 54–58. [Google Scholar] [PubMed]
- de Albuquerque, M.A.V.; Abath Neto, O.; da Silva, F.M.A.; Zanoteli, E.; Reed, U.C. Limb-girdle muscular dystrophy type 2A in Brazilian children. Arq. Neuropsiquiatr. 2015, 73, 993–997. [Google Scholar] [CrossRef]
- Weekly Steroids in Muscular Dystrophy-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04054375 (accessed on 12 October 2020).
- Merlini, L.; Cicognani, A.; Malaspina, E.; Gennari, M.; Gnudi, S.; Talim, B.; Franzoni, E. Early prednisone treatment in Duchenne muscular dystrophy. Muscle Nerve 2003, 27, 222–227. [Google Scholar] [CrossRef]
- Mesa, L.E.; Dubrovsky, A.L.; Corderi, J.; Marco, P.; Flores, D. Steroids in duchenne muscular dystrophy—Deflazacort trial. Neuromuscul. Disord. 1991, 1, 261–266. [Google Scholar] [CrossRef]
- Hussein, M.R.; Hamed, S.A.; Mostafa, M.G.; Abu-Dief, E.E.; Kamel, N.F.; Kandil, M.R. The effects of glucocorticoid therapy on the inflammatory and Dendritic cells in muscular dystrophies. Int. J. Exp. Pathol. 2006, 87, 451–461. [Google Scholar] [CrossRef]
- Spencer, M.J.; Tidball, J.G. Do immune cells promote the pathology of dystrophin-deficient myopathies? Neuromuscul. Disord. 2001, 11, 556–564. [Google Scholar] [CrossRef]
- Gaud, A.; Simon, J.M.; Witzel, T.; Carre-Pierrat, M.; Wermuth, C.G.; Ségalat, L. Prednisone reduces muscle degeneration in dystrophin-deficient Caenorhabditis elegans. Neuromuscul. Disord. 2004, 14, 365–370. [Google Scholar] [CrossRef]
- Wagner, K.R.; Fleckenstein, J.L.; Amato, A.A.; Barohn, R.J.; Bushby, K.; Escolar, D.M.; Flanigan, K.M.; Pestronk, A.; Tawil, R.; Wolfe, G.I.; et al. A phase I/II trial of MYO-029 in adult subjects with muscular dystrophy. Ann. Neurol. 2008, 63, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Bartoli, M.; Poupiot, J.; Vulin, A.; Fougerousse, F.; Arandel, L.; Daniele, N.; Roudaut, C.; Noulet, F.; Garcia, L.; Danos, O.; et al. AAV-mediated delivery of a mutated myostatin propeptide ameliorates calpain 3 but not α-sarcoglycan deficiency. Gene Ther. 2007, 14, 733–740. [Google Scholar] [CrossRef][Green Version]
- Kramerova, I.; Marinov, M.; Owens, J.; Lee, S.J.; Becerra, D.; Spencer, M.J. Myostatin inhibition promotes fast fibre hypertrophy but causes loss of AMP-activated protein kinase signalling and poor exercise tolerance in a model of limb-girdle muscular dystrophy R1/2A. J. Physiol. 2020, 598, 3927–3939. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, M.; Ishida, J.; Ebner, N.; Anker, S.D.; Von Haehling, S. Myostatin inhibitors as pharmacological treatment for muscle wasting and muscular dystrophy. JCSM Clin. Rep. 2017, 2, 1–10. [Google Scholar] [CrossRef]
- Mariot, V.; Joubert, R.; Hourdé, C.; Féasson, L.; Hanna, M.; Muntoni, F.; Maisonobe, T.; Servais, L.; Bogni, C.; Le Panse, R.; et al. Downregulation of myostatin pathway in neuromuscular diseases may explain challenges of anti-myostatin therapeutic approaches. Nat. Commun. 2017, 8, 1–8. [Google Scholar] [CrossRef]
- Roudaut, C.; Le Roy, F.; Suel, L.; Poupiot, J.; Charton, K.; Bartoli, M.; Richard, I. Restriction of calpain3 expression to the skeletal muscle prevents cardiac toxicity and corrects pathology in a murine model of limb-girdle muscular dystrophy. Circulation 2013, 128, 1094–1104. [Google Scholar] [CrossRef]
