In Silico Identification and Validation of Cuproptosis-Related LncRNA Signature as a Novel Prognostic Model and Immune Function Analysis in Colon Adenocarcinoma

 ,
,
Abstract
1. Introduction
2. Materials and Methods
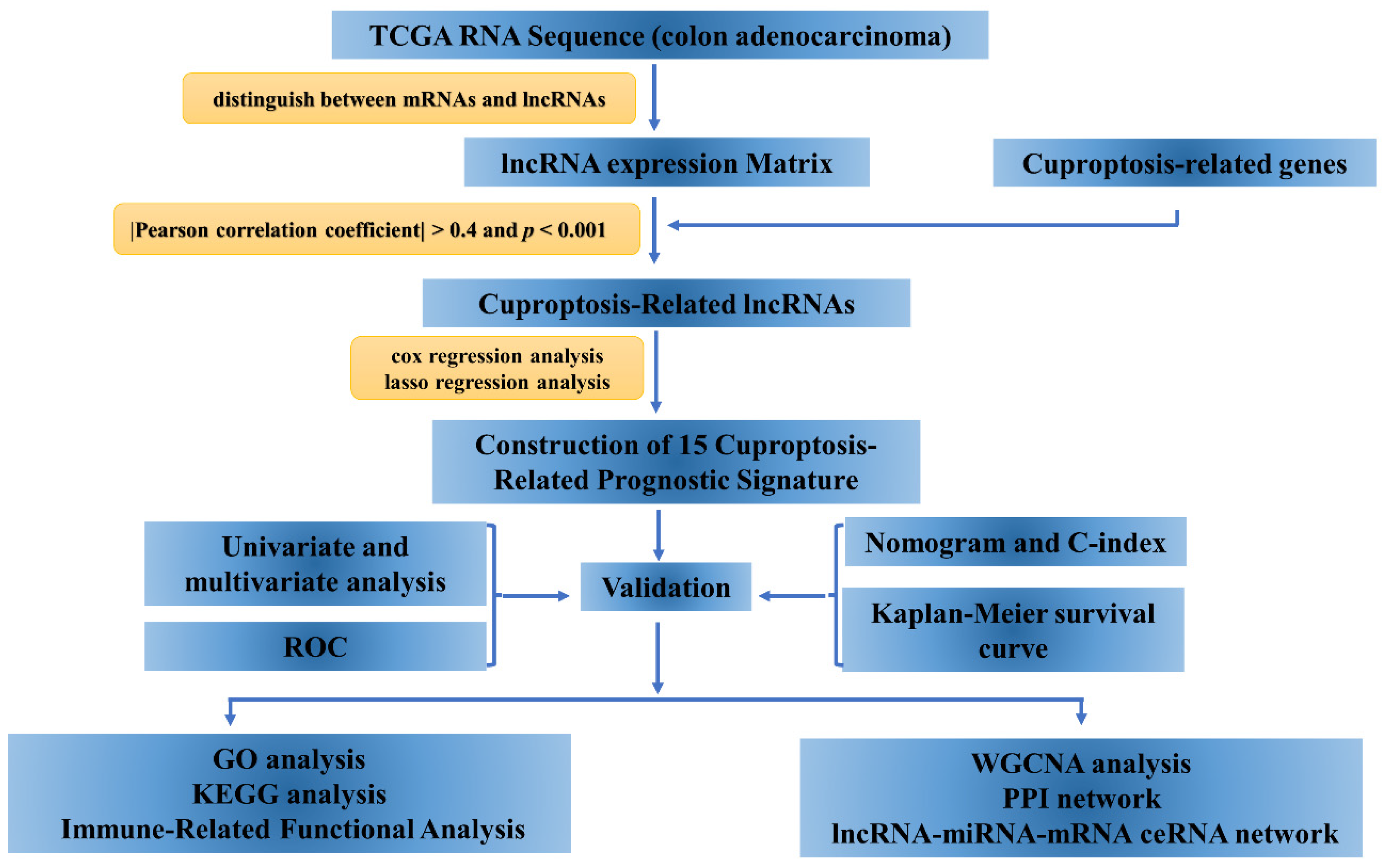
2.1. Data Collection and Preparation
2.2. Identification of Cuproptosis-Related lncRNAs
2.3. Construction of Cuproptosis-Related Prognostic Signature
2.4. Construction of Nomogram
2.5. Functional Enrichment Analysis and Immune-Related Functional Analysis
2.6. Tumor Mutational Burden Analysis and Sensitivity Assessment of Potential Drugs
2.7. Construction of Competing Endogenous RNA (ceRNA) Networks and Protein–Protein Interaction Network (PPI) Based on Weighted Gene Co-Expression Network Analysis (WGCNA)
2.8. Statistical Analysis
3. Results
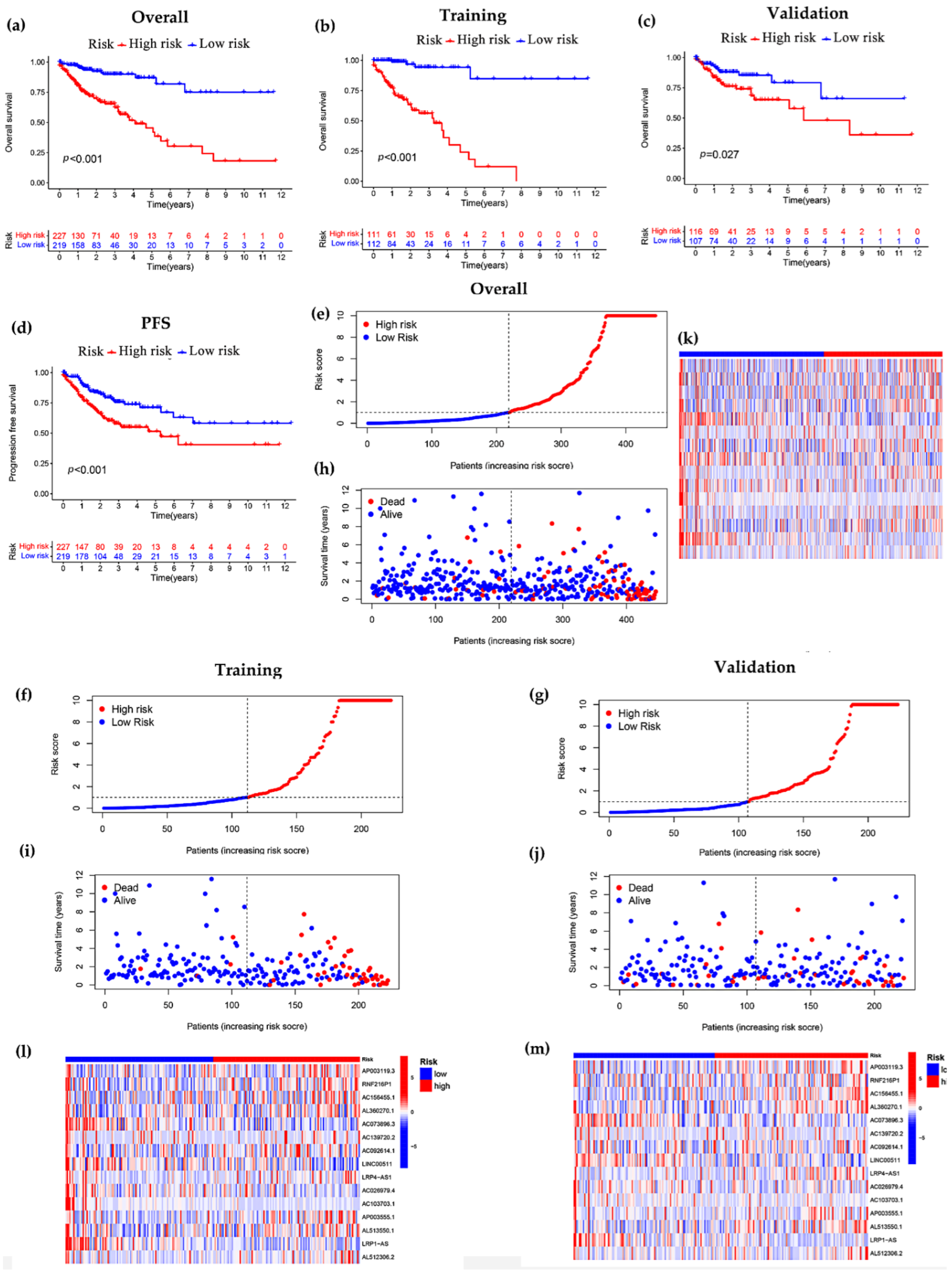
3.1. Derivation of lncRNA Prognostic Model
3.2. Constructing a 15-lncRNA Prediction Signature of COAD
3.3. Independent Prognostic Analysis of Predictive Model and Stratified Analysis of Clinicopathological Features
3.4. Pathway Enrichment Analysis and Immune-Related Functional Analysis
3.5. Correlation between Risk Score Models and Somatic Cell Variants
3.6. Screening of Potential Drugs for COAD
3.7. Construction of ceRNA Network and PPI Network and Gene Set Enrichment Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Barresi, V.; Bonetti, L.R.; Ieni, A.; Caruso, R.A.; Tuccari, G. Histological grading in colorectal cancer: New insights and perspectives. Histol. Histopathol. 2015, 30, 1059–1067. [Google Scholar] [CrossRef]
- Pita-Fernández, S.; González-Sáez, L.; López-Calviño, B.; Seoane-Pillado, T.; Rodríguez-Camacho, E.; Pazos-Sierra, A.; González-Santamaría, P.; Pértega-Díaz, S. Effect of diagnostic delay on survival in patients with colorectal cancer: A retrospective cohort study. BMC Cancer 2016, 16, 664. [Google Scholar] [CrossRef]
