Multimodal Cancer Therapy and Accelerated Brain Aging: Mechanisms, Biomarkers, and Clinical Consequences

 ,
,  , ,
, , 
Simple Summary
Abstract
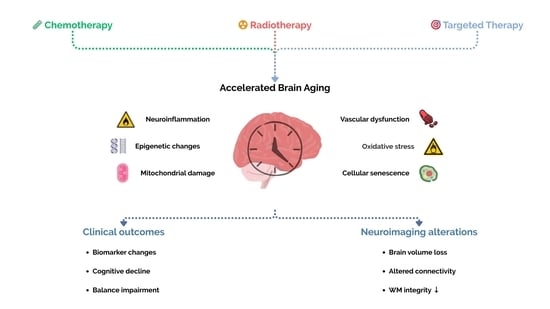
1. Introduction
2. Molecular and Cellular Mechanisms of Chemotherapy-Induced Brain Aging
2.1. Oxidative Stress and Mitochondrial Dysfunction
2.2. Neuroinflammation and Cytokine Signaling
2.3. Blood–Brain Barrier Dysfunction
2.4. Impaired Neurogenesis and Synaptic Plasticity
2.5. DNA Damage and Disrupted Repair Mechanisms
2.6. Cellular Senescence and SASP-Mediated Effects
2.7. Epigenetic Alterations and Aging Pathways
3. Biomarkers of Chemotherapy-Induced Brain Aging
4. Radiotherapy and Accelerated Brain Aging
4.1. Indirect Effects on the Brain During Extracranial Radiotherapy
4.2. Direct Effects of Cranial Radiotherapy
5. Targeted Therapy as a Modifier of Brain Aging
6. Clinical Evidence of Cancer Treatment-Related Cognitive Impairment and Brain Aging
6.1. Clinical Manifestations and Cognitive Domains Affected by Cancer Treatment
- -
- Processing speed and attention;
- -
- Executive functions (e.g., planning, task switching);
- -
- Working memory and short-term recall;
- -
- Episodic memory.
6.2. Neuroimaging Evidence of Cancer Treatment-Related Cognitive Impairment
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CNS | Central Nervous System |
| CRCI | Cancer Treatment-Related Cognitive Impairment |
| ROS | Reactive Oxygen Species |
| BBB | Blood–Brain Barrier |
| SASP | Senescence-Associated Secretory Phenotype |
| MRI | Magnetic Resonance Imaging |
| ICAM-1 | Intercellular Adhesion Molecule-1 |
| PECAM-1 | Platelet Endothelial Cell Adhesion Molecule-1 |
| NSE | Neuron-Specific Enolase |
| NR2 | N-Methyl-D-Aspartate Receptor Subunit 2 |
| IL-6 | Interleukin-6 |
| IL-8 | Interleukin-8 |
| TNF-α | Tumor Necrosis Factor-Alpha |
| CRP | C-Reactive Protein |
| BDNF | Brain-Derived Neurotrophic Factor |
| CAR-T | Chimeric Antigen Receptor T-cell |
| Anti-HER2 | Anti-Human Epidermal Growth Factor Receptor 2 |
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global Cancer Statistics 2022: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef]
- Siegel, R.L.; Kratzer, T.B.; Giaquinto, A.N.; Sung, H.; Jemal, A. Cancer Statistics, 2025. CA Cancer J. Clin. 2025, 75, 10–45. [Google Scholar] [CrossRef]
- Li, T.; Zhang, H.; Lian, M.; He, Q.; Lv, M.; Zhai, L.; Zhou, J.; Wu, K.; Yi, M. Global Status and Attributable Risk Factors of Breast, Cervical, Ovarian, and Uterine Cancers from 1990 to 2021. J. Hematol. Oncol. 2025, 18, 5. [Google Scholar] [CrossRef]
- Obeagu, E.I.; Obeagu, G.U. Breast Cancer: A Review of Risk Factors and Diagnosis. Medicine 2024, 103, E36905. [Google Scholar] [CrossRef]
- Fukata, K.; Akiyoshi, T.; Numao, N.; Komai, Y.; Mukai, T.; Hiyoshi, Y.; Yamaguchi, T.; Nagasaki, T.; Konishi, T.; Fukunaga, Y. Robotic-Assisted Laparoscopic Surgery for Synchronous Primary Rectal and Prostate Cancer: Initial Case Series. Asian J. Endosc. Surg. 2022, 15, 678–682. [Google Scholar] [CrossRef]
- Vendrely, V.; Rivin Del Campo, E.; Modesto, A.; Jolnerowski, M.; Meillan, N.; Chiavassa, S.; Serre, A.A.; Gérard, J.P.; Créhanges, G.; Huguet, F.; et al. Rectal Cancer Radiotherapy. Cancer/Radiother. 2022, 26, 272–278. [Google Scholar] [CrossRef]
- Escande, A.; Leblanc, J.; Hannoun-Levi, J.M.; Renard, S.; Ducassou, A.; Hennequin, C.; Chargari, C. Place of Radiotherapy for Treatment of Metastatic Cervical, Vaginal and Endometrial Uterine Cancer. Cancer/Radiother. 2024, 28, 15–21. [Google Scholar] [CrossRef]
- Jeon, H.; Wang, S.; Song, J.; Gill, H.; Cheng, H. Update 2025: Management of Non-Small-Cell Lung Cancer. Lung 2025, 203, 53. [Google Scholar] [CrossRef]
- Heater, N.K.; Warrior, S.; Lu, J. Current and Future Immunotherapy for Breast Cancer. J. Hematol. Oncol. 2024, 17, 131. [Google Scholar] [CrossRef]
- Yoo, H.C.; Lee, S.; Park, J.Y.; Lee, E.J. AAV for Ovarian Cancer Gene Therapy. Cancer Gene Ther. 2025, 32, 817–830. [Google Scholar] [CrossRef]
- Soares do Brito, J.; Santos, R.; Sarmento, M.; Fernandes, P.; Portela, J. Chemotherapy Regimens for Non-Metastatic Conventional Appendicular Osteosarcoma: A Literature Review Based on the Outcomes. Curr. Oncol. 2023, 30, 6148–6165. [Google Scholar] [CrossRef]
- Massimo, G.; Fortunato, M.; Massimo, M.; Ernesto, V.; Enrica, A.M.; Francesco, M.; Giovanni, M.; Claudio, C. Chemotherapy-Based Regimens in Multiple Myeloma in 2020. Panminerva Med. 2021, 63, 7–12. [Google Scholar] [CrossRef]