- Yalvac, M.E.; Amornvit, J.; Braganza, C.; Chen, L.; Hussain, S.R.A.; Shontz, K.M.; Montgomery, C.L.; Flanigan, K.M.; Lewis, S.; Sahenk, Z. Impaired regeneration in calpain-3 null muscle is associated with perturbations in mTORC1 signaling and defective mitochondrial biogenesis. Skelet. Muscle 2017, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Bartoli, M.; Roudaut, C.; Martin, S.; Fougerousse, F.; Suel, L.; Poupiot, J.; Gicquel, E.; Noulet, F.; Danos, O.; Richard, I. Safety and efficacy of AAV-mediated calpain 3 gene transfer in a mouse model of limb-girdle muscular dystrophy Type 2A. Mol. Ther. 2006, 13, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Lostal, W.; Roudaut, C.; Faivre, M.; Charton, K.; Suel, L.; Bourg, N.; Best, H.; Smith, J.E.; Gohlke, J.; Corre, G.; et al. Titin splicing regulates cardiotoxicity associated with calpain 3 gene therapy for limb-girdle muscular dystrophy type 2A. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Chicoine, L.G.; Al-Zaidy, S.A.; Sahenk, Z.; Lehman, K.; Lowes, L.; Miller, N.; Alfano, L.; Galliers, B.; Lewis, S.; et al. Gene Delivery for Limb-Girdle Muscular Dystrophy Type 2D by Isolated Limb Infusion. Hum. Gene Ther. 2019, 30, 794–801. [Google Scholar] [CrossRef]
- Chicoine, L.G.; Rodino-Klapac, L.R.; Shao, G.; Xu, R.; Bremer, W.G.; Camboni, M.; Golden, B.; Montgomery, C.L.; Shontz, K.; Heller, K.N.; et al. Vascular delivery of rAAVrh74.MCK.GALGT2 to the gastrocnemius muscle of the rhesus macaque stimulates the expression of dystrophin and laminin α2 surrogates. Mol. Ther. 2014, 22, 713–724. [Google Scholar] [CrossRef]
- 1135—Biopotency and Biodistribution/ Toxicology Studies Following Systemic Gene Therapy with AAVrh74.tMCK.hCAPN3 in the Mouse Model for LGMD2A-ASGCT 23rd Annual Meeting. Available online: https://cslide-us.ctimeetingtech.com/asgct23/attendee/eposter/poster/466 (accessed on 16 October 2020).
- Li, C.; Samulski, R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020, 21, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Jaka, O.; Casas-Fraile, L.; Azpitarte, M.; Aiastui, A.; López de Munain, A.; Sáenz, A. FRZB and melusin, overexpressed in LGMD2A, regulate integrin β1D isoform replacement altering myoblast fusion and the integrin-signalling pathway. Expert Rev. Mol. Med. 2017, 19. [Google Scholar] [CrossRef]
- Kramerova, I.; Kudryashova, E.; Wu, B.; Spencer, M.J. Regulation of the M-Cadherin-β-Catenin Complex by Calpain 3 during Terminal Stages of Myogenic Differentiation. Mol. Cell. Biol. 2006, 26, 8437–8447. [Google Scholar] [CrossRef]
- Liu, J.; Wu, X.; Mitchell, B.; Kintner, C.; Ding, S.; Schultz, P.G. A small-molecule agonist of the Wnt signaling pathway. Angew. Chem. Int. Ed. 2005, 44, 1987–1990. [Google Scholar] [CrossRef]
- Lim, J.C.; Kania, K.D.; Wijesuriya, H.; Chawla, S.; Sethi, J.K.; Pulaski, L.; Romero, I.A.; Couraud, P.O.; Weksler, B.B.; Hladky, S.B.; et al. Activation of β-catenin signalling by GSK-3 inhibition increases p-glycoprotein expression in brain endothelial cells. J. Neurochem. 2008, 106, 1855–1865. [Google Scholar] [CrossRef]
- Liu, J.; Campagna, J.; John, V.; Damoiseaux, R.; Mokhonova, E.; Becerra, D.; Meng, H.; McNally, E.M.; Pyle, A.D.; Kramerova, I.; et al. A Small-Molecule Approach to Restore a Slow-Oxidative Phenotype and Defective CaMKIIβ Signaling in Limb Girdle Muscular Dystrophy. Cell Rep. Med. 2020, 1. [Google Scholar] [CrossRef]
- Alenzi, F.Q.; Lotfy, M.; Tamimi, W.G.; Wyse, R.K.H. Review: Stem cells and gene therapy. Lab. Hematol. 2010, 16, 53–73. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, S.; Dhoke, N.R.; Kiley, J.; Mateos-Aierdi, A.J.; Tungtur, S.; Mondragon-Gonzalez, R.; Killeen, G.; Oliveira, V.K.P.; López de Munain, A.; Perlingeiro, R.C.R. Gene Correction of LGMD2A Patient-Specific iPSCs for the Development of Targeted Autologous Cell Therapy. Mol. Ther. 2019, 27, 2147–2157. [Google Scholar] [CrossRef]