- Voli, F.; Valli, E. Intratumoral Copper Modulates PD-L1 Expression and Influences Tumor Immune Evasion. Cancer Res. 2020, 80, 4129–4144. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, Q. Association between serum copper levels and lung cancer risk: A meta-analysis. J. Int. Med. Res. 2018, 46, 4863–4873. [Google Scholar] [CrossRef]
- Pavithra, V.; Sathisha, T.G.; Kasturi, K.; Mallika, D.S.; Amos, S.J.; Ragunatha, S. Serum levels of metal ions in female patients with breast cancer. J. Clin. Diagn. Res. JCDR 2015, 9, BC25–BC27. [Google Scholar] [CrossRef]
- Baltaci, A.K.; Dundar, T.K.; Aksoy, F.; Mogulkoc, R. Changes in the Serum Levels of Trace Elements Before and After the Operation in Thyroid Cancer Patients. Biol. Trace Elem. Res. 2017, 175, 57–64. [Google Scholar] [CrossRef]
- Shanbhag, V.C.; Gudekar, N.; Jasmer, K.; Papageorgiou, C.; Singh, K.; Petris, M.J. Copper metabolism as a unique vulnerability in cancer. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118893. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Coy, S. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef]
- Ulitsky, I.; Bartel, D.P. lincRNAs: Genomics, evolution, and mechanisms. Cell 2013, 154, 26–46. [Google Scholar] [CrossRef]
- Alexander, R.P.; Fang, G.; Rozowsky, J.; Snyder, M.; Gerstein, M.B. Annotating non-coding regions of the genome. Nat. Rev. Genet. 2010, 11, 559–571. [Google Scholar] [CrossRef]
- Peng, W.X.; Koirala, P.; Mo, Y.Y. LncRNA-mediated regulation of cell signaling in cancer. Oncogene 2017, 36, 5661–5667. [Google Scholar] [CrossRef]
- Chai, X.K.; Qi, W.; Zou, C.Y.; He, C.X.; Su, M.; Zhao, D.Q. Potential Prognostic Value of a Seven m6A-Related LncRNAs Signature and the Correlative Immune Infiltration in Colon Adenocarcinoma. Front. Genet. 2021, 12, 774010. [Google Scholar] [CrossRef]