- Mollaei, M.; Hassan, Z.M.; Khorshidi, F.; Langroudi, L. Chemotherapeutic Drugs: Cell Death- and Resistance-Related Signaling Pathways. Are They Really as Smart as the Tumor Cells? Transl. Oncol. 2021, 14, 101056. [Google Scholar] [CrossRef]
- Knezevic, C.E.; Clarke, W. Cancer Chemotherapy: The Case for Therapeutic Drug Monitoring. Ther. Drug Monit. 2020, 42, 6–19. [Google Scholar] [CrossRef]
- Ying, Q.; Fan, R.; Shen, Y.; Chen, B.; Zhang, J.; Li, Q.; Shi, X. Small Cell Lung Cancer-An Update on Chemotherapy Resistance. Curr. Treat. Options Oncol. 2024, 25, 1112–1123. [Google Scholar] [CrossRef]
- Ruiz-Cordero, R.; Devine, W.P. Targeted Therapy and Checkpoint Immunotherapy in Lung Cancer. Surg. Pathol. Clin. 2020, 13, 17–33. [Google Scholar] [CrossRef]
- Mahumud, R.A.; Shahjalal, M.; Dahal, P.K.; Mosharaf, M.P.; Hoque, M.E.; Wawryk, O. Systemic Therapy and Radiotherapy Related Complications and Subsequent Hospitalisation Rates: A Systematic Review. BMC Cancer 2024, 24, 826. [Google Scholar] [CrossRef]
- Shahrokni, A.; Wu, A.J.; Carter, J.; Lichtman, S.M. Long-Term Toxicity of Cancer Treatment in Older Patients. Clin. Geriatr. Med. 2016, 32, 63–80. [Google Scholar] [CrossRef]
- Marco, E.; Trépanier, G.; Chang, E.; Mauti, E.; Jones, J.M.; Zhong, T. Postmastectomy Functional Impairments. Curr. Oncol. Rep. 2023, 25, 1445–1453. [Google Scholar] [CrossRef]
- Rao, V.; Bhushan, R.; Kumari, P.; Cheruku, S.P.; Ravichandiran, V.; Kumar, N. Chemobrain: A Review on Mechanistic Insight, Targets and Treatments. Adv. Cancer Res. 2022, 155, 29–76. [Google Scholar] [CrossRef]
- Bennedsgaard, K.; Grosen, K.; Attal, N.; Bouhassira, D.; Crombez, G.; Jensen, T.S.; Bennett, D.L.; Ventzel, L.; Andersen, I.S.; Finnerup, N.B. Neuropathy and Pain after Breast Cancer Treatment: A Prospective Observational Study. Scand. J. Pain 2022, 23, 49–58. [Google Scholar] [CrossRef]
- Bae, E.H.; Greenwald, M.K.; Schwartz, A.G. Chemotherapy-Induced Peripheral Neuropathy: Mechanisms and Therapeutic Avenues. Neurotherapeutics 2021, 18, 2384–2396. [Google Scholar] [CrossRef]
- Grusdat, N.P.; Stäuber, A.; Tolkmitt, M.; Schnabel, J.; Schubotz, B.; Wright, P.R.; Schulz, H. Routine Cancer Treatments and Their Impact on Physical Function, Symptoms of Cancer-Related Fatigue, Anxiety, and Depression. Support. Care Cancer 2022, 30, 3733–3744. [Google Scholar] [CrossRef]
- Carreira, H.; Williams, R.; Dempsey, H.; Stanway, S.; Smeeth, L.; Bhaskaran, K. Quality of Life and Mental Health in Breast Cancer Survivors Compared with Non-Cancer Controls: A Study of Patient-Reported Outcomes in the United Kingdom. J. Cancer Surviv. 2021, 15, 564–575. [Google Scholar] [CrossRef]
- Wefel, J.S.; Lenzi, R.; Theriault, R.; Buzdar, A.U.; Cruickshank, S.; Meyers, C.A. “Chemobrain” in Breast Carcinoma? A Prologue. Cancer 2004, 101, 466–475. [Google Scholar] [CrossRef]
- Onzi, G.R.; D’Agustini, N.; Garcia, S.C.; Guterres, S.S.; Pohlmann, P.R.; Rosa, D.D.; Pohlmann, A.R. Chemobrain in Breast Cancer: Mechanisms, Clinical Manifestations, and Potential Interventions. Drug Saf. 2022, 45, 601–621. [Google Scholar] [CrossRef]
- Fleming, B.; Edison, P.; Kenny, L. Cognitive Impairment after Cancer Treatment: Mechanisms, Clinical Characterization, and Management. BMJ 2023, 380, e071726. [Google Scholar] [CrossRef]
- Blinkouskaya, Y.; Caçoilo, A.; Gollamudi, T.; Jalalian, S.; Weickenmeier, J. Brain Aging Mechanisms with Mechanical Manifestations. Mech. Ageing Dev. 2021, 200, 111575. [Google Scholar] [CrossRef]
- Lee, J.; Kim, H.J. Normal Aging Induces Changes in the Brain and Neurodegeneration Progress: Review of the Structural, Biochemical, Metabolic, Cellular, and Molecular Changes. Front. Aging Neurosci. 2022, 14, 931536. [Google Scholar] [CrossRef]
- Gaspar-Silva, F.; Trigo, D.; Magalhaes, J. Ageing in the Brain: Mechanisms and Rejuvenating Strategies. Cell. Mol. Life Sci. 2023, 80, 190. [Google Scholar] [CrossRef]
- Ionescu-Tucker, A.; Cotman, C.W. Emerging Roles of Oxidative Stress in Brain Aging and Alzheimer’s Disease. Neurobiol. Aging 2021, 107, 86–95. [Google Scholar] [CrossRef]
- Nguyen, L.D.; Ehrlich, B.E. Cellular Mechanisms and Treatments for Chemobrain: Insight from Aging and Neurodegenerative Diseases. EMBO Mol. Med. 2020, 12, e12075. [Google Scholar] [CrossRef]
- Ren, X.; Boriero, D.; Chaiswing, L.; Bondada, S.; St. Clair, D.K.; Butterfield, D.A. Plausible Biochemical Mechanisms of Chemotherapy-Induced Cognitive Impairment (“Chemobrain”), a Condition That Significantly Impairs the Quality of Life of Many Cancer Survivors. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1088–1097. [Google Scholar] [CrossRef]
- Cauli, O. Oxidative Stress and Cognitive Alterations Induced by Cancer Chemotherapy Drugs: A Scoping Review. Antioxidants 2021, 10, 1116. [Google Scholar] [CrossRef]
- Jaiswara, P.K.; Shukla, S.K. Chemotherapy-Mediated Neuronal Aberration. Pharmaceuticals 2023, 16, 1165. [Google Scholar] [CrossRef]
- Oliveros, A.; Poleschuk, M.; Cole, P.D.; Boison, D.; Jang, M.H. Chemobrain: An Accelerated Aging Process Linking Adenosine A2A Receptor Signaling in Cancer Survivors. Int. Rev. Neurobiol. 2023, 170, 267. [Google Scholar] [CrossRef]
- Patai, R.; Csik, B.; Nyul-Toth, A.; Gulej, R.; Vali Kordestan, K.; Sai Chandragiri, S.; Shanmugarama, S.; Tarantini, S.; Mukli, P.; Ungvari, A.; et al. Persisting Blood–Brain Barrier Disruption Following Cisplatin Treatment in a Mouse Model of Chemotherapy-Associated Cognitive Impairment. GeroScience 2025, 47, 3835–3847. [Google Scholar] [CrossRef]
- Csik, B.; Nyúl-Tóth, Á.; Gulej, R.; Patai, R.; Kiss, T.; Delfavero, J.; Nagaraja, R.Y.; Balasubramanian, P.; Shanmugarama, S.; Ungvari, A.; et al. Senescent Endothelial Cells in Cerebral Microcirculation Are Key Drivers of Age-Related Blood–Brain Barrier Disruption, Microvascular Rarefaction, and Neurovascular Coupling Impairment in Mice. Aging Cell 2025, 24, e70048. [Google Scholar] [CrossRef]
- Sekeres, M.J.; Bradley-Garcia, M.; Martinez-Canabal, A.; Winocur, G. Chemotherapy-Induced Cognitive Impairment and Hippocampal Neurogenesis: A Review of Physiological Mechanisms and Interventions. Int. J. Mol. Sci. 2021, 22, 12697. [Google Scholar] [CrossRef]
- Wang, S.; Prizment, A.; Thyagarajan, B.; Blaes, A. Cancer Treatment-Induced Accelerated Aging in Cancer Survivors: Biology and Assessment. Cancers 2021, 13, 427. [Google Scholar] [CrossRef]
- Wang, S.; El Jurdi, N.; Thyagarajan, B.; Prizment, A.; Blaes, A.H. Accelerated Aging in Cancer Survivors: Cellular Senescence, Frailty, and Possible Opportunities for Interventions. Int. J. Mol. Sci. 2024, 25, 3319. [Google Scholar] [CrossRef]
- Ma, Y.; Erb, M.L.; Moore, D.J. Aging, Cellular Senescence and Parkinson’s Disease. J. Park. Dis. 2025, 15, 239–254. [Google Scholar] [CrossRef]
- Alhowail, A.H.; Aldubayan, M. Recent Progress in the Elucidation of the Mechanisms of Chemotherapy-Induced Cognitive Impairment. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 5807–5817. [Google Scholar] [CrossRef]
- Williams, A.L.M.; Phillips, N.S.; Dong, Q.; Ehrhardt, M.J.; Gilmore, N.; Loh, K.P.; Meng, X.; Ness, K.K.; Hudson, M.M.; Robison, L.L.; et al. Epigenetic Age Acceleration, Telomere Length, and Neurocognitive Function in Long-Term Survivors of Childhood Cancer. Nat. Commun. 2025, 16, 10655. [Google Scholar] [CrossRef]
- Pérez, R.F.; Tejedor, J.R.; Santamarina-Ojeda, P.; Martínez, V.L.; Urdinguio, R.G.; Villamañán, L.; Candiota, A.P.; Sarró, N.M.V.; Barradas, M.; Fernandez-Marcos, P.J.; et al. Conservation of Aging and Cancer Epigenetic Signatures across Human and Mouse. Mol. Biol. Evol. 2021, 38, 3415–3435. [Google Scholar] [CrossRef]
- Sehl, M.E.; Carroll, J.E.; Horvath, S.; Bower, J.E. The Acute Effects of Adjuvant Radiation and Chemotherapy on Peripheral Blood Epigenetic Age in Early Stage Breast Cancer Patients. npj Breast Cancer 2020, 6, 23. [Google Scholar] [CrossRef]
- Guida, J.L.; Ahles, T.A.; Belsky, D.; Campisi, J.; Cohen, H.J.; DeGregori, J.; Fuldner, R.; Ferrucci, L.; Gallicchio, L.; Gavrilov, L.; et al. Measuring Aging and Identifying Aging Phenotypes in Cancer Survivors. JNCI J. Natl. Cancer Inst. 2019, 111, 1245. [Google Scholar] [CrossRef]
- Fulop, T.; Larbi, A.; Pawelec, G.; Khalil, A.; Cohen, A.A.; Hirokawa, K.; Witkowski, J.M.; Franceschi, C. Immunology of Aging: The Birth of Inflammaging. Clin. Rev. Allergy Immunol. 2023, 64, 109–122. [Google Scholar] [CrossRef]
- Behranvand, N.; Nasri, F.; Zolfaghari Emameh, R.; Khani, P.; Hosseini, A.; Garssen, J.; Falak, R. Chemotherapy: A Double-Edged Sword in Cancer Treatment. Cancer Immunol. Immunother. 2022, 71, 507–526. [Google Scholar] [CrossRef]
- Gallicchio, L.; Guida, J.L.; Green, P.A. Introduction to the Special Section on Cancer Survivors and Treatment-Related Accelerated Aging. J. Cancer Surviv. 2024, 18, 1085–1088. [Google Scholar] [CrossRef]
- Abraham, S.; Parekh, J.; Lee, S.; Afrin, H.; Rozenblit, M.; Blenman, K.R.M.; Perry, R.J.; Ferrucci, L.M.; Liu, J.; Irwin, M.L.; et al. Accelerated Aging in Cancer and Cancer Treatment: Current Status of Biomarkers. Cancer Med. 2025, 14, e70929. [Google Scholar] [CrossRef]
- Gaman, A.M.; Uzoni, A.; Popa-Wagner, A.; Andrei, A.; Petcu, E.B. The Role of Oxidative Stress in Etiopathogenesis of Chemotherapy Induced Cognitive Impairment (CICI)-“Chemobrain”. Aging Dis. 2016, 7, 307. [Google Scholar] [CrossRef]
- Moruno-Manchon, J.F.; Uzor, N.E.; Kesler, S.R.; Wefel, J.S.; Townley, D.M.; Nagaraja, A.S.; Pradeep, S.; Mangala, L.S.; Sood, A.K.; Tsvetkov, A.S. Peroxisomes Contribute to Oxidative Stress in Neurons during Doxorubicin-Based Chemotherapy. Mol. Cell. Neurosci. 2018, 86, 65–71. [Google Scholar] [CrossRef]
- Budamagunta, V.; Kumar, A.; Rani, A.; Manohar Sindhu, S.; Yang, Y.; Zhou, D.; Foster, T.C. Senolytic Treatment Alleviates Doxorubicin-Induced Chemobrain. Aging Cell 2024, 23, e14037. [Google Scholar] [CrossRef]
- Shachar, S.S.; Deal, A.M.; Reeder-Hayes, K.E.; Nyrop, K.A.; Mitin, N.; Anders, C.K.; Carey, L.A.; Claire Dees, E.; Jolly, T.A.; Kimmick, G.G.; et al. Effects of Breast Cancer Adjuvant Chemotherapy Regimens on Expression of the Aging Biomarker, P16INK4a. JNCI Cancer Spectr. 2020, 4, pkaa082. [Google Scholar] [CrossRef]
- Nikolaeva, A.; Pospelova, M.; Krasnikova, V.; Makhanova, A.; Tonyan, S.; Krasnopeev, Y.; Kayumova, E.; Vasilieva, E.; Efimtsev, A.; Levchuk, A.; et al. Elevated Levels of Serum Biomarkers Associated with Damage to the CNS Neurons and Endothelial Cells Are Linked with Changes in Brain Connectivity in Breast Cancer Patients with Vestibulo-Atactic Syndrome. Pathophysiology 2023, 30, 260–274. [Google Scholar] [CrossRef]
- Nikolaeva, A.T.; Pospelova, M.L.; Krasnikova, V.V.; Makhanova, A.M.; Tonyan, S.N.; Fionik, O.V.; Efimtsev, A.Y.; Levchuk, A.G.; Krasnopeev, Y.I. Clinical and Neuroimaging Laboratory Possibilities of Diagnostics of Vestibulo-Atactic Syndrome in Patients with Postmastectomic Syndrome. Transl. Med. 2023, 10, 25–35. [Google Scholar] [CrossRef]
- Pospelova, M.; Krasnikova, V.; Fionik, O.; Alekseeva, T.; Samochernykh, K.; Ivanova, N.; Trofimov, N.; Vavilova, T.; Vasilieva, E.; Topuzova, M.; et al. Adhesion Molecules ICAM-1 and PECAM-1 as Potential Biomarkers of Central Nervous System Damage in Women Breast Cancer Survivors. Pathophysiology 2022, 29, 52–65. [Google Scholar] [CrossRef]
- Patel, C.; Glytsou, C.; Jang, M.H.; Cole, P.D. The Effects of Doxorubicin on Blood-Brain Barrier Integrity in HCMEC/D3. Neurotoxicology 2025, 111, 103355. [Google Scholar] [CrossRef]
- Trudeau, J.; Ng, D.Q.; Sayer, M.; Tan, C.J.; Ke, Y.; Chan, R.J.; Chan, A. Brain-Derived Neurotrophic Factor and Cytokines as Predictors of Cognitive Impairment in Adolescent and Young Adult Cancer Patients Receiving Chemotherapy: A Longitudinal Study. BMC Cancer 2025, 25, 1045. [Google Scholar] [CrossRef]
- Ng, D.Q.; Chan, D.; Agrawal, P.; Zhao, W.; Xu, X.; Acharya, M.; Chan, A. Evidence of Brain-Derived Neurotrophic Factor in Ameliorating Cancer-Related Cognitive Impairment: A Systematic Review of Human Studies. Crit. Rev. Oncol. Hematol. 2022, 176, 103748. [Google Scholar] [CrossRef]
- Sayer, M.; Ng, D.Q.; Trudeau, J.; Chan, R.J.; Acharya, M.M.; Kober, K.; Chan, A. Epigenetic Age Acceleration and Neurotrophin Signaling Pathways in Cancer-Related Cognitive Impairment: A Longitudinal, Prospective Cohort Study. Front. Aging 2025, 6, 1667638. [Google Scholar] [CrossRef]
- Ng, D.Q.; Hudson, C.; Nguyen, T.; Gupta, S.K.; Koh, Y.Q.; Acharya, M.M.; Chan, A. Dynamin-1 Is a Potential Mediator in Cancer-Related Cognitive Impairment. Neurotherapeutics 2025, 22, e00480. [Google Scholar] [CrossRef]
- Pospelova, M.; Krasnikova, V.; Fionik, O.; Alekseeva, T.; Samochernykh, K.; Ivanova, N.; Trofimov, N.; Vavilova, T.; Vasilieva, E.; Topuzova, M.; et al. Potential Molecular Biomarkers of Central Nervous System Damage in Breast Cancer Survivors. J. Clin. Med. 2022, 11, 1215. [Google Scholar] [CrossRef]
- Claessens, A.; Manchart, A.; Boufraine, M.; Guignard, A.; Bergeot, A.; Kieffer, A.; Lambert, A. Neuron-Specific Enolase as a Biomarker in Ifosfamide-Induced Encephalopathy: A Case Report. Case Rep. Oncol. 2025, 18, 800. [Google Scholar] [CrossRef]
- Hadjiagapiou, M.S.; Krashias, G.; Christodoulou, C.; Pantzaris, M.; Lambrianides, A. Serum Reactive Antibodies against the N-Methyl-D-Aspartate Receptor NR2 Subunit—Could They Act as Potential Biomarkers? Int. J. Mol. Sci. 2023, 24, 16170. [Google Scholar] [CrossRef]
- Ren, X.; Keeney, J.T.R.; Miriyala, S.; Noel, T.; Powell, D.K.; Chaiswing, L.; Bondada, S.; St. Clair, D.K.; Butterfield, D.A. The Triangle of Death of Neurons: Oxidative Damage, Mitochondrial Dysfunction, and Loss of Choline-Containing Biomolecules in Brains of Mice Treated with Doxorubicin. Advanced Insights into Mechanisms of Chemotherapy Induced Cognitive Impairment (“chemobrain”) involving TNF-α. Free Radic. Biol. Med. 2019, 134, 1–8. [Google Scholar] [CrossRef]
- Murillo, L.C.; Sutachan, J.J.; Albarracín, S.L. An Update on Neurobiological Mechanisms Involved in the Development of Chemotherapy-Induced Cognitive Impairment (CICI). Toxicol. Rep. 2023, 10, 544–553. [Google Scholar] [CrossRef]
- Demos-Davies, K.; Lawrence, J.; Rogich, A.; Lind, E.; Seelig, D. Cancer Treatment Induces Neuroinflammation and Behavioral Deficits in Mice. Front. Behav. Neurosci. 2023, 16, 1067298. [Google Scholar] [CrossRef]
- McGinnis, G.J.; Friedman, D.; Young, K.H.; Torres, E.R.S.; Thomas, C.R.; Gough, M.J.; Raber, J. Neuroinflammatory and Cognitive Consequences of Combined Radiation and Immunotherapy in a Novel Preclinical Model. Oncotarget 2016, 8, 9155. [Google Scholar] [CrossRef]
- Demos-Davies, K.; Lawrence, J.; Coffey, J.; Morgan, A.; Ferreira, C.; Hoeppner, L.H.; Seelig, D. Longitudinal Neuropathological Consequences of Extracranial Radiation Therapy in Mice. Int. J. Mol. Sci. 2024, 25, 5731. [Google Scholar] [CrossRef]
- Feiock, C.; Yagi, M.; Maidman, A.; Rendahl, A.; Hui, S.; Seelig, D. Central Nervous System Injury—A Newly Observed Bystander Effect of Radiation. PLoS ONE 2016, 11, e0163233. [Google Scholar] [CrossRef]
- Turnquist, C.; Harris, B.T.; Harris, C.C. Radiation-Induced Brain Injury: Current Concepts and Therapeutic Strategies Targeting Neuroinflammation. Neurooncol. Adv. 2020, 2, vdaa057. [Google Scholar] [CrossRef]
- Li, M.; Tong, F.; Wu, B.; Dong, X. Radiation-Induced Brain Injury: Mechanistic Insights and the Promise of Gut–Brain Axis Therapies. Brain Sci. 2024, 14, 1295. [Google Scholar] [CrossRef]
- Constanzo, J.; Midavaine, É.; Fouquet, J.; Lepage, M.; Descoteaux, M.; Kirby, K.; Tremblay, L.; Masson-Côté, L.; Geha, S.; Longpré, J.M.; et al. Brain Irradiation Leads to Persistent Neuroinflammation and Long-Term Neurocognitive Dysfunction in a Region-Specific Manner. Prog. Neuropsychopharmacol. Biol. Psychiatry 2020, 102, 109954. [Google Scholar] [CrossRef]
- Greene-Schloesser, D.; Robbins, M.E.; Peiffer, A.M.; Shaw, E.G.; Wheeler, K.T.; Chan, M.D. Radiation-Induced Brain Injury: A Review. Front. Oncol. 2012, 2, 73. [Google Scholar] [CrossRef]
- Li, X.; Ding, Z. Cognitive Dysfunction Induced by Cranial Radiotherapy: Mechanisms and Therapeutic Methods. Brain Res. Bull. 2024, 218, 111106. [Google Scholar] [CrossRef]
- Sterpi, A.E.; Triantafyllou, A.S.; Tzanetakos, D.; Ampantzi, E.; Kitsos, D.; Theodorou, A.; Koutsouraki, E.; Maili, M.; Stefanou, M.I.; Moschovos, C.; et al. Multiple Sclerosis-like Lesions Induced by Radiation: A Case Report and Systematic Review of the Literature. J. Clin. Med. 2024, 13, 7554. [Google Scholar] [CrossRef]
- Kim, J.H.; Brown, S.L.; Gordon, M.N. Radiation-Induced Senescence: Therapeutic Opportunities. Radiat. Oncol. 2023, 18, 10. [Google Scholar] [CrossRef]
- Markarian, M.; Krattli, R.P.; Baddour, J.D.; Alikhani, L.; Giedzinski, E.; Usmani, M.T.; Agrawal, A.; Baulch, J.E.; Tenner, A.J.; Acharya, M.M. Glia-Selective Deletion of Complement C1q Prevents Radiation-Induced Cognitive Deficits and Neuroinflammation. Cancer Res. 2021, 81, 1732–1744. [Google Scholar] [CrossRef]
- Krattli, R.P.; Do, A.H.; El-Khatib, S.M.; Alikhani, L.; Markarian, M.; Vagadia, A.R.; Usmani, M.T.; Madan, S.; Baulch, J.E.; Clark, R.J.; et al. C5aR1 Inhibition Alleviates Cranial Radiation-Induced Cognitive Decline. Cancer Res. 2026, 86, 255. [Google Scholar] [CrossRef]
- Joly, F.; Giffard, B.; Rigal, O.; De Ruiter, M.B.; Small, B.J.; Dubois, M.; Lefel, J.; Schagen, S.B.; Ahles, T.A.; Wefel, J.S.; et al. Impact of Cancer and Its Treatments on Cognitive Function: Advances in Research From the Paris International Cognition and Cancer Task Force Symposium and Update Since 2012. J. Pain Symptom Manag. 2015, 50, 830–841. [Google Scholar] [CrossRef]
- Chen, W.; Hu, X.; Yao, S.; Bi, Z.; Chen, M.; Cheng, H. Relationship Between Cognitive Disorder and First-Line Targeted Therapy for Oncogene Driver-Positive Patients With Non-Small Cell Lung Cancer: Prospective Cohort Study. JMIR Cancer 2025, 11, e59647. [Google Scholar] [CrossRef]
- Mayford, M. Protein Kinase Signaling in Synaptic Plasticity and Memory. Curr. Opin. Neurobiol. 2007, 17, 313–317. [Google Scholar] [CrossRef]
- Hyland, K.A.; Eisel, S.L.; Hoogland, A.I.; Root, J.C.; Bowles, K.; James, B.; Nelson, A.M.; Booth-Jones, M.; Jacobsen, P.B.; Ahles, T.A.; et al. Cognition in Patients Treated with Targeted Therapy for Chronic Myeloid Leukemia: A Controlled Comparison. Leuk. Lymphoma 2022, 64, 415. [Google Scholar] [CrossRef]
- Chamoun, K.; Rabinovich, E.; Baer, L.; Fastenau, P.; de Lima, M. A Case of Neurocognitive Deficit Strongly Related to Dasatinib Therapy. Hematol. Transfus. Cell Ther. 2020, 42, 80–82. [Google Scholar] [CrossRef]
- Jones, A.; Bonomi, S.; Stockerl-Goldstein, K.E.; Bateman, R.J. Neuropsychiatric Symptoms Mimicking Dementia in a Patient Treated With Imatinib. Ann. Clin. Transl. Neurol. 2025. Epub ahead of printing. [Google Scholar] [CrossRef]
- Gust, J.; Hay, K.A.; Hanafi, L.A.; Li, D.; Myerson, D.; Gonzalez-Cuyar, L.F.; Yeung, C.; Liles, W.C.; Wurfel, M.; Lopez, J.A.; et al. Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017, 7, 1404–1419. [Google Scholar] [CrossRef]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASBMT Consensus Grading for Cytokine Release Syndrome and Neurological Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transplant. 2018, 25, 625. [Google Scholar] [CrossRef]
- García-Sánchez, J.; Torregrosa, M.D.; Cauli, O. Cognitive Functions under Anti-HER2 Targeted Therapy in Cancer Patients: A Scoping Review. Endocr. Metab. Immune Disord. Drug Targets 2021, 21, 1163–1170. [Google Scholar] [CrossRef]
- Stangler, L.T.B.; de Almeida Robatto, A.A.; Freire, P.J.G.; de Castro Junior, G. The Challenge of Chemotherapy-Related Cognitive Impairment: Assessing and Managing Cognitive Decline after Cancer Treatment. Ecancermedicalscience 2025, 19, 1958. [Google Scholar] [CrossRef]
- Das, A.; Ranadive, N.; Kinra, M.; Nampoothiri, M.; Arora, D.; Mudgal, J. An Overview on Chemotherapy-Induced Cognitive Impairment and Potential Role of Antidepressants. Curr. Neuropharmacol. 2020, 18, 838. [Google Scholar] [CrossRef]
- Yi, J.C.; Syrjala, K.L. Anxiety and Depression in Cancer Survivors. Med. Clin. N. Am. 2017, 101, 1099–1113. [Google Scholar] [CrossRef]
- Perez-Tejada, J.; Labaka, A.; Vegas, O.; Larraioz, A.; Pescador, A.; Arregi, A. Anxiety and Depression after Breast Cancer: The Predictive Role of Monoamine Levels. Eur. J. Oncol. Nurs. 2021, 52, 101953. [Google Scholar] [CrossRef]
- Bock, K.; Peltzer, J.; Liu, W.; Colgrove, Y.; Smirnova, I.; Siengsukon, C. Sleep Quality and Lymphedema in Breast Cancer Survivors: A Mixed Method Analysis. J. Cancer Surviv. 2025, 19, 978–992. [Google Scholar] [CrossRef] [PubMed]
- Noal, S.; Levy, C.; Hardouin, A.; Rieux, C.; Heutte, N.; Ségura, C.; Collet, F.; Allouache, D.; Switsers, O.; Delcambre, C.; et al. One-Year Longitudinal Study of Fatigue, Cognitive Functions, and Quality of Life after Adjuvant Radiotherapy for Breast Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, 795–803. [Google Scholar] [CrossRef]
- Müller, J.; Ringhof, S.; Vollmer, M.; Jäger, L.B.; Stein, T.; Weiler, M.; Wiskemann, J. Out of Balance—Postural Control in Cancer Patients before and after Neurotoxic Chemotherapy. Gait Posture 2020, 77, 156–163. [Google Scholar] [CrossRef]
- Piper, K.S.; Myhre, K.K.; Jensen, H.E.; Madsen, K.; Mikkelsen, M.K.; Lund, C. Dizziness and Impaired Walking Balance in Aging Patients during Chemotherapy. J. Geriatr. Oncol. 2024, 15, 102059. [Google Scholar] [CrossRef]
- Simó, M.; Rifà-Ros, X.; Rodriguez-Fornells, A.; Bruna, J. Chemobrain: A Systematic Review of Structural and Functional Neuroimaging Studies. Neurosci. Biobehav. Rev. 2013, 37, 1311–1321. [Google Scholar] [CrossRef]
- Wefel, J.S.; Vardy, J.; Ahles, T.; Schagen, S.B. International Cognition and Cancer Task Force Recommendations to Harmonise Studies of Cognitive Function in Patients with Cancer. Lancet Oncol. 2011, 12, 703–708. [Google Scholar] [CrossRef]
- Lange, M.; Joly, F.; Vardy, J.; Ahles, T.; Dubois, M.; Tron, L.; Winocur, G.; De Ruiter, M.B.; Castel, H. Cancer-Related Cognitive Impairment: An Update on State of the Art, Detection, and Management Strategies in Cancer Survivors. Ann. Oncol. 2019, 30, 1925–1940. [Google Scholar] [CrossRef]
- Bai, L.; Yu, E. A Narrative Review of Risk Factors and Interventions for Cancer-Related Cognitive Impairment. Ann. Transl. Med. 2021, 9, 72. [Google Scholar] [CrossRef]
- Fernandes, H.A.; Richard, N.M.; Edelstein, K. Cognitive Rehabilitation for Cancer-Related Cognitive Dysfunction: A Systematic Review. Support. Care Cancer 2019, 27, 3253–3279. [Google Scholar] [CrossRef]
- Vardy, J.L.; Pond, G.R.; Bell, M.L.; Renton, C.; Dixon, A.; Dhillon, H.M. A Randomised Controlled Trial Evaluating Two Cognitive Rehabilitation Approaches for Cancer Survivors with Perceived Cognitive Impairment. J. Cancer Surviv. 2022, 17, 1583. [Google Scholar] [CrossRef]
- Campbell, K.L.; Zadravec, K.; Bland, K.A.; Chesley, E.; Wolf, F.; Janelsins, M.C. The Effect of Exercise on Cancer-Related Cognitive Impairment and Applications for Physical Therapy: Systematic Review of Randomized Controlled Trials. Phys. Ther. 2020, 100, 523. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, S.J.; Gómez, C.S.; Gutiérrez, S.S.; García-Tizón, S.J.; González, J.L.S.; Galve, M.I.R.; Sánchez, E.F.; Rodríguez, E.J.F. A Cognitive Training Programme on Cancer-Related Cognitive Impairment (CRCI) in Breast Cancer Patients Undergoing Active Treatment: A RCT Study Protocol. J. Clin. Med. 2025, 14, 5047. [Google Scholar] [CrossRef]
- Kiesl, D.; Kuzdas-Sallaberger, M.; Fuchs, D.; Brunner, S.; Kommenda, R.; Tischler, C.; Hornich, H.; Akbari, K.; Kellermair, J.; Blessberger, H.; et al. Protocol for the Exercise, Cancer and Cognition—The ECCO-Study: A Randomized Controlled Trial of Simultaneous Exercise During Neo-/Adjuvant Chemotherapy in Breast Cancer Patients and Its Effects on Neurocognition. Front. Neurol. 2022, 13, 777808. [Google Scholar] [CrossRef]
- Gates, P.; Green, H.J.; Gough, K.; Dhillon, H.; Vardy, J.L.; Dickinson, M.; Guarnera, J.; Krishnasamy, M.; Livingston, P.M.; White, V.; et al. Web-Based Cognitive Rehabilitation Intervention for Cancer-Related Cognitive Impairment Following Chemotherapy for Aggressive Lymphoma: Protocol for a Randomised Pilot Trial. BMJ Open 2024, 14, e081084. [Google Scholar] [CrossRef]
- Rapp, S.R.; Dressler, E.V.M.; Brown, W.M.; Wade, J.L.; Le-Lindqwister, N.; King, D.M.; Rowland, K.M.; Weaver, K.E.; Klepin, H.D.; Shaw, E.G.; et al. Phase 3 Randomized Placebo-Controlled Trial of Donepezil for Late Cancer-Related Cognitive Impairment in Breast Cancer Survivors Exposed to Chemotherapy from the Wake Forest NCORP Research Base REMEMBER Trial (WF97116). J. Clin. Oncol. 2023, 41, 12004. [Google Scholar] [CrossRef]
- Henneghan, A.; Rao, V.; Harrison, R.A.; Karuturi, M.; Blayney, D.W.; Palesh, O.; Kesler, S.R. Cortical Brain Age from Pre-Treatment to Post-Chemotherapy in Patients with Breast Cancer. Neurotox. Res. 2020, 37, 788. [Google Scholar] [CrossRef] [PubMed]
- Daniel, E.; Deng, F.; Patel, S.K.; Sedrak, M.S.; Kim, H.; Razavi, M.; Sun, C.-L.; Root, J.C.; Ahles, T.A.; Dale, W.; et al. Altered Gyrification in Chemotherapy-Treated Older Long-Term Breast Cancer Survivors. Res. Sq. 2023. [Google Scholar] [CrossRef]
- Mohammadi, M.; Banisharif, S.; Moradi, F.; Zamanian, M.; Tanzifi, G.; Ghaderi, S. Brain Diffusion MRI Biomarkers after Oncology Treatments. Rep. Pract. Oncol. Radiother. 2024, 28, 823. [Google Scholar] [CrossRef]
- Alexander, S.; Nikolaeva, A.; Pospelova, M.; Voynov, M.; Krasnikova, V.; Makhanova, A.; Tonyan, S.; Efimtsev, A.; Olga, F.; Levchuk, A.; et al. Diffusion Tensor Tractography Shows White Matter Tract Changes in Breast Cancer Survivors with Balance Impairment. Pathophysiology 2025, 32, 63. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Jung, C.O.; Jeon, H.R.; Sung, L.H. Rehabilitation for Ataxia Following Chemotherapy for Burkitt Lymphoma Involving the Rectum. Ann. Rehabil. Med. 2012, 36, 578–583. [Google Scholar] [CrossRef]
- Land, S.R.; Kopec, J.A.; Cecchini, R.S.; Ganz, P.A.; Wieand, H.S.; Colangelo, L.H.; Murphy, K.; Kuebler, J.P.; Seay, T.E.; Needles, B.M.; et al. Neurotoxicity from Oxaliplatin Combined with Weekly Bolus Fluorouracil and Leucovorin as Surgical Adjuvant Chemotherapy for Stage II and III Colon Cancer: NSABP C-07. J. Clin. Oncol. 2007, 25, 2205–2211. [Google Scholar] [CrossRef]
- Magge, R.S.; DeAngelis, L.M. The Double-Edged Sword: Neurotoxicity of Chemotherapy. Blood Rev. 2015, 29, 93–100. [Google Scholar] [CrossRef]
- Chen, B.T.; Chen, Z.; Deng, F.; Patel, S.K.; Sedrak, M.S.; Root, J.C.; Ahles, T.A.; Razavi, M.; Kim, H.; Sun, C.L.; et al. Signal Variability and Cognitive Function in Older Long-Term Survivors of Breast Cancer with Exposure to Chemotherapy: A Prospective Longitudinal Resting-State FMRI Study. Brain Sci. 2022, 12, 1283. [Google Scholar] [CrossRef] [PubMed]
- Bukkieva, T.; Pospelova, M.; Efimtsev, A.; Fionik, O.; Alekseeva, T.; Samochernych, K.; Gorbunova, E.; Krasnikova, V.; Makhanova, A.; Levchuk, A.; et al. Functional Network Connectivity Reveals the Brain Functional Alterations in Breast Cancer Survivors. J. Clin. Med. 2022, 11, 617. [Google Scholar] [CrossRef]
- Nikolaeva, A.; Pospelova, M.; Krasnikova, V.; Makhanova, A.; Tonyan, S.; Efimtsev, A.; Levchuk, A.; Trufanov, G.; Voynov, M.; Sklyarenko, M.; et al. MRI Voxel Morphometry Shows Brain Volume Changes in Breast Cancer Survivors: Implications for Treatment. Pathophysiology 2025, 32, 11. [Google Scholar] [CrossRef]
- Hatchard, T.; Penta, S.; Mioduzsewski, O.; Correia, S.; Tissera, T.; Brown, O.; Haefner, S.A.; Poulin, P.; Smith, A.M. Increased Gray Matter Following Mindfulness-Based Stress Reduction in Breast Cancer Survivors with Chronic Neuropathic Pain: Preliminary Evidence Using Voxel-Based Morphometry. Acta Neurol. Belg. 2022, 122, 735–743. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Biomarker Category | Representative Biomarkers | Biological Process Reflected | Key Findings in Chemotherapy-Treated Patients | References |
|---|---|---|---|---|
| Inflammatory biomarkers | IL-6, TNF-α, IL-1β, CRP | Chronic low-grade inflammation ("inflammaging") | Persistent elevation of pro-inflammatory cytokines after chemotherapy, associated with cognitive complaints and systemic aging phenotypes | [32,41,50,51,67] |
| Oxidative stress markers | Malondialdehyde, 4-hydroxynonenal, 8-hydroxy-2′-deoxyguanosine; ↓ glutathione | Oxidative damage and redox imbalance | Increased oxidative stress and reduced antioxidant capacity following chemotherapy, paralleling mechanisms of brain aging | [52,53,68] |
| Epigenetic aging markers | DNA methylation-based epigenetic clocks | Epigenetic age acceleration | Chemotherapy induces acceleration of epigenetic age relative to chronological age | [44,45,46,47] |
| Cellular senescence markers | p16INK4a, p21, IL-6, IL-8, MCP-1, MMPs | Therapy-induced cellular senescence | Increased senescence marker expression after cytotoxic therapy | [40,54,55] |
| Endothelial dysfunction markers | ICAM-1, PECAM-1 | Endothelial activation, vascular inflammation, BBB vulnerability | Significantly increased circulating ICAM-1 and PECAM-1 levels in breast cancer survivors after therapy, indicating persistent endothelial dysfunction | [56,58,59] |
| Neuronal injury markers | NSE | Neuronal stress and metabolic injury | Altered NSE levels in chemotherapy-treated patients, reflecting ongoing neuronal stress | [64,65] |
| Autoantibodies to neuronal antigens | Anti-NR-2 antibodies | NMDA receptor-related neuronal alterations | Altered levels after therapy indicating long-term CNS changes | [64,66] |
| Neurotrophic biomarkers | BDNF | ↓Synaptic plas-ticity, neurogene-sis, neuronal sur-vival | Reduced BDNF levels have been associated with cognitive im-pairment follow-ing chemothera-py | [60,61] |
| Synaptic function biomarkers | Dynamin-1 | ↓Synaptic vesicle trafficking, neurotransmission | Reduced Dynamin-1 levels has been associated with CRCI, suggesting synaptic dysfunction as a contributing mechanism | [63] |
| Clinical Domain | Key Manifestations |
|---|---|
| Cognitive function | Impaired attention, reduced processing speed, executive dysfunction |
| Memory | Short-term and working memory impairment, reduced verbal and visuospatial recall |
| Executive function | Difficulties in planning, multitasking, cognitive flexibility, decision-making |
| Processing speed and attention | Slowed information processing, reduced sustained attention |
| Affective symptoms | Anxiety, depressive symptoms, emotional lability |
| Fatigue and sleep disturbances | Persistent fatigue, insomnia, altered sleep–wake cycle |
| Vestibular and balance symptoms | Dizziness, gait instability, impaired balance |
| Subjective cognitive complaints | Self-reported “brain fog”, forgetfulness, reduced mental clarity |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Voynov, M.; Pospelova, M.; Nikolaeva, A.; Krasnikova, V.; Makhanova, A.; Fionik, O.; Samochernykh, K.; Alekseeva, T.; Combs, S.E.; Shevtsov, M. Multimodal Cancer Therapy and Accelerated Brain Aging: Mechanisms, Biomarkers, and Clinical Consequences. Curr. Oncol. 2026, 33, 121. https://doi.org/10.3390/curroncol33020121
Voynov M, Pospelova M, Nikolaeva A, Krasnikova V, Makhanova A, Fionik O, Samochernykh K, Alekseeva T, Combs SE, Shevtsov M. Multimodal Cancer Therapy and Accelerated Brain Aging: Mechanisms, Biomarkers, and Clinical Consequences. Current Oncology. 2026; 33(2):121. https://doi.org/10.3390/curroncol33020121
Chicago/Turabian StyleVoynov, Mark, Maria Pospelova, Alexandra Nikolaeva, Varvara Krasnikova, Albina Makhanova, Olga Fionik, Konstantin Samochernykh, Tatyana Alekseeva, Stephanie E. Combs, and Maxim Shevtsov. 2026. "Multimodal Cancer Therapy and Accelerated Brain Aging: Mechanisms, Biomarkers, and Clinical Consequences" Current Oncology 33, no. 2: 121. https://doi.org/10.3390/curroncol33020121
APA StyleVoynov, M., Pospelova, M., Nikolaeva, A., Krasnikova, V., Makhanova, A., Fionik, O., Samochernykh, K., Alekseeva, T., Combs, S. E., & Shevtsov, M. (2026). Multimodal Cancer Therapy and Accelerated Brain Aging: Mechanisms, Biomarkers, and Clinical Consequences. Current Oncology, 33(2), 121. https://doi.org/10.3390/curroncol33020121