- Peng, G.-Y.; Lin, Y.; Li, J.-J.; Wang, Y.; Huang, H.-Y.; Shen, Z.-Y. The Application of Induced Pluripotent Stem Cells in Pathogenesis Study and Gene Therapy for Vascular Disorders: Current Progress and Future Challenges. Stem Cells Int. 2019, 2019, 9613258. [Google Scholar] [CrossRef]
- Nilsson, M.I.; Macneil, L.G.; Kitaoka, Y.; Alqarni, F.; Suri, R.; Akhtar, M.; Haikalis, M.E.; Dhaliwal, P.; Saeed, M.; Tarnopolsky, M.A. Redox state and mitochondrial respiratory chain function in skeletal muscle of LGMD2A patients. PLoS ONE 2014, 9, e102549. [Google Scholar] [CrossRef] [PubMed]
- Kramerova, I.; Kudryashova, E.; Wu, B.; Germain, S.; Vandenborne, K.; Romain, N.; Haller, R.G.; Verity, M.A.; Spencer, M.J. Mitochondrial abnormalities, energy deficit and oxidative stress are features of calpain 3 deficiency in skeletal muscle. Hum. Mol. Genet. 2009, 18, 3194–3205. [Google Scholar] [CrossRef] [PubMed]
- Moyer, A.L.; Wagner, K.R. Mammalian Mss51 is a Skeletal Muscle-Specific Gene Modulating Cellular Metabolism. J. Neuromuscul. Dis. 2015, 2, 371–385. [Google Scholar] [CrossRef]
- Research–Coalition to Cure Calpain 3. Available online: https://www.curecalpain3.org/research/ (accessed on 16 October 2020).
- Fanin, M.; Nascimbeni, A.C.; Angelini, C. Muscle atrophy in Limb Girdle Muscular Dystrophy 2A: A morphometric and molecular study. Neuropathol. Appl. Neurobiol. 2013, 39, 762–771. [Google Scholar] [CrossRef]
- CAPN3 Protein (Human)-STRING İnteraction Network. Available online: https://string-db.org/network/9606.ENSP00000380349 (accessed on 6 February 2021).
- Kaneko, M.; Imaißumi, K.; Saito, A.; Kanemoto, S.; Asada, R.; Matsuhisa, K.; Ohtake, Y. ER stress and disease: Toward prevention and treatment. Biol. Pharm. Bull. 2017, 40, 1337–1343. [Google Scholar] [CrossRef]
- Zhang, K.; Kaufman, R.J. Signaling the unfolded protein response from the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 25935–25938. [Google Scholar] [CrossRef]
- Kincaid, M.M.; Cooper, A.A. ERADicate ER stress or die trying. Antioxid. Redox Signal. 2007, 9, 2373–2387. [Google Scholar] [CrossRef]
- Toral-Ojeda, I.; Aldanondo, G.; Lasa-Elgarresta, J.; Lasa-Fernández, H.; Fernández-Torrón, R.; De Munain, A.L.; Vallejo-Illarramendi, A. Calpain 3 deficiency affects SERCA expression and function in the skeletal muscle. Expert Rev. Mol. Med. 2016, 18. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Kaufman, R.J. The endoplasmic reticulum and the unfolded protein response. Semin. Cell Dev. Biol. 2007, 18, 716–731. [Google Scholar] [CrossRef]
- Nakka, V.P.; Prakash-babu, P.; Vemuganti, R. Crosstalk Between Endoplasmic Reticulum Stress, Oxidative Stress, and Autophagy: Potential Therapeutic Targets for Acute CNS Injuries. Mol. Neurobiol. 2016, 53, 532–544. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, S.J.; Ron, D. Endoplasmic reticulum stress signaling in disease. Physiol. Rev. 2006, 86, 1133–1149. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, N. ER and aging-Protein folding and the ER stress response. Ageing Res. Rev. 2009, 8, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Sarvani, C.; Sireesh, D.; Ramkumar, K.M. Unraveling the role of ER stress inhibitors in the context of metabolic diseases. Pharmacol. Res. 2017, 119, 412–421. [Google Scholar] [CrossRef]
- Liu, M.Q.; Chen, Z.; Chen, L.X. Endoplasmic reticulum stress: A novel mechanism and therapeutic target for cardiovascular diseases. Acta Pharmacol. Sin. 2016, 37, 425–443. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Zhou, C.; Chi, J.; Pan, S.; Lin, H.; Gao, F.; Ni, T.; Meng, L.; Zhang, J.; Jiang, C.; et al. The Role of Tauroursodeoxycholic Acid on Dedifferentiation of Vascular Smooth Muscle Cells by Modulation of Endoplasmic Reticulum Stress and as an Oral Drug Inhibiting In-Stent Restenosis. Cardiovasc. Drugs Ther. 2019, 33, 25–33. [Google Scholar] [CrossRef]
- Mesbah Moosavi, Z.S.; Hood, D.A. The unfolded protein response in relation to mitochondrial biogenesis in skeletal muscle cells. Am. J. Physiol. Cell Physiol. 2017, 312, C583–C594. [Google Scholar] [CrossRef]
- Romero-Ramírez, L.; Nieto-Sampedro, M.; Barreda-Manso, M.A. All roads go to salubrinal: Endoplasmic reticulum stress, neuroprotection and glial scar formation. Neural Regen. Res. 2015, 10, 1926–1927. [Google Scholar] [CrossRef]
- Foltz, S.J.; Luan, J.; Call, J.A.; Patel, A.; Peissig, K.B.; Fortunato, M.J.; Beedle, A.M. Four-week rapamycin treatment improves muscular dystrophy in a fukutin-deficient mouse model of dystroglycanopathy. Skelet. Muscle 2016, 6. [Google Scholar] [CrossRef]
- Bibee, K.P.; Cheng, Y.J.; Ching, J.K.; Marsh, J.N.; Li, A.J.; Keeling, R.M.; Connolly, A.M.; Golumbek, P.T.; Myerson, J.W.; Hu, G.; et al. Rapamycin nanoparticles target defective autophagy in muscular dystrophy to enhance both strength and cardiac function. FASEB J. 2014, 28, 2047–2061. [Google Scholar] [CrossRef]
- Kawakami, Y.; Hambright, W.S.; Takayama, K.; Mu, X.; Lu, A.; Cummins, J.H.; Matsumoto, T.; Yurube, T.; Kuroda, R.; Kurosaka, M.; et al. Rapamycin Rescues Age-Related Changes in Muscle-Derived Stem/Progenitor Cells from Progeroid Mice. Mol. Ther. Methods Clin. Dev. 2019, 14, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, E.V.; Gurevich, V.V. Therapeutic potential of small molecules and engineered proteins. Handb. Exp. Pharmacol. 2014, 219, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Dahl, R.; Hsieh, W.; Shin, A.; Zsebo, K.M.; Buettner, C.; Hajjar, R.J.; Lebeche, D. Small molecular allosteric activator of the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) attenuates diabetes and metabolic disorders. J. Biol. Chem. 2016, 291, 5185–5198. [Google Scholar] [CrossRef]
- Dahl, R. A new target for Parkinson’s disease: Small molecule SERCA activator CDN1163 ameliorates dyskinesia in 6-OHDA-lesioned rats. Bioorg. Med. Chem. 2017, 25, 53–57. [Google Scholar] [CrossRef]
- Nelson, B.R.; Makarewich, C.A.; Anderson, D.M.; Winders, B.R.; Troupes, C.D.; Wu, F.; Reese, A.L.; McAnally, J.R.; Chen, X.; Kavalali, E.T.; et al. Muscle physiology: A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science 2016, 351, 271–275. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Type | Administration | Expectation | Stage | Comment | Ref. | |
|---|---|---|---|---|---|---|
| Drug Therapy | ||||||
| Prednisone | Glucocorticoid steroid | Taking orally | Reduce inflammatory response | Phase I/II study | Undesirable situations may occur due to suppressing the immune system. | [20] |
| MYO-029 | Antibody | Injected intravenously | Neutralize myostatin protein | Phase I/II study | Myostatin inhibition resulted in a minor improvement in muscle. | [26] |
| Anti-myostatin antibody | Antibody | Injected intraperitoneally | Inhibition of follistatin, which is an endogenous inhibitor of myostatin | Experimental study on a murine model | Increase in muscle mass but not in functional muscle. | [28] |
| AMBMP | Small molecule | Injected intraperitoneally | As a Wnt agonist activates CaMKII | Experimental study on a murine model | Induction of slow oxidative genes. | [43] |
| Gene Therapy | ||||||
| pAAV-CMV-mSeAPpropmyoD76A vector | Plasmid DNA | Injected intramuscularly | Inhibition of myostatin | Experimental study on a murine model | Increase in muscle mass and absolute power | [27] |
| CAPN3 gene transfer via AAV vector, | Plasmid DNA | Systemic injection | Replacement of functional CAPN3 gene | Experimental study on a murine model | CAPN3 overexpression caused cardiac toxicity. | [31] |
| CAPN3 gene, and cardiac-specific microRNA-208a transfer via AAV | Plasmid DNA | Systemic injection | Replacement of functional CAPN3 gene and overcoming cardiac toxicity | Experimental study on a murine model | CAPN3 expression and no cardiac toxicity were achieved. | [31] |
| AAVrh74.tMCK.hCAPN3 vector | Plasmid DNA | Injected intravenously | Replacement of functional CAPN3 gene, overcoming off-target and toxic effects | Experimental study on a primate model | CAPN3 expression, no toxicity, and skeletal-muscle-specific vector were achieved. | [37] |
| rAAV-C3+miRT and rAAV-C3 | Plasmid DNA | Injected intravascularly and intramuscularly | Replacement of functional CAPN3 gene and overcoming cardiac toxicity | Experimental study on a primate model | In murine models, overexpression of CAPN3 is more prone to cardiac toxicity than in primates, due to physiological differences. CAPN3 expression increased in both applications and no cardiac toxicity was observed. | [34] |
| Combined Therapy (Cell- and Gene-Based) | ||||||
| IPSCs | CRISPR-Cas9 and stem cell | Injected intramuscularly | Replacement of functional CAPN3 in myogenic progenitor and mature muscle cells expressing CAPN3 | Experimental study on a murine model | CAPN3 mRNA levels were increased. | [44] |
| Type | Application | Expectation | Ref. | |
|---|---|---|---|---|
| Mss51 | Muscle-specific protein | Inhibition of Mss51 gene | Energy production increases and mitochondrial activity improves | [50] |
| TUDCA | The chemical chaperone mimetic drug | Different applications of TUDCA | Reduces effects on ER stress-related molecules | [65] |
| Salubrinal | A small molecule for selective inhibition of eIF2α | Different applications of salubrinal | Induces degradation of non-translated ER-targeted protein mRNAs | [66] |
| Rapamycin | Drug | Oral gavage | Provides inhibition of mTORC1, decrease in ER stress and inflammation, Improves muscle strength | [67] |
| CDN1163 | A small molecule as a SERCA2 activator | Injected intraperitoneally | Reduces ER stress and maintains Ca+2 homeostasis | [71] |
| DWORF | Muscle-specific long non-coding RNA | Upregulate of DWORF gene | Inhibits SERCA inhibitors and increases SERCA activity | [73] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Şahin, İ.O.; Özkul, Y.; Dündar, M. Current and Future Therapeutic Strategies for Limb Girdle Muscular Dystrophy Type R1: Clinical and Experimental Approaches. Pathophysiology 2021, 28, 238-249. https://doi.org/10.3390/pathophysiology28020016
Şahin İO, Özkul Y, Dündar M. Current and Future Therapeutic Strategies for Limb Girdle Muscular Dystrophy Type R1: Clinical and Experimental Approaches. Pathophysiology. 2021; 28(2):238-249. https://doi.org/10.3390/pathophysiology28020016
Chicago/Turabian StyleŞahin, İzem Olcay, Yusuf Özkul, and Munis Dündar. 2021. "Current and Future Therapeutic Strategies for Limb Girdle Muscular Dystrophy Type R1: Clinical and Experimental Approaches" Pathophysiology 28, no. 2: 238-249. https://doi.org/10.3390/pathophysiology28020016
APA StyleŞahin, İ. O., Özkul, Y., & Dündar, M. (2021). Current and Future Therapeutic Strategies for Limb Girdle Muscular Dystrophy Type R1: Clinical and Experimental Approaches. Pathophysiology, 28(2), 238-249. https://doi.org/10.3390/pathophysiology28020016