- Li, J.; Xiang, R.; Song, W.; Wu, J.; Kong, C.; Fu, T. A Novel Ferroptosis-Related LncRNA Pair Prognostic Signature Predicts Immune Landscapes and Treatment Responses for Gastric Cancer Patients. Front. Genet. 2022, 13, 899419. [Google Scholar] [CrossRef]
- Liu, L.; Huang, L.; Chen, W.; Zhang, G.; Li, Y.; Wu, Y.; Xiong, J.; Jie, Z. Comprehensive Analysis of Necroptosis-Related Long Noncoding RNA Immune Infiltration and Prediction of Prognosis in Patients With Colon Cancer. Front. Mol. Biosci. 2022, 9, 811269. [Google Scholar] [CrossRef]
- Zhou, W.; Zhang, S. Development of Prognostic Indicator Based on Autophagy-Related lncRNA Analysis in Colon Adenocarcinoma. BioMed Res. Int. 2020, 2020, 9807918. [Google Scholar] [CrossRef]
- Sun, J.; Li, L.; Chen, H.; Gan, L.; Guo, X.; Sun, J. Identification and Validation of an m7G-Related lncRNAs Signature for Prognostic Prediction and Immune Function Analysis in Endometrial Cancer. Genes 2022, 13, 1301. [Google Scholar] [CrossRef]
- Meier, L.; Van De Geer, S.; Bühlmann, P. The group lasso for logistic regression. J. R. Stat. Soc. Ser. B (Stat. Methodol.) 2008, 70, 53–71. [Google Scholar] [CrossRef]
- Long, J.; Zhang, L.; Wan, X.; Lin, J.; Bai, Y.; Xu, W.; Xiong, J.; Zhao, H. A four-gene-based prognostic model predicts overall survival in patients with hepatocellular carcinoma. J. Cell. Mol. Med. 2018, 22, 5928–5938. [Google Scholar] [CrossRef]
- Li, H.; Gao, C.; Liu, L.; Zhuang, J.; Yang, J.; Liu, C.; Zhou, C.; Feng, F.; Sun, C. 7-lncRNA Assessment Model for Monitoring and Prognosis of Breast Cancer Patients: Based on Cox Regression and Co-expression Analysis. Front. Oncol. 2019, 9, 1348. [Google Scholar] [CrossRef]
- Park, S.Y. Nomogram: An analogue tool to deliver digital knowledge. J. Thorac. Cardiovasc. Surg. 2018, 155, 1793. [Google Scholar] [CrossRef]
- Harrