Mechanisms and Functions of γδ T Cells in Tumor Cell Recognition

 and
and
Abstract
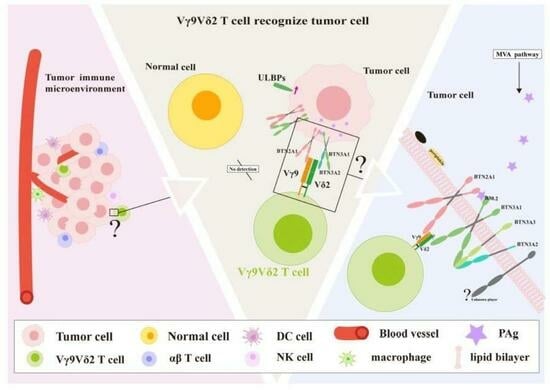
1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| γδ T-Cell Subset | Paired Vγ Gene Usage | Distribution | Features | Tumor-Related Functions | References |
| Vδ1+T cells | Vγ2/3/4/5/8/9 | PB, skin, gut, Spleen, liver | Express NK cell receptors, Toll-like receptors, co-stimulatory factors; exhibit cytotoxicity against tumor cells via IFN-γ, and IL-10; low levels of IL-4, perforin, and granzyme | 1. Promote tumor growth by secreting cytokines like IL-17 that induce vascular endothelial growth factor (VEGF) secretion from tumor cells 2. Antitumor effect: In some hematological malignancies (such as leukemia), the clonal expansion and cytotoxicity of adult Vδ1 cells may enhance the tumor-killing ability, which is associated with a better prognosis | [18,19,20,21,22,23] |
| Vδ2+T cells | Vγ9 | PB | Mainly Vγ9Vδ2 T cells responding to phosphorylated non-peptide “PAgs”; categorized into subgroups based on CD27 and CD45RA expression; naive and central memory cells respond to isopentenyl pyrophosphate (IPP), effector memory cells produce high IFN-γ, and terminally differentiated cells secrete perforin and granzyme | Effector memory Vδ2+ T cells have strong antitumor capacity, while terminally differentiated cells exert cytotoxic effects; activated Vδ2+ T cells can serve as antigen-presenting cells (APCs) | [24,25,26,27] |
| Vδ3+T cells | Vγ2/3 | PB, liver | Express CD56, CD161, NKG2D; enhance CD1d recognition and act on CD1d target cells expressing CD107a | Limited investigations: functional role in tumor-related studies is not well defined | [28,29] |
| Vδ5+T cells | Vγ4 | PB | EPCR | [16] | |
| Functional Subsets | Source | Secreted Cytokines | Function | References | |
| IFN-γ+ γδ T cell | thymus origin | IFN-γ | Functionally diverse: autoimmune diseases and tumor surveillance | [30] | |
| IL-17+ γδ T cell | Vδ1γδ T-cell subpopulation of thymus origin | IL-17; | Rapid induction of IL-8-mediated migration and phagocytosis of neutrophils | [31] | |
| γδ Treg | Vδ1γδ T-cell subpopulation | IFN-γ; GM-CSF | Inhibitory effect on the proliferation of autologous innate CD4+T cells | [32] | |
| γδ T-APC | Initiated a specific immune response | [13] | |||
| TIGIT + γδ T | Vδ1γδ T cells | Dysfunctional effector state | [33] | ||
2. Mechanisms Underlying Tumor Cell Recognition and the Stimulation of γδ T Cells
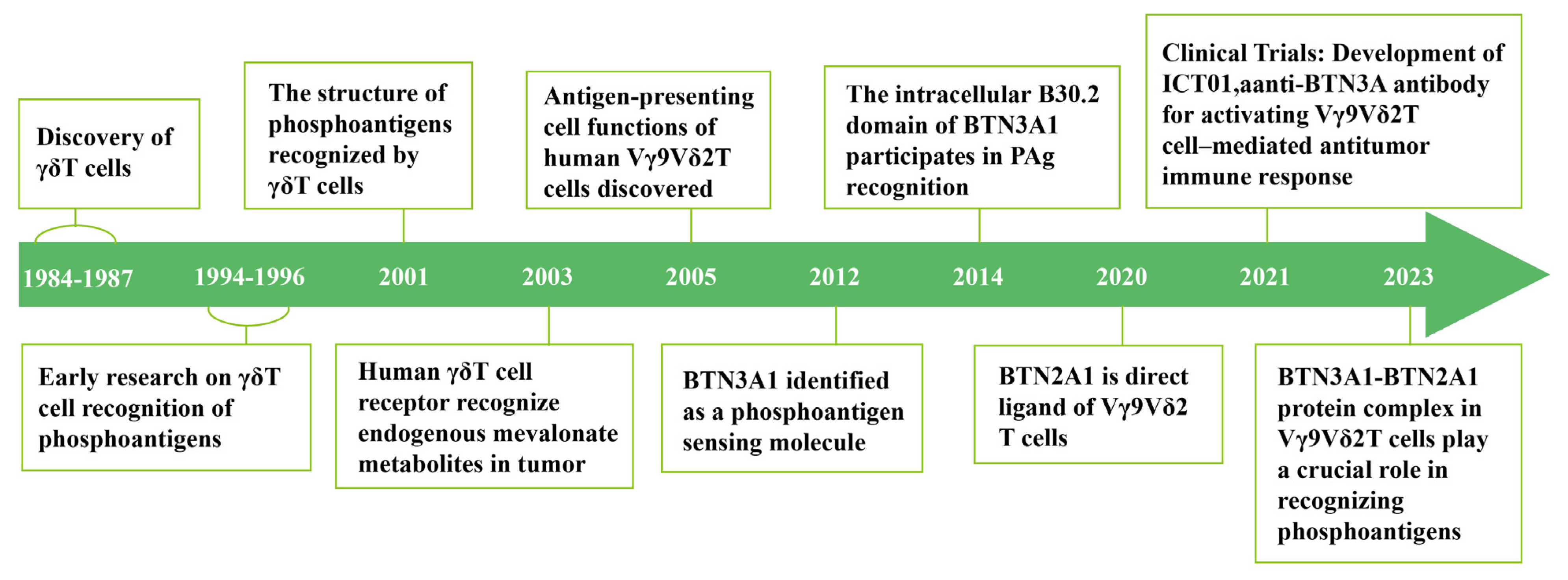
2.1. Early Studies on γδ T-Cell Activation: The Initial Discovery of PAgs
| Name | Specific Source | Biological Activity | Research Progress | References |
|---|---|---|---|---|
| TUBag4 | Mycobacterium tuberculosis H37RV strain | Stimulates Vγ9Vδ2 T cell expansion supports the hypothesis of γδ T cell recognition of non-peptide ligands | First isolated from M. tuberculosis, confirming non-peptide ligands can activate Vγ9Vδ2 T cells. | [9] |
| IPP | Tumor cells, bacteria (e.g., Mycobacterium smegmatis), eukaryotic mevalonate pathway | Weak agonist requires higher concentrations to activate Vγ9Vδ2 T cells | Natural intermediate of MVP in eukaryotic cells, increased expression in tumor cells. | [12] |
| DMAPP | Tumor cells, bacteria, eukaryotic mevalonate pathway | Weak agonist, similar to IPP, requires higher concentrations to activate Vγ9Vδ2 T cells | Like IPP, an intermediate of MVP, increased expression in tumor cells. | [12] |
| HMBPP | Bacteria (e.g., E. coli, M. tuberculosis), parasites (e.g., Plasmodium) via MEP pathway | Strongest natural agonist, activates Vγ9Vδ2 T cells at very low concentrations | In 2023, its hydroxyl group was found to form hydrogen bonds with BTN3A1, explaining its high potency. | [17] |
| BrHPP | Synthetic compound (modified from natural phosphoantigen structures) | Highly efficient synthetic activator, activity close to HMBPP | Widely used in clinical research as a substitute for HMBPP. | [11] |
| Zoledronic Acid (ZOL) | Synthetic amino bisphosphonate (originally developed for osteoporosis treatment) | Indirectly activates Vγ9Vδ2 T cells by inhibiting the MVP pathway and increasing IPP levels | Used in immunotherapy, confirming its immunomodulatory effects. | [43] |
| Pamidronate | Synthetic amino bisphosphonate | Indirectly activates Vγ9Vδ2 T cells by inhibiting the MVP pathway and increasing IPP levels | Similar to ZOL, it is used in cancer treatment research. | [44] |
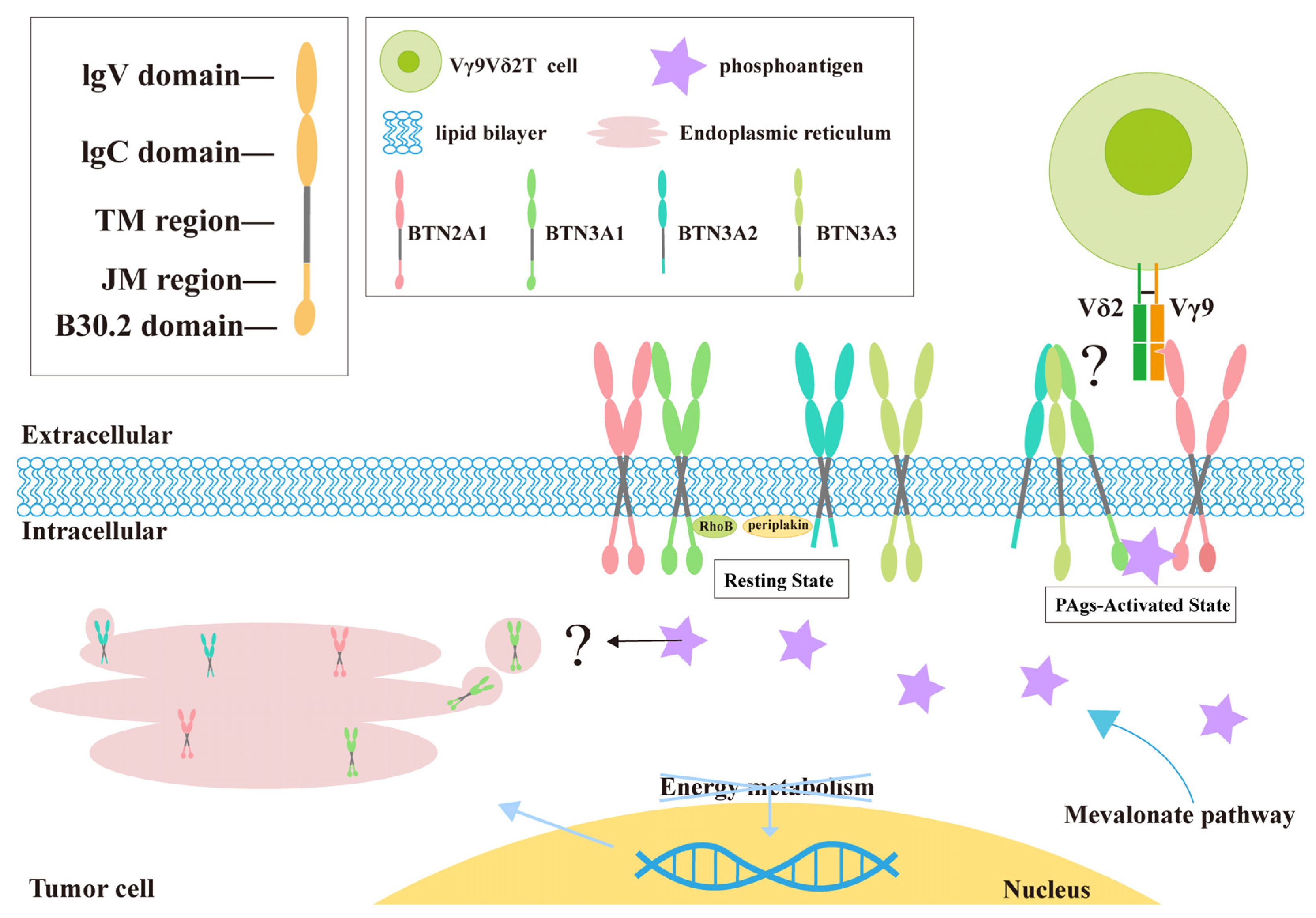
2.2. How γδ T Cells Detect PAg: BTN3A Family
2.3. Various Factors Induce Changes in Cell Membrane Fluidity
2.4. BTN2A1 Serves as a Direct Ligand for Vγ9Vδ2 T Cells
3. The Two-Fold Function of γδ T Cells in the TME
3.1. The Direct Antitumor Effect of γδ T Cells
3.2. Coordination of Additional Cells by γδ T Cells in Antitumor Activity
3.3. The Promoting Effect of γδ T Cells on Tumors
3.4. Coordination of Additional Cells by γδ T Cells in Promoting Tumors
4. Immunotherapy with γδ T Cells
| Subset | Tumor Type | Function and Prognostic Association | Role in TME | Potential Therapeutic Strategies | References |
|---|---|---|---|---|---|
| Vδ1+ γδ T Cells | Colorectal Cancer | Poor prognosis (MSS type): this comprises 74.4% of γδ T cells with impaired function (reduced levels of cytotoxic molecules such as perforin, granzyme B, and IFN-γ). | Inflammatory fibroblasts overexpress NECTIN2, which binds to TIGIT on Vδ1+ cells, thereby suppressing their activity. | Anti-TIGIT antibodies or NECTIN2 blockade. | [84] |
| Favorable prognosis (MSI type): maintains strong cytotoxicity (granzyme B and IFN-γ). | Direct tumor cell killing. | PD-1 inhibitors are effective, but only in a minority of cases. | [84] | ||
| Non-Small Cell Lung Cancer | Favorable prognosis: high abundance of intratumoral Vδ1+ T cells is associated with recurrence-free survival. TCGA data indicate a longer overall survival in patients with high TRDV1 expression. | CD103+ Vδ1+ T cells colonize lung tissue and recognize early tumor stress signals independent of MHC restriction. | Expand CD103+ Vδ1+ T cells ex vivo for adoptive transfer to enhance tumor targeting. | [21] | |
| Merkel Cell Carcinoma | 1. Enriched in MHC-I-deficient tumors, compensating for CD8+ T cell limitations. 2. Vδ1+ clonal expansion correlates with prolonged survival. | 1. NKG2D-mediated killing of MHC-I-deficient tumors. 2. Direct recognition of MCPyV viral peptides via TCR. | Design MCPyV peptide vaccines to enhance Vδ1+ T cell expansion. | [106] | |
| Ovarian Cancer | CD3+Vδ1+ T cells are significantly elevated in ovarian cancer patients and correlate with advanced FIGO stage and metastasis. | High Foxp3 and Vδ1 expression, low CD28, maintaining immunosuppressive function and promoting progression. | Target Vδ1+ surface markers (e.g., Vδ1, Foxp3) to block immunosuppression. | [107] | |
| Hepatocellular Carcinoma (HCC) | 1. Increased Vδ1+/Vδ2+ ratio correlates with shorter survival. 2. CD69+ Vδ1+ T cells are antitumor subpopulations linked to smaller tumor size and prolonged survival. | 1. Synergizes with apoptosis, ferroptosis, and pyroptosis pathways; PD-1/PD-L1 overexpression. 2. CD69+ Vδ1+ T cells localize to tumor sites for direct cytotoxicity. | 1. Combine PD-1/PD-L1 inhibitors to reverse T cell exhaustion. 2. Expand CD69+ Vδ1+ T cells ex vivo for adoptive transfer. | [108,109] | |
| Vδ2+ γδ T Cells | Renal Cell Carcinoma | No direct prognostic correlation, but γδ T cell models (including Vδ2) predict immunotherapy response. | Functionally restricted in high TGF-β or IL-10 environments; requires combination therapy. | Zoledronic acid or BTN3A1 agonists to enhance activity. | [110] |
| Colon Adenocarcinoma | Vδ2+ infiltration correlates with inflammation but lacks standalone prognostic value. | Activity depends on tumor BTN3A1 expression and phosphoantigen availability; suppressed in TGF-β-rich TME. | Pre-treat tumor cells with zoledronic acid to increase IPP release and activate Vδ2+ cells. | [111] | |
| Breast Cancer | Reduced peripheral Vδ2+ T cell levels correlate with tumor progression. | Vγ9Vδ2 TCR recognizes tumor metabolic stress via BTN3A1. | Adoptive transfer of ex vivo-expanded Vδ2+ cells combined with IL-2 to sustain activity. | [112] | |
| Ovarian Cancer | No significant difference in CD3+Vδ2+ T cell proportions between benign and malignant tumors. | Likely not directly involved in immunosuppression. | Not recommended as a therapeutic target. | [107] | |
| Multiple Myeloma | Reduced peripheral Vδ2+ T cells correlate with advanced disease; bone marrow Vδ2+ T cell infiltration links to relapse/refractory MM. | CXCL10 recruits γδ T cells via CXCR3 into hypoxic bone marrow, promoting IL-17+ polarization. | Restore Vδ2+ function with PD-1 inhibitors combined with SRC-3 inhibitors. | [113] |
4.1. Research on BTN3A1 and Vγ9Vδ2 T-Cell Immunotherapy
4.2. Types of Immunotherapeutic γδ T Cells
| Clinical Trials Gov Identifier | Interventions | Cancers/Tumors | Phase | Outcomes/Preliminary Findings |
|---|---|---|---|---|
| Autologous/Allogeneic γδ T cells | ||||
| NCT02418481 | γδ T cells with or without DC-CIK cells | Breast Cancer | I/II | No published results (Study Completion June 2016). |
| NCT02425735 | Vγ9Vδ2 T cells with or without DC-CIK cells | Cholangiocarcinoma | I/II | Modulated immune functions, reduced tumor activity, enhanced quality of life, and extended lifespan. Following eight γδ T cell treatments, there was a significant reduction in lymph node size along with diminished activity [123]. |
| NCT02425748 | γδ T cells with or without DC-CIK cells | Lung Cancer | I/II | No published results (Study Completion 20 June 2019). Offer another promising immunotherapy approach. |
| NCT02585908 | Vγ9Vδ2 T cells with or without CIK cells | Gastric Cancer | I/II | No published results (Study Completion December 2022). |
| NCT03180437 | Vγ9Vδ2 T cells with IRE surgery | Locally Advanced Pancreatic Cancer | I/II | Strengthened immune response, inhibited tumor expansion, and prolonged the survival of liver and pancreas cancer patients [139]. |
| NCT03183232 | γδ T cells with Cryosurgery or IRE | Liver Cancer Lung Cancer | I/II | Decreased tumor volume and increased survival in mice. Allogeneic Vγ9Vδ2 T cells have shown clinical safety and initial evidence of therapeutic effectiveness in patients with solid tumors [122]. |
| NCT03533816 | Ex-vivo Expanded/Activated γδ T-cell Infusion | Hematological Malignancies | I | Assessing the maximum tolerated dose and safety profile of autologous gamma-delta T cells in leukemia patients who have undergone a partially matched bone marrow transplant. |
| NCT03790072 | Ex-vivo Expanded Allogeneic γδ T-lymphocytes (OmnImmune®) | Acute Myeloid Leukemia | I | Allogeneic Vγ9Vδ2 T-cell infusion was shown to be safe and feasible up to a cell dose of 108/kg [140]. |
| NCT04764513 | Ex-vivo expanded γδ T-cell infusion | Acute Myeloid Leukemia Acute Lymphoblastic Leukemia Myelodysplastic Syndromes Lymphoma | I/II | Recruiting (Study Completion December 2025). |
| NCT04518774 | Ex-vivo expanded Allogeneic γδ T cells | Hepatocellular Carcinoma | Early Phase I | No published results (Study Completion 15 August 2021). |
| NCT04696705 | Ex-vivo expanded Allogeneic γδ T cells | Non-Hodgkin’s Lymphoma and Peripheral T-Cell Lymphomas | Early phase I | No published results (Study Completion 25 December 2023). |
| NCT04765462 | Allogeneic γδ T cells | Malignant Solid Tumors | I/II | No published results (Study Completion 31 December 2024). |
| NCT05015426 | γδ T-Cell Infusion | Acute Myeloid Leukemia | I | Not Recruiting (Study Completion September 2026). |
| NCT05358808 | Ex-Vivo expanded Allogeneic γδ T-lymphocytes (TCB008) | Acute Myeloid Leukemia Myelodysplastic Syndromes | II | Recruiting (Study Completion December 2025). |
| NCT05628545 | Allogeneic γδ-T Cells (GDKM-100) | Advanced Hepatocellular Carcinoma | I/II | No published results (Study Completion 31 October 2024). |
| NCT05886491 | Allogeneic Vδ1 T cells | Acute Myeloid Leukemia | I/II | Recruiting (Study Completion 30 June 2027). |
| CAR-γδ T cell | ||||
| NCT02656147 | Anti-CD19-CAR-γδ T cell | Leukemia and Lymphoma | I | No published results (Study Completion April 2020). |
| NCT04107142 | NKG2DL-targeting CAR-γδ T cell | Solid Cancer | I | NKG2DL-targeting CAR-γδ T cells enhanced cytotoxicity against tumor cell lines, with Vγ9Vδ2 T cells modified by NKG2D RNA-based CAR showing notable therapeutic effects in mouse tumor models [141]. |
| NCT04702841 | CAR-γδ T cell | Relapsed and Refractory CD7 positive T | I | No published results (Study Completion December 2022). |
| NCT04735471/ NCT04911478/ NCT06375993 | ADI-001 Anti-CD20 CAR-engineered Allogeneic γδ T Cells | Lymphoma, Follicular Lymphoma, Mantle-Cell Marginal Zone Lymphoma Primary Mediastinal B-cell Lymphoma/Lupus Nephritis Autoimmune Diseases | I | CD20 CAR-modified Vδ1 γδ T cells did not cause xenogeneic graft-versus-host disease in immunodeficient mice. They demonstrated tumor cell lysis in vitro and proinflammatory cytokine release, as well as inhibition of B-cell lymphoma xenograft growth in immunodeficient mice [142]. |
| NCT05388305 | CAR-γδ T cell | Acute myeloid leukemia | Not applicable | No published results (Study Completion 30 May 2023). |
| NCT05302037 | Allogeneic NKG2DL-targeting CAR-grafted γδ T cells (CTM-N2D) | Malignancy Refractory Cancer | I | Recruiting (Study Completion December 2026). |
| NCT05554939 | Allogeneic CD19-CAR-γδ T cell | Non-Hodgkin’s Lymphoma | I/II | Recruiting (Study Completion 31 December 2026). |
| NCT05653271 | Allogeneic CD20-conjugated γδ T-cell | B-cell Lymphoma Non-Hodgkin’s Lymphoma Primary Mediastinal Large B Cell Lymphoma | I | Recruiting (Study Completion September 2027). |
| NCT06106893 | CD19 Universal CAR-γδ T cells | Systemic Lupus Erythematosus | I/II | Recruiting (Study Completion December 2027). |
| NCT06150885 | CAR-γδ T cells CAR001 | Solid Tumor | I/II | Recruiting (Study Completion 30 September 2027). |
| NCT06404281 | γδ T-PD-1 Ab cells | Advanced Solid Tumors | I | Recruiting (Study Completion 1 June 2026). |
| NCT06480565 | ADI-270 (engineered γδ Chimeric Receptor CAR Vδ1 T cells Targeting CD70) | Clear Cell Renal Cell Carcinoma | I/II | Recruiting (Study Completion June 2027). |
| Antibodies with Autologous/Allogeneic γδ T cells | ||||
| NCT04243499 | Anti-BTN3A | Hematological and Solid Ttumors | I/II | Good tolerability and pharmacodynamic activity in initial patients, with the potential to enhance immune cell infiltration into the tumor microenvironment [8]. |
| NCT06364800 | Allogeneic γδ T cells combined with targeted therapy and PD-1 | Hepatocellular Carcinoma | Early Phase 1 | Recruiting (Study Completion 26 September 2026). |
| NCT06212388 | Allogeneic γδ T cells Combined with Interferon-alpha1b or PD-1 | Melanoma | Early Phase 1 | Recruiting (Study Completion 30 October 2028). |
| Drug with Autologous/Allogeneic γδ T cells | ||||
| NCT04165941 | Drug Resistant Immunotherapy with Activated, Gene-Modified γδ T cells | Glioblastoma Multiforme | I | Increased median survival in mice [143]. |
| NCT05400603 | Ex Vivo Expanded Allogeneic γδ T cells in Combination with Dinutuximab, Temozolomide, Irinotecan, and Zoledronate | Neuroblastoma Refractory Neuroblastoma Relapsed Neuroblastoma Relapsed Osteosarcoma Refractory Osteosarcoma | I | Recruiting (Study Completion December 2025). |
| NCT05664243 | Gene-Modified Allogeneic or Autologous γδ T cells | Glioblastoma | I/II | Recruiting (Study Completion December 2025). |
| NCT06364787 | Allogeneic Gamma-delta T cells combined with targeted therapy and immunotherapy | Hepatocellular Carcinoma | I | Recruiting (Study Completion September 2026). |
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| PAg | Phosphoantigen |
| MVP | Mevalonate pathway |
| BTN3A1 | Butyrophilin Subfamily 3 Member A1 |
| BTN2A1 | Butyrophilin Subfamily 2 Member A1 |
| TME | Tumor microenvironment |
| MHC | Major histocompatibility complex |
| αβT | Alpha-Beta T |
| γδ T | Gamma-Delta T |
| TCR | T Cell Receptor |
| VEGF | Vascular endothelial growth factor |
| GM-CSF | Granulocyte-Macrophage Colony-Stimulating Factor |
| IPP | Isopentenyl Pyrophosphate |
| APCs | Antigen-Presenting Cells |
| DMAPP | Dimethylallyl Pyrophosphate |
| HMBPP | Hydroxymethylbutenyl Diphosphate |
| HMGR | 3-Hydroxy-3-Methylglutaryl-CoA Reductase |
| MEP | methylerythritol phosphate pathway |
| β2M | β2-microglobulin |
| JM | juxtamembrane |
| NK cell | Natural killer cell |
| DC | Dendritic cell |
| TAAs | Tumor-associated antigens |
References
- de Visser, K.E.; Joyce, J.A. The Evolving Tumor Microenvironment: From Cancer Initiation to Metastatic Outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.; Agrawal, A.; Borthakur, G.; Battula, V.L.; Maiti, A. Gamma Delta T Cells in Acute Myeloid Leukemia: Biology and Emerging Therapeutic Strategies. J. Immunother. Cancer 2024, 12, e007981. [Google Scholar] [CrossRef]
- Bellio, M.; Lone, Y.C.; de la Calle-Martin, O.; Malissen, B.; Abastado, J.P.; Kourilsky, P. The V beta complementarity determining region 1 of a major histocompatibility complex (MHC) class I-restricted T cell receptor is involved in the recognition of peptide/MHC I and superantigen/MHC II complex. J. Exp. Med. 1994, 179, 1087–1097. [Google Scholar] [CrossRef]
- Sandoz, P.A.; Kuhnigk, K.; Szabo, E.K.; Thunberg, S.; Erikson, E.; Sandström, N.; Verron, Q.; Brech, A.; Watzl, C.; Wagner, A.K.; et al. Modulation of Lytic Molecules Restrain Serial Killing in γδ T Lymphocytes. Nat. Commun. 2023, 14, 6035. [Google Scholar] [CrossRef]
- Silva-Santos, B. γδ T Cells in Cancer. Nat. Rev. Immunol. 2015, 15, 683–691. [Google Scholar] [CrossRef]
- Sebestyen, Z. Translating Gammadelta (γδ) T Cells and Their Receptors into Cancer Cell Therapies. Nat. Rev. Drug Discov. 2020, 19, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Hu, Q.; Li, Y.; Lu, L.; Xiang, Z.; Yin, Z.; Kabelitz, D.; Wu, Y. γδ T Cells: Origin and Fate, Subsets, Diseases and Immunotherapy. Signal Transduct. Target. Ther. 2023, 8, 434. [Google Scholar] [PubMed]
- De Gassart, A. Development of ICT01, a First-in-Class, Anti-BTN3A Antibody for Activating Vγ9Vδ2 T Cell–Mediated Antitumor Immune Response. Sci. Transl. Med. 2021, 13, eabj0835. [Google Scholar] [CrossRef]
- Constant, P.; Davodeau, F.; Peyrat MAPoquet, Y.; Puzo, G.; Bonneville, M.; Fournié, J.J. Stimulation of Human Gamma Delta T Cells by Nonpeptidic Mycobacterial Ligands. Science 1994, 264, 267–270. [Google Scholar] [CrossRef]
- Morita, C.T.; Beckman, E.M.; Bukowski, J.F.; Band, H.; Bloom, B.R.; Golan, D.E.; Brenner’, B. Direct Presentation of Nonpeptide Prenyl Pyrophosphate Antigens to Human Gamma Delta T Cells. Immunity 1996, 3, 495–507. [Google Scholar] [CrossRef]
- Espinosa, E.; Belmant, C.; Pont, F.; Luciani, B.; Poupot, R.; Romagné, F.; Brailly, H.; Bonneville, M.; Fournié, J.-J. Chemical synthesis and biological activity of bromohydrin pyrophosphate, a potent stimulator of human gamma delta T cells. J. Biol. Chem. 2001, 276, 18337–18344. [Google Scholar] [CrossRef] [PubMed]
- Gober, H.-J.; Kistowska, M.; Angman, L.; Jenö, P.; Mori, L.; Libero, G.D. Human T Cell Receptor Gammadelta Cells Recognize Endogenous Mevalonate Metabolites in Tumor Cells. J. Exp. Med. 2003, 197, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Moser, B.; Brandes, M. γδ T Cells: An Alternative Type of Professional APC. Trends Immunol. 2006, 27, 112–118. [Google Scholar] [CrossRef]
- Harly, C.; Guillaume, Y.; Nedellec, S.; Peigné, C.-M.; Mönkkönen, H.; Li, J.; Kuball, J.; Adams, E.J.; Netzer, S.; Déchanet-Merville, J.; et al. Key Implication of CD277/Butyrophilin-3 (BTN3A) in Cellular Stress Sensing by a Major Human γδ T-Cell Subset. Blood 2012, 120, 2269–2279. [Google Scholar] [CrossRef]
- Sandstrom, A.; Peigné, C.-M.; Léger, A.; Crooks, J.E.; Konczak, F.; Gesnel, M.-C.; Breathnach, R.; Bonneville, M.; Scotet, E.; Adams, E.J. The Intracellular B30.2 Domain of Butyrophilin 3A1 Binds Phosphoantigens to Mediate Activation of Human Vγ9Vδ2 T Cells. Immunity 2014, 40, 490–500. [Google Scholar] [CrossRef]
- Cano, C.E.; Pasero, C.; De Gassart, A.; Kerneur, C.; Gabriac, M.; Fullana, M.; Granarolo, E.; Hoet, R.; Scotet, E.; Rafia, C.; et al. BTN2A1, an Immune Checkpoint Targeting Vγ9Vδ2 T Cell Cytotoxicity against Malignant Cells. Cell Rep. 2021, 36, 109359. [Google Scholar] [CrossRef]
- Yuan, L.; Ma, X.; Yang, Y.; Qu, Y.; Li, X.; Zhu, X.; Ma, W.; Duan, J.; Xue, J.; Yang, H.; et al. Phosphoantigens Glue Butyrophilin 3A1 and 2A1 to Activate Vγ9Vδ2 T Cells. Nature 2023, 621, 840–848. [Google Scholar] [CrossRef]
- Zakeri, N.; Hall, A.; Swadling, L.; Pallett, L.J.; Schmidt, N.M.; Diniz, M.O.; Kucykowicz, S.; Amin, O.E.; Gander, A.; Pinzani, M.; et al. Characterisation and Induction of Tissue-Resident Gamma Delta T-Cells to Target Hepatocellular Carcinoma. Nat. Commun. 2022, 13, 1372. [Google Scholar] [CrossRef] [PubMed]
- Jandke, A.; Melandri, D.; Monin, L.; Ushakov, D.S.; Laing, A.G.; Vantourout, P.; East, P.; Nitta, T.; Narita, T.; Takayanagi, H.; et al. Butyrophilin-like Proteins Display Combinatorial Diversity in Selecting and Maintaining Signature Intraepithelial γδ T Cell Compartments. Nat. Commun. 2020, 11, 3769. [Google Scholar] [CrossRef]
- Wu, Y.; Kyle-Cezar, F.; Woolf, R.T.; Naceur-Lombardelli, C.; Owen, J.; Biswas, D.; Lorenc, A.; Vantourout, P.; Gazinska, P.; Grigoriadis, A.; et al. An Innate-like Vδ1+ γδ T Cell Compartment in the Human Breast Is Associated with Remission in Triple-Negative Breast Cancer. Sci. Transl. Med. 2019, 11, eaax9364. [Google Scholar] [CrossRef]
- Wu, Y.; Biswas, D.; Usaite, I.; Angelova, M.; Boeing, S.; Morton, C.; Joseph, M.; Hessey, S.; Georgiou, A.; Al-Bakir, M.; et al. A Local Human Vδ1 T Cell Population Is Associated with Survival in Nonsmall-Cell Lung Cancer. Nat. Cancer 2022, 3, 696–709. [Google Scholar] [CrossRef]
- Foord, E.; Arruda, L.C.M.; Gaballa, A.; Klynning, C.; Uhlin, M. Characterization of ascites- and tumor-infiltrating γδ T cells reveals distinct repertoires and a beneficial role in ovarian cancer. Sci. Transl. Med. 2021, 13, eabb0192. [Google Scholar] [CrossRef]
- Wu, D.; Wu, P.; Qiu, F.; Wei, Q.; Huang, J. Human γδ T-Cell Subsets and Their Involvement in Tumor Immunity. Cell Mol. Immunol. 2017, 14, 245–253. [Google Scholar] [CrossRef]
- Fiala, G.J.; Gomes, A.Q.; Silva-Santos, B. From Thymus to Periphery: Molecular Basis of Effector γδ-T Cell Differentiation. Immunol. Rev. 2020, 298, 47–60. [Google Scholar] [CrossRef]
- Ng, J.W.K.; Tan, K.W.; Guo, D.Y.; Lai, J.J.H.; Fan, X.; Poon, Z.; Lim, T.H.; Lim, A.S.T.; Lim, T.K.H.; Hwang, W.Y.K.; et al. Cord Blood-Derived Vδ2+ and Vδ2–T Cells Acquire Differential Cell State Compositions upon in Vitro Expansion. Sci. Adv. 2023, 9, eadf3120. [Google Scholar] [CrossRef]
- Hsu, H.; Boudova, S.; Mvula, G.; Divala, T.H.; Rach, D.; Mungwira, R.G.; Boldrin, F.; Degiacomi, G.; Manganelli, R.; Laufer, M.K.; et al. Age-Related Changes in PD-1 Expression Coincide with Increased Cytotoxic Potential in Vδ2 T Cells during Infancy. Cell Immunol. 2021, 359, 104244. [Google Scholar] [CrossRef]
- Harmon, C.; Zaborowski, A.; Moore, H.; St. Louis, P.; Slattery, K.; Duquette, D.; Scanlan, J.; Kane, H.; Kunkemoeller, B.; McIntyre, C.L.; et al. γδ T Cell Dichotomy with Opposing Cytotoxic and Wound Healing Functions in Human Solid Tumors. Nat. Cancer 2023, 4, 1122–1137. [Google Scholar] [CrossRef]
- Petrasca, A.; Melo, A.M.; Breen, E.P.; Doherty, D.G. Human Vδ3+ γδ T Cells Induce Maturation and IgM Secretion by B Cells. Immunol. Lett. 2018, 196, 126–134. [Google Scholar] [CrossRef]
- Rice, M.T.; Von Borstel, A.; Chevour, P.; Awad, W.; Howson, L.J.; Littler, D.R.; Gherardin, N.A.; Le Nours, J.; Giles, E.M.; Berry, R.; et al. Recognition of the Antigen-Presenting Molecule MR1 by a Vδ3+ γδ T Cell Receptor. Proc. Natl. Acad. Sci. USA 2021, 118, e2110288118. [Google Scholar] [CrossRef]
- Muñoz-Ruiz, M.; Llorian, M.; D’Antuono, R.; Pavlova, A.; Mavrigiannaki, A.M.; McKenzie, D.; García-Cassani, B.; Iannitto, M.L.; Wu, Y.; Dart, R.; et al. IFN-γ–Dependent Interactions between Tissue-Intrinsic γδ T Cells and Tissue-Infiltrating CD8 T Cells Limit Allergic Contact Dermatitis. J. Allergy Clin. Immunol. 2023, 152, 1520–1540. [Google Scholar] [CrossRef]
- Papotto, P.H.; Reinhardt, A.; Prinz, I.; Silva-Santos, B. Innately Versatile: γδ17 T Cells in Inflammatory and Autoimmune Diseases. J. Autoimmun. 2018, 87, 26–37. [Google Scholar] [CrossRef]
- Si, F.; Liu, X.; Tao, Y.; Zhang, Y.; Ma, F.; Hsueh, E.C.; Puram, S.V.; Peng, G. Blocking Senescence and Tolerogenic Function of Dendritic Cells Induced by γδ Treg Cells Enhances Tumor-Specific Immunity for Cancer Immunotherapy. J. Immunother. Cancer 2024, 12, e008219. [Google Scholar] [CrossRef]
- Jin, Z.; Lan, T.; Zhao, Y.; Du, J.; Chen, J.; Lai, J.; Xu, L.; Chen, S.; Zhong, X.; Wu, X.; et al. Higher TIGIT+CD226-γδ T cells in Patients with Acute Myeloid Leukemia. Immunol. Investig. 2022, 51, 40–50. [Google Scholar] [CrossRef]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The Immunobiology of Cancer Immunosurveillance and Immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef]
- Peters, B.; Nielsen, M.; Sette, A. T Cell Epitope Predictions. Annu. Rev. Immunol. 2020, 38, 123–145. [Google Scholar] [CrossRef]
- Uldrich, A.P.; Rigau, M.; Godfrey, D.I. Immune Recognition of Phosphoantigen-butyrophilin Molecular Complexes by γδ T Cells. Immunol. Rev. 2020, 298, 74–83. [Google Scholar] [CrossRef]
- Tanaka, Y. Cancer Immunotherapy Harnessing γδ T Cells and Programmed Death-1. Immunol. Rev. 2020, 298, 237–253. [Google Scholar] [CrossRef]
- Rigau, M.; Uldrich, A.P.; Behren, A. Targeting Butyrophilins for Cancer Immunotherapy. Trends Immunol. 2021, 42, 670–680. [Google Scholar] [CrossRef]
- Fichtner, A.S.; Karunakaran, M.M.; Gu, S.; Boughter, C.T.; Borowska, M.T.; Starick, L.; Nöhren, A.; Göbel, T.W.; Adams, E.J.; Herrmann, T. Alpaca (Vicugna Pacos), the First Nonprimate Species with a Phosphoantigen-Reactive Vγ9Vδ2 T Cell Subset. Proc. Natl. Acad. Sci. USA 2020, 117, 6697–6707. [Google Scholar] [CrossRef]
- Laplagne, C.; Ligat, L.; Foote, J.; Lopez, F.; Fournié, J.-J.; Laurent, C.; Valitutti, S.; Poupot, M. Self-Activation of Vγ9Vδ2 T Cells by Exogenous Phosphoantigens Involves TCR and Butyrophilins. Cell Mol. Immunol. 2021, 18, 1861–1870. [Google Scholar] [CrossRef]
- Mullen, P.J.; Yu, R.; Longo, J.; Archer, M.C.; Penn, L.Z. The Interplay between Cell Signalling and the Mevalonate Pathway in Cancer. Nat. Rev. Cancer 2016, 16, 718–731. [Google Scholar] [CrossRef]
- Kabelitz, D.; Serrano, R.; Kouakanou, L.; Peters, C.; Kalyan, S. Cancer Immunotherapy with γδ T Cells: Many Paths Ahead of Us. Cell Mol. Immunol. 2020, 17, 925–939. [Google Scholar] [CrossRef]
- Kuroda, J.; Kimura, S.; Segawa, H.; Kobayashi, Y.; Yoshikawa, T. The third-generation bisphosphonate zoledronate synergistically augments the anti-Ph+ leukemia activity of imatinib mesylate. Blood 2003, 102, 2229–2235. [Google Scholar] [CrossRef]
- Li, H.; Xiang, Z.; Feng, T.; Li, J.; Liu, Y. Human Vγ9Vδ2-T cells efficiently kill influenza virus-infected lung alveolar epithelial cells. Cell Mol. Immunol. 2013, 10, 159–164. [Google Scholar] [CrossRef]
- Burke, K.P.; Chaudhri, A.; Freeman, G.J.; Sharpe, A.H. The B7:CD28 Family and Friends: Unraveling Coinhibitory Interactions. Immunity 2024, 57, 223–244. [Google Scholar] [CrossRef]
- Herrmann, T. Caveat: Monoclonal Antibodies 20.1 and 103.2 Bind All Human BTN3A Proteins and Are Not Suited to Study BTN3A1-Specific Features. Proc. Natl. Acad. Sci. USA 2023, 120, e2304065120. [Google Scholar] [CrossRef]
- Yang, W.; Cheng, B.; Chen, P.; Sun, X.; Wen, Z.; Cheng, Y. BTN3A1 Promotes Tumor Progression and Radiation Resistance in Esophageal Squamous Cell Carcinoma by Regulating ULK1-Mediated Autophagy. Cell Death Dis. 2022, 13, 984. [Google Scholar] [CrossRef]
- De Vries, N.L.; Van De Haar, J.; Veninga, V.; Chalabi, M.; Ijsselsteijn, M.E.; Van Der Ploeg, M.; Van Den Bulk, J.; Ruano, D.; Van Den Berg, J.G.; Haanen, J.B.; et al. γδ T Cells Are Effectors of Immunotherapy in Cancers with HLA Class I Defects. Nature 2023, 613, 743–750. [Google Scholar] [CrossRef]
- Ribot, J.C.; Lopes, N.; Silva-Santos, B. γδ T Cells in Tissue Physiology and Surveillance. Nat. Rev. Immunol. 2021, 21, 221–232. [Google Scholar] [CrossRef]
- Déchanet-Merville, J.; Prinz, I. From Basic Research to Clinical Application of γδ T Cells. Immunol. Rev. 2020, 298, 5–9. [Google Scholar] [CrossRef]
- Yang, Y.; Li, L.; Yuan, L.; Zhou, X.; Duan, J.; Xiao, H.; Cai, N.; Han, S.; Ma, X.; Liu, W.; et al. A Structural Change in Butyrophilin upon Phosphoantigen Binding Underlies Phosphoantigen-Mediated Vγ9Vδ2 T Cell Activation. Immunity 2019, 50, 1043–1053. [Google Scholar] [CrossRef]
- Karunakaran, M.M.; Subramanian, H.; Jin, Y.; Mohammed, F.; Kimmel, B.; Juraske, C.; Starick, L.; Nöhren, A.; Länder, N.; Willcox, C.R.; et al. A Distinct Topology of BTN3A IgV and B30.2 Domains Controlled by Juxtamembrane Regions Favors Optimal Human γδ T Cell Phosphoantigen Sensing. Nat. Commun. 2023, 14, 7617. [Google Scholar] [CrossRef] [PubMed]
- Vantourout, P.; Laing, A.; Woodward, M.J.; Zlatareva, I.; Apolonia, L.; Jones, A.W.; Snijders, A.P.; Malim, M.H.; Hayday, A.C. Heteromeric Interactions Regulate Butyrophilin (BTN) and BTN-like Molecules Governing γδ T Cell Biology. Proc. Natl. Acad. Sci. USA 2018, 115, 1039–1044. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, M.M.; Willcox, C.R.; Salim, M.; Paletta, D.; Fichtner, A.S.; Noll, A.; Starick, L.; Nöhren, A.; Begley, C.R.; Berwick, K.A.; et al. Butyrophilin-2A1 Directly Binds Germline-Encoded Regions of the Vγ9Vδ2 TCR and Is Essential for Phosphoantigen Sensing. Immunity 2020, 52, 487–498.e6. [Google Scholar] [CrossRef]
- Vavassori, S.; Kumar, A.; Wan, G.S.; Ramanjaneyulu, G.S.; Cavallari, M.; Daker, S.E.; Beddoe, T.; Theodossis, A.; Williams, N.K.; Gostick, E.; et al. Butyrophilin 3A1 Binds Phosphorylated Antigens and Stimulates Human γδ T Cells. Nat. Immunol. 2013, 14, 908–916. [Google Scholar] [CrossRef]
- Riaño, F.; Karunakaran, M.M.; Starick, L.; Li, J.; Scholz, C.J.; Kunzmann, V.; Olive, D.; Amslinger, S.; Herrmann, T. Vγ9Vδ2 TCR-activation by Phosphorylated Antigens Requires Butyrophilin 3 A1 (BTN3A1) and Additional Genes on Human Chromosome 6. Eur. J. Immunol. 2014, 44, 2571–2576. [Google Scholar] [CrossRef]
- A Rhodes, D.; Chen, H.-C.; Price, A.J.; Keeble, A.H.; Davey, M.S.; James, L.C.; Eberl, M.; Trowsdale, J. Activation of human γδ T cells by cytosolic interactions of BTN3A1 with soluble phosphoantigens and the cytoskeletal adaptor periplakin. J. Immunol. 2015, 194, 2390–2398. [Google Scholar] [CrossRef]
- Sebestyen, Z.; Scheper, W.; Vyborova, A.; Gu, S.; Rychnavska, Z.; Schiffler, M.; Cleven, A.; Chéneau, C.; van Noorden, M.; Peigné, C.-M.; et al. RhoB Mediates Phosphoantigen Recognition by Vγ9Vδ2 T Cell Receptor. Cell Rep. 2016, 15, 1973–1985. [Google Scholar] [CrossRef]
- Vyborova, A.; Beringer, D.X.; Fasci, D.; Karaiskaki, F.; Van Diest, E.; Kramer, L.; De Haas, A.; Sanders, J.; Janssen, A.; Straetemans, T.; et al. γ9δ2T Cell Diversity and the Receptor Interface with Tumor Cells. J. Clin. Investig. 2020, 130, 4637–4651. [Google Scholar] [CrossRef]
- Rigau, M. Butyrophilin 2A1 Is Essential for Phosphoantigen Reactivity by γδ T Cells. Science 2020, 367, eaay5516. [Google Scholar] [CrossRef]
- Willcox, C.R.; Salim, M.; Begley, C.R.; Karunakaran, M.M.; Easton, E.J.; Von Klopotek, C.; Berwick, K.A.; Herrmann, T.; Mohammed, F.; Jeeves, M.; et al. Phosphoantigen Sensing Combines TCR-Dependent Recognition of the BTN3A IgV Domain and Germline Interaction with BTN2A1. Cell Rep. 2023, 42, 112321. [Google Scholar] [CrossRef]
- Nguyen, K.; Jin, Y.; Howell, M.; Hsiao, C.-H.C.; Wiemer, A.J.; Vinogradova, O. Mutations to the BTN2A1 Linker Region Impact Its Homodimerization and Its Cytoplasmic Interaction with Phospho-Antigen–Bound BTN3A1. J. Immunol. 2023, 211, 23–33. [Google Scholar] [CrossRef]
- Poe, M.M.; Agabiti, S.S.; Liu, C.; Li, V.; Teske, K.A.; Hsiao, C.-H.C.; Wiemer, A.J. Probing the Ligand-Binding Pocket of BTN3A1. J. Med. Chem. 2019, 62, 6814–6823. [Google Scholar] [CrossRef]
- Hernández-López, P.; Van Diest, E.; Brazda, P.; Heijhuurs, S.; Meringa, A.; Hoorens Van Heyningen, L.; Riillo, C.; Schwenzel, C.; Zintchenko, M.; Johanna, I.; et al. Dual Targeting of Cancer Metabolome and Stress Antigens Affects Transcriptomic Heterogeneity and Efficacy of Engineered T Cells. Nat. Immunol. 2024, 25, 88–101. [Google Scholar] [CrossRef]
- Mamedov, M.R. CRISPR Screens Decode Cancer Cell Pathways That Trigger γδ T Cell Detection. Nature 2023, 621, 188–195. [Google Scholar] [CrossRef]
- Mu, X.; Xiang, Z.; Xu, Y.; He, J.; Lu, J.; Chen, Y.; Wang, X.; Tu, C.R.; Zhang, Y.; Zhang, W.; et al. Glucose Metabolism Controls Human γδ T-Cell-Mediated Tumor Immunosurveillance in Diabetes. Cell Mol. Immunol. 2022, 19, 944–956. [Google Scholar] [CrossRef]
- Dang, A.T.; Strietz, J.; Zenobi, A.; Khameneh, H.J.; Brandl, S.M.; Lozza, L.; Conradt, G.; Kaufmann, S.H.E.; Reith, W.; Kwee, I.; et al. NLRC5 Promotes Transcription of BTN3A1-3 Genes and Vγ9Vδ2 T Cell-Mediated Killing. iScience 2020, 24, 101900. [Google Scholar] [CrossRef]
- Li, J.; Feng, H.; Zhu, J.; Yang, K.; Zhang, G.; Gu, Y.; Shi, T.; Chen, W. Gastric Cancer Derived Exosomal THBS1 Enhanced Vγ9Vδ2 T-Cell Function through Activating RIG-I-like Receptor Signaling Pathway in a N6-Methyladenosine Methylation Dependent Manner. Cancer Lett. 2023, 576, 216410. [Google Scholar] [CrossRef]
- Fiala, G.J.; Lücke, J.; Huber, S. Pro- and Antitumorigenic Functions of γδ T Cells. Eur. J. Immunol. 2024, 54, e2451070. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.; Zanettini, C.; Coker, M.; Boudova, S.; Rach, D.; Mvula, G.; Divala, T.H.; Mungwira, R.G.; Boldrin, F.; Degiacomi, G.; et al. Concomitant Assessment of PD-1 and CD56 Expression Identifies Subsets of Resting Cord Blood Vδ2 T Cells with Disparate Cytotoxic Potential. Cell Immunol. 2024, 395–396, 104797. [Google Scholar] [CrossRef] [PubMed]
- Lopes, N.; McIntyre, C.; Martin, S.; Raverdeau, M.; Sumaria, N.; Kohlgruber, A.C.; Fiala, G.J.; Agudelo, L.Z.; Dyck, L.; Kane, H.; et al. Distinct Metabolic Programs Established in the Thymus Control Effector Functions of γδ T Cell Subsets in Tumor Microenvironments. Nat. Immunol. 2021, 22, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Rosso, D.A.; Rosato, M.; Iturrizaga, J.; González, N.; Shiromizu, C.M.; Keitelman, I.A.; Coronel, J.V.; Gómez, F.D.; Amaral, M.M.; Rabadan, A.T.; et al. Glioblastoma Cells Potentiate the Induction of the Th1-like Profile in Phosphoantigen-Stimulated γδ T Lymphocytes. J. Neurooncol 2021, 153, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Nezhad Shamohammadi, F.; Yazdanifar, M.; Oraei, M.; Kazemi, M.H.; Roohi, A.; Mahya Shariat Razavi, S.; Rezaei, F.; Parvizpour, F.; Karamlou, Y.; Namdari, H. Controversial Role of γδ T Cells in Pancreatic Cancer. Int. Immunopharmacol. 2022, 108, 108895. [Google Scholar] [CrossRef] [PubMed]
- Dhar, P.; Wu, J.D. NKG2D and Its Ligands in Cancer. Curr. Opin. Immunol. 2018, 51, 55–61. [Google Scholar] [CrossRef]
- Wesch, D.; Kabelitz, D.; Oberg, H. Tumor Resistance Mechanisms and Their Consequences on γδ T Cell Activation. Immunol. Rev. 2020, 298, 84–98. [Google Scholar] [CrossRef]
- Yi, M.; Li, T.; Niu, M.; Mei, Q.; Zhao, B.; Chu, Q.; Dai, Z.; Wu, K. Exploiting Innate Immunity for Cancer Immunotherapy. Mol. Cancer 2023, 22, 187. [Google Scholar] [CrossRef]
- Kabelitz, D. A Ménage à Trois of Cytotoxic Effector Cells: γδ T Cells Suppress NK Cells but Not CTLs. Cancer Immunol. Res. 2022, 10, 543. [Google Scholar] [CrossRef]
- Payne, K.K.; Mine, J.A.; Biswas, S.; Chaurio, R.A.; Perales-Puchalt, A.; Anadon, C.M.; Costich, T.L.; Harro, C.M.; Walrath, J.; Ming, Q.; et al. BTN3A1 Governs Antitumor Responses by Coordinating Aβ and γδ T Cells. Science 2020, 369, 942–949. [Google Scholar] [CrossRef]
- Suzuki, T.; Hayman, L.; Kilbey, A.; Edwards, J.; Coffelt, S.B. Gut γδ T Cells as Guardians, Disruptors, and Instigators of Cancer. Immunol. Rev. 2020, 298, 198–217. [Google Scholar] [CrossRef]
- Röring, R.J.; Debisarun, P.A.; Botey-Bataller, J.; Suen, T.K.; Bulut, Ö.; Kilic, G.; Koeken, V.A.C.M.; Sarlea, A.; Bahrar, H.; Dijkstra, H.; et al. MMR Vaccination Induces Trained Immunity via Functional and Metabolic Reprogramming of γδ T Cells. J. Clin. Investig. 2024, 134, e170848. [Google Scholar] [CrossRef]
- Medina, B.D.; Liu, M.; Vitiello, G.A.; Seifert, A.M.; Zeng, S.; Bowler, T.; Zhang, J.Q.; Cavnar, M.J.; Loo, J.K.; Param, N.J.; et al. Oncogenic Kinase Inhibition Limits Batf3-Dependent Dendritic Cell Development and Antitumor Immunity. J. Exp. Med. 2019, 216, 1359–1376. [Google Scholar] [CrossRef] [PubMed]
- Silva-Santos, B.; Mensurado, S.; Coffelt, S.B. γδ T Cells: Pleiotropic Immune Effectors with Therapeutic Potential in Cancer. Nat. Rev. Cancer 2019, 19, 392–404. [Google Scholar] [CrossRef]
- Wu, P.; Wu, D.; Ni, C.; Ye, J.; Chen, W.; Hu, G.; Wang, Z.; Wang, C.; Zhang, Z.; Xia, W.; et al. γδ T17 Cells Promote the Accumulation and Expansion of Myeloid-Derived Suppressor Cells in Human Colorectal Cancer. Immunity 2014, 40, 785–800. [Google Scholar] [CrossRef] [PubMed]
- Meraviglia, S.; Lo Presti, E.; Tosolini, M.; La Mendola, C.; Orlando, V.; Todaro, M.; Catalano, V.; Stassi, G.; Cicero, G.; Vieni, S.; et al. Distinctive features of tumor-infiltrating γδ T lymphocytes in human colorectal cancer. Oncoimmunology 2017, 6, e1347742. [Google Scholar] [CrossRef]
- Jin, C.; Lagoudas, G.K.; Zhao, C.; Bullman, S.; Bhutkar, A.; Hu, B.; Ameh, S.; Sandel, D.; Liang, X.S.; Mazzilli, S.; et al. Commensal Microbiota Promote Lung Cancer Development via γδ T Cells. Cell 2019, 176, 998–1013.e16. [Google Scholar] [CrossRef]
- Van Hede, D.; Polese, B.; Humblet, C.; Wilharm, A.; Renoux, V.; Dortu, E.; De Leval, L.; Delvenne, P.; Desmet, C.J.; Bureau, F.; et al. Human Papillomavirus Oncoproteins Induce a Reorganization of Epithelial-Associated γδ T Cells Promoting Tumor Formation. Proc. Natl. Acad. Sci. USA 2017, 114, E9056–E9065. [Google Scholar] [CrossRef]
- Lo Presti, E.; Toia, F.; Oieni, S.; Buccheri, S.; Turdo, A.; Mangiapane, L.R.; Campisi, G.; Caputo, V.; Todaro, M.; Stassi, G.; et al. Squamous Cell Tumors Recruit γδ T Cells Producing Either IL17 or IFNγ Depending on the Tumor Stage. Cancer Immunol. Res. 2017, 5, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, K.; Shirai, M.; Taniguchi, K.; Hosoi, A.; Sun, C.; Kobayashi, Y.; Maejima, K.; Fujita, M.; Nakagawa, H.; Nomura, S.; et al. Deep Immunophenotyping at the Single-Cell Level Identifies a Combination of Anti-IL-17 and Checkpoint Blockade as an Effective Treatment in a Preclinical Model of Data-Guided Personalized Immunotherapy. J. Immunother. Cancer 2020, 8, e001358. [Google Scholar] [CrossRef]
- Liu, J.; Duan, Y.; Cheng, X.; Chen, X.; Xie, W.; Long, H.; Lin, Z.; Zhu, B. IL-17 Is Associated with Poor Prognosis and Promotes Angiogenesis via Stimulating VEGF Production of Cancer Cells in Colorectal Carcinoma. Biochem. Biophys. Res. Commun. 2011, 407, 348–354. [Google Scholar] [CrossRef]
- Coffelt, S.B.; Kersten, K.; Doornebal, C.W.; Weiden, J.; Vrijland, K.; Hau, C.-S.; Verstegen, N.J.M.; Ciampricotti, M.; Hawinkels, L.J.A.C.; Jonkers, J.; et al. IL-17-Producing γδ T Cells and Neutrophils Conspire to Promote Breast Cancer Metastasis. Nature 2015, 522, 345–348. [Google Scholar] [CrossRef]
- He, M.; Peng, A.; Huang, X.-Z.; Shi, D.-C.; Wang, J.-C.; Zhao, Q.; Lin, H.; Kuang, D.-M.; Ke, P.-F.; Lao, X.-M. Peritumoral Stromal Neutrophils Are Essential for C-Met-Elicited Metastasis in Human Hepatocellular Carcinoma. OncoImmunology 2016, 5, e1219828. [Google Scholar] [CrossRef] [PubMed]
- Li, T.-J.; Jiang, Y.-M.; Hu, Y.-F.; Huang, L.; Yu, J.; Zhao, L.-Y.; Deng, H.-J.; Mou, T.-Y.; Liu, H.; Yang, Y.; et al. Interleukin-17–Producing Neutrophils Link Inflammatory Stimuli to Disease Progression by Promoting Angiogenesis in Gastric Cancer. Clin. Cancer Res. 2017, 23, 1575–1585. [Google Scholar] [CrossRef]
- He, D.; Li, H.; Yusuf, N.; Elmets, C.A.; Li, J.; Mountz, J.D.; Xu, H. IL-17 Promotes Tumor Development through the Induction of Tumor Promoting Microenvironments at Tumor Sites and Myeloid-Derived Suppressor Cells. J. Immunol. 2010, 184, 2281–2288. [Google Scholar] [CrossRef]
- Li, L.; Cao, B.; Liang, X.; Lu, S.; Luo, H.; Wang, Z.; Wang, S.; Jiang, J.; Lang, J.; Zhu, G. Microenvironmental Oxygen Pressure Orchestrates an Anti- and pro-Tumoral γδ T Cell Equilibrium via Tumor-Derived Exosomes. Oncogene 2019, 38, 2830–2843. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, J.R.; Lloyd, C.M. Resolution of Allergic Airway Inflammation and Airway Hyperreactivity Is Mediated by IL-17–Producing γδ T Cells. Am. J. Respir. Crit. Care Med. 2010, 182, 464–476. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhang, G.; Wan, X. Challenges and New Technologies in Adoptive Cell Therapy. J. Hematol. Oncol. 2023, 16, 97. [Google Scholar] [CrossRef]
- Mazinani, M.; Rahbarizadeh, F. New Cell Sources for CAR-Based Immunotherapy. Biomark. Res. 2023, 11, 49. [Google Scholar] [CrossRef]
- Robert, C.; Carlino, M.S.; McNeil, C.; Ribas, A.; Grob, J.-J.; Schachter, J.; Nyakas, M.; Kee, D.; Petrella, T.M.; Blaustein, A.; et al. Seven-Year Follow-Up of the Phase III KEYNOTE-006 Study: Pembrolizumab Versus Ipilimumab in Advanced Melanoma. JCO 2023, 41, 3998–4003. [Google Scholar] [CrossRef]
- Bagley, S.J.; O’Rourke, D.M. Clinical Investigation of CAR T Cells for Solid Tumors: Lessons Learned and Future Directions. Pharmacol. Ther. 2020, 205, 107419. [Google Scholar] [CrossRef]
- Sánchez Martínez, D.; Tirado, N.; Mensurado, S.; Martínez-Moreno, A.; Romecín, P.; Gutiérrez Agüera, F.; Correia, D.V.; Silva-Santos, B.; Menéndez, P. Generation and Proof-of-Concept for Allogeneic CD123 CAR-Delta One T (DOT) Cells in Acute Myeloid Leukemia. J. Immunother. Cancer 2022, 10, e005400. [Google Scholar] [CrossRef]
- Tang, C.; Zhang, Y. Potential alternatives to αβ-T cells to prevent graft-versus-host disease (GvHD) in allogeneic chimeric antigen receptor (CAR)-based cancer immunotherapy: A comprehensive review. Pathol. Res. Pract. 2024, 262, 155518. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Kohler, M.E.; Chien, C.D.; Sauter, C.T.; Jacoby, E.; Yan, C.; Hu, Y.; Wanhainen, K.; Qin, H.; Fry, T.J. TCR engagement negatively affects CD8 but not CD4 CAR T cell expansion and leukemic clearance. Sci. Transl. Med. 2017, 9, eaag1209. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, N.; Feng, K.; Chen, M.; Zhang, Y.; Liu, Y.; Yang, Q.; Nie, J.; Tang, N.; Zhang, X.; et al. Phase I study of CAR-T cells with PD-1 and TCR disruption in mesothelin-positive solid tumors. Cell Mol. Immunol. 2021, 18, 2188–2198. [Google Scholar] [CrossRef]
- Moradi, V.; Khodabandehloo, E.; Alidadi, M.; Omidkhoda, A.; Ahmadbeigi, N. Progress and pitfalls of gene editing technology in CAR-T cell therapy: A state-of-the-art review. Front. Oncol. 2024, 14, 1388475. [Google Scholar] [CrossRef]
- Lerner, E.C.; Woroniecka, K.I.; D’anniballe, V.M.; Wilkinson, D.S.; Mohan, A.A.; Lorrey, S.J.; Waibl-Polania, J.; Wachsmuth, L.P.; Miggelbrink, A.M.; Jackson, J.D.; et al. CD8+ T cells maintain killing of MHC-I-negative tumor cells through the NKG2D-NKG2DL axis. Nat. Cancer 2023, 4, 1258–1272. [Google Scholar] [CrossRef]
- Reinstein, Z.Z.; Zhang, Y.; Ospina, O.E.; Nichols, M.D.; Chu, V.A.; Pulido, A.d.M.; Prieto, K.; Nguyen, J.V.; Yin, R.; Segura, C.M.; et al. Preexisting Skin-Resident CD8 and γδ T-cell Circuits Mediate Immune Response in Merkel Cell Carcinoma and Predict Immunotherapy Efficacy. Cancer Discov. 2024, 14, 1631–1652. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Xu, J.; Wu, M.; Liu, S.; Fu, X.; Shang, W.; Wang, T.; Jia, X.; Wang, F. Circulating CD4+ Treg, CD8+ Treg, and CD3+ γδ T Cell Subpopulations in Ovarian Cancer. Medicina 2023, 59, 205. [Google Scholar] [CrossRef]
- Giraud, J.; Chalopin, D.; Blanc, J.F.; Saleh, M. Hepatocellular Carcinoma Immune Landscape and the Potential of Immunotherapies. Front. Immunol. 2021, 12, 655697. [Google Scholar] [CrossRef]
- You, H.; Wang, Y.; Wang, X.; Zhu, H.; Zhao, Y.; Qin, P.; Liu, X.; Zhang, M.; Fu, X.; Xu, B.; et al. CD69+ Vδ1γδ T cells are anti-tumor subpopulations in hepatocellular carcinoma. Mol. Immunol. 2024, 175, 164. [Google Scholar] [CrossRef]
- Wang, Y.; Suarez, E.R.; Kastrunes, G.; de Campos, N.S.P.; Abbas, R.; Pivetta, R.S.; Murugan, N.; Chalbatani, G.M.; D’andrea, V.; Marasco, W.A. Evolution of cell therapy for renal cell carcinoma. Mol. Cancer 2024, 23, 8. [Google Scholar] [CrossRef]
- Rodin, W.; Szeponik, L.; Rangelova, T.; Kebede, F.T.; Österlund, T.; Sundström, P.; Hogg, S.; Wettergren, Y.; Cosma, A.; Ståhlberg, A.; et al. γδ T cells in human colon adenocarcinomas comprise mainly Vδ1, Vδ2, and Vδ3 cells with distinct phenotype and function. Cancer Immunol. Immunother. 2024, 73, 174. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Wei, C.; Li, X. Association between γδ T cells and clinicopathological features of breast cancer. Int. Immunopharmacol. 2022, 103, 108457. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Peng, Z.; Guo, J.; Wang, Y.; Wang, S.; Jiang, H.; Wang, M.; Xie, Y.; Li, X.; Hu, M.; et al. CXCL10 Recruitment of γδ T Cells into the Hypoxic Bone Marrow Environment Leads to IL17 Expression and Multiple Myeloma Progression. Cancer Immunol. Res. 2023, 11, 1384–1399. [Google Scholar] [CrossRef] [PubMed]
- Kabelitz, D. Novel Insights into Regulation of Butyrophilin Molecules: Critical Components of Cancer Immunosurveillance by γδ T Cells. Cell Mol. Immunol. 2024, 21, 409–411. [Google Scholar] [CrossRef]
- Fulford, T.S.; Soliman, C.; Castle, R.G.; Rigau, M.; Ruan, Z.; Dolezal, O.; Seneviratna, R.; Brown, H.G.; Hanssen, E.; Hammet, A.; et al. Vγ9Vδ2 T Cells Recognize Butyrophilin 2A1 and 3A1 Heteromers. Nat. Immunol. 2024, 25, 1355–1366. [Google Scholar] [CrossRef]
- Benelli, R.; Costa, D.; Salvini, L.; Tardito, S.; Tosetti, F.; Villa, F.; Zocchi, M.R.; Poggi, A. Targeting of Colorectal Cancer Organoids with Zoledronic Acid Conjugated to the Anti-EGFR Antibody Cetuximab. J. Immunother. Cancer 2022, 10, e005660. [Google Scholar] [CrossRef]
- Herrmann, T.; Karunakaran, M.M. Phosphoantigen recognition by Vγ9Vδ2 T cells. Eur. J. Immunol. 2024, 54, e2451068. [Google Scholar] [CrossRef]
- Starick, L.; Riano, F.; Karunakaran, M.M.; Kunzmann, V.; Li, J.; Kreiss, M.; Amslinger, S.; Scotet, E.; Olive, D.; De Libero, G.; et al. Butyrophilin 3A (BTN3A, CD277)-specific antibody 20.1 differentially activates Vγ9Vδ2 TCR clonotypes and interferes with phosphoantigen activation. Eur. J. Immunol. 2017, 47, 982–992. [Google Scholar] [CrossRef]
- Lo Presti, V.; Meringa, A.; Dunnebach, E.; Van Velzen, A.; Moreira, A.V.; Stam, R.W.; Kotecha, R.S.; Krippner-Heidenreich, A.; Heidenreich, O.T.; Plantinga, M.; et al. Combining CRISPR-Cas9 and TCR Exchange to Generate a Safe and Efficient Cord Blood-Derived T Cell Product for Pediatric Relapsed AML. J. Immunother. Cancer 2024, 12, e008174. [Google Scholar] [CrossRef]
- Johanna, I.; Straetemans, T.; Heijhuurs, S.; Aarts-Riemens, T.; Norell, H.; Bongiovanni, L.; De Bruin, A.; Sebestyen, Z.; Kuball, J. Evaluating in Vivo Efficacy—Toxicity Profile of TEG001 in Humanized Mice Xenografts against Primary Human AML Disease and Healthy Hematopoietic Cells. J. Immunother. Cancer 2019, 7, 69. [Google Scholar] [CrossRef]
- Van Diest, E.; Hernández López, P.; Meringa, A.D.; Vyborova, A.; Karaiskaki, F.; Heijhuurs, S.; Gumathi Bormin, J.; Van Dooremalen, S.; Nicolasen, M.J.T.; Gatti, L.C.D.E.; et al. Gamma Delta TCR Anti-CD3 Bispecific Molecules (GABs) as Novel Immunotherapeutic Compounds. J. Immunother. Cancer 2021, 9, e003850. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Xiang, Z.; Alnaggar, M.; Kouakanou, L.; Li, J.; He, J.; Yang, J.; Hu, Y.; Chen, Y.; Lin, L.; et al. Allogeneic Vγ9Vδ2 T-Cell Immunotherapy Exhibits Promising Clinical Safety and Prolongs the Survival of Patients with Late-Stage Lung or Liver Cancer. Cell Mol. Immunol. 2021, 18, 427–439. [Google Scholar] [CrossRef]
- Alnaggar, M.; Xu, Y.; Li, J.; He, J.; Chen, J.; Li, M.; Wu, Q.; Lin, L.; Liang, Y.; Wang, X.; et al. Allogenic Vγ9Vδ2 T Cell as New Potential Immunotherapy Drug for Solid Tumor: A Case Study for Cholangiocarcinoma. J. Immunother. Cancer 2019, 7, 36. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.R.; Correia, D.V.; Fernandes-Platzgummer, A.; da Silva, C.L.; da Silva, M.G.; Anjos, D.R.; Silva-Santos, B. Delta One T Cells for Immunotherapy of Chronic Lymphocytic Leukemia: Clinical-Grade Expansion/Differentiation and Preclinical Proof of Concept. Clin. Cancer Res. 2016, 22, 5795–5804. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Domínguez, R.; Barros, L.; Carreira, M.; van der Ploeg, M.; Condeço, C.; Marsères, G.; Ferreira, C.; Costa, C.; Ferreira, C.M.; Déchanet-Merville, J.; et al. Dual modulation of cytotoxic and checkpoint receptors tunes the efficacy of adoptive Delta One T cell therapy against colorectal cancer. Nat Cancer. 2025. [Google Scholar] [CrossRef]
- Marcu-Malina, V.; Heijhuurs, S.; Van Buuren, M.; Hartkamp, L.; Strand, S.; Sebestyen, Z.; Scholten, K.; Martens, A.; Kuball, J. Redirecting αβ T Cells against Cancer Cells by Transfer of a Broadly Tumor-Reactive γδ T-Cell Receptor. Blood 2011, 118, 50–59. [Google Scholar] [CrossRef]
- Li, C. Novel CD19-Specific γ/δ TCR-T Cells in Relapsed or Refractory Diffuse Large B-Cell Lymphoma. J. Hematol. Oncol. 2023, 16, 5. [Google Scholar] [CrossRef]
- Maalej, K.M.; Merhi, M.; Inchakalody, V.P.; Mestiri, S.; Alam, M.; Maccalli, C.; Cherif, H.; Uddin, S.; Steinhoff, M.; Marincola, F.M.; et al. CAR-Cell Therapy in the Era of Solid Tumor Treatment: Current Challenges and Emerging Therapeutic Advances. Mol. Cancer 2023, 22, 20. [Google Scholar] [CrossRef]
- Deniger, D.C.; Switzer, K.; Mi, T.; Maiti, S.; Hurton, L.; Singh, H.; Huls, H.; Olivares, S.; A Lee, D.; E Champlin, R.; et al. Bispecific T-cells expressing polyclonal repertoire of endogenous γδ T-cell receptors and introduced CD19-specific chimeric antigen receptor. Mol. Ther. 2013, 21, 638–647. [Google Scholar] [CrossRef]
- Harrer, D.C.; Simon, B.; Fujii, S.-I.; Shimizu, K.; Uslu, U.; Schuler, G.; Gerer, K.F.; Hoyer, S.; Dörrie, J.; Schaft, N. RNA-transfection of γ/δ T cells with a chimeric antigen receptor or an α/β T-cell receptor: A safer alternative to genetically engineered α/β T cells for the immunotherapy of melanoma. BMC Cancer 2017, 17, 551. [Google Scholar] [CrossRef]
- Fisher, J.; Abramowski, P.; Wisidagamage Don, N.D.; Flutter, B.; Capsomidis, A.; Cheung, G.W.; Gustafsson, K.; Anderson, J. Avoidance of On-Target Off-Tumor Activation Using a Co-stimulation-Only Chimeric Antigen Receptor. Mol. Ther. 2017, 25, 1234–1247. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Li, H.; Lu, Q.; Zhang, Z.; Li, J.; Wang, Z.; Yang, N.; Yu, Z.; Yang, C.; Chen, Y.; et al. mRNA-Engineered CD5-CAR-γδTCD5- Cells for the Immunotherapy of T-Cell Acute Lymphoblastic Leukemia. Adv. Sci. 2024, 11, e2400024. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wu, M.; Yang, Y.; Wang, Z.; He, S.; Tian, X.; Wang, H. γδ T cells and the PD-1/PD-L1 axis: A love-hate relationship in the tumor microenvironment. J. Transl. Med. 2024, 22, 553. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Han, J.; Wang, D.; Cai, M.; Xu, Y.; Hu, Y.; Chen, H.; He, W.; Zhang, J. Anti-PD-1 antibody armored γδ T cells enhance anti-tumor efficacy in ovarian cancer. Signal Transduct. Target. Ther. 2023, 8, 399. [Google Scholar] [CrossRef]
- Leane, C.M.; Sutton, C.E.; Moran, B.; Mills, K.H.G. PD-1 regulation of pathogenic IL-17-secreting γδ T cells in experimental autoimmune encephalomyelitis. Eur. J. Immunol. 2024, 54, e2451212. [Google Scholar] [CrossRef]
- Obajdin, J.; Larcombe-Young, D.; Glover, M.; Kausar, F.; Hull, C.M.; Flaherty, K.R.; Tan, G.; Beatson, R.E.; Dunbar, P.; Mazza, R.; et al. Solid Tumor Immunotherapy Using NKG2D-Based Adaptor CAR T Cells. Cell Rep. Med. 2024, 5, 101827. [Google Scholar] [CrossRef]
- Vaghari-Tabari, M.; Ferns, G.A.; Qujeq, D.; Andevari, A.N.; Sabahi, Z.; Moein, S. Signaling, Metabolism, and Cancer: An Important Relationship for Therapeutic Intervention. J. Cell Physiol. 2021, 236, 5512–5532. [Google Scholar] [CrossRef]
- Garber, K. γδ T cells bring unconventional cancer-targeting to the clinic—Again. Nat. Biotechnol. 2020, 38, 389–391. [Google Scholar] [CrossRef]
- Lin, M.; Zhang, X.; Liang, S.; Luo, H.; Alnaggar, M.; Liu, A.; Yin, Z.; Chen, J.; Niu, L.; Jiang, Y. Irreversible Electroporation Plus Allogenic Vγ9Vδ2 T Cells Enhances Antitumor Effect for Locally Advanced Pancreatic Cancer Patients. Signal Transduct. Target. Ther. 2020, 5, 215. [Google Scholar] [CrossRef]
- Vydra, J.; Cosimo, E.; Lesný, P.; Wanless, R.S.; Anderson, J.; Clark, A.G.; Scott, A.; Nicholson, E.K.; Leek, M. A Phase I Trial of Allogeneic γδ T Lymphocytes from Haploidentical Donors in Patients with Refractory or Relapsed Acute Myeloid Leukemia. Clin. Lymphoma Myeloma Leuk. 2023, 23, e232–e239. [Google Scholar] [CrossRef]
- Ang, W.X.; Ng, Y.Y.; Xiao, L.; Chen, C.; Li, Z.; Chi, Z.; Tay, J.C.-K.; Tan, W.K.; Zeng, J.; Toh, H.C.; et al. Electroporation of NKG2D RNA CAR Improves Vγ9Vδ2 T Cell Responses against Human Solid Tumor Xenografts. Mol. Ther. Oncolytics 2020, 17, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, K.P.; Barca, T.; Azameera, A.; Makkouk, A.; Romero, J.M.; Bai, L.; Brodey, M.M.; Kennedy-Wilde, J.; Shao, H.; Papaioannou, S. Allogeneic CD20-targeted γδ T cells exhibit innate and adaptive antitumor activities in preclinical B-cell lymphoma models. Clin. Transl. Immunol. 2022, 11, e1373. [Google Scholar] [CrossRef] [PubMed]
- Lamb, L.S.; Pereboeva, L.; Youngblood, S.; Gillespie, G.Y.; Nabors, L.B.; Markert, J.M.; Dasgupta, A.; Langford, C.; Spencer, H.T. A combined treatment regimen of MGMT-modified γδ T cells and temozolomide chemotherapy is effective against primary high-grade gliomas. Sci. Rep. 2021, 11, 21133. [Google Scholar] [CrossRef] [PubMed]
- Kabelitz, D. The Vγ4/Butyrophilin Conspiracy: Novel Role of Intraepithelial γδ T Cells in Chronic Inflammatory Bowel Disease. Signal Transduct. Target. Ther. 2023, 8, 433. [Google Scholar] [CrossRef]
- Beatson, R.E.; Parente-Pereira, A.C.; Halim, L.; Cozzetto, D.; Hull, C.; Whilding, L.M.; Martinez, O.; Taylor, C.A.; Obajdin, J.; Luu Hoang, K.N.; et al. TGF-Β1 Potentiates Vγ9Vδ2 T Cell Adoptive Immunotherapy of Cancer. Cell Rep. Med. 2021, 2, 100473. [Google Scholar] [CrossRef]
- Costa, G.P.; Mensurado, S.; Silva, B. Therapeutic Avenues for γδ T Cells in Cancer. J. Immunother. Cancer 2023, 11, e007955. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, J.; Wu, C.; Na, J.; Deng, Y.; Qin, S.; Zhong, L.; Zhao, Y. Mechanisms and Functions of γδ T Cells in Tumor Cell Recognition. Curr. Oncol. 2025, 32, 329. https://doi.org/10.3390/curroncol32060329
Tang J, Wu C, Na J, Deng Y, Qin S, Zhong L, Zhao Y. Mechanisms and Functions of γδ T Cells in Tumor Cell Recognition. Current Oncology. 2025; 32(6):329. https://doi.org/10.3390/curroncol32060329
Chicago/Turabian StyleTang, Jing, Chen Wu, Jintong Na, Yamin Deng, Simin Qin, Liping Zhong, and Yongxiang Zhao. 2025. "Mechanisms and Functions of γδ T Cells in Tumor Cell Recognition" Current Oncology 32, no. 6: 329. https://doi.org/10.3390/curroncol32060329
APA StyleTang, J., Wu, C., Na, J., Deng, Y., Qin, S., Zhong, L., & Zhao, Y. (2025). Mechanisms and Functions of γδ T Cells in Tumor Cell Recognition. Current Oncology, 32(6), 329. https://doi.org/10.3390/curroncol32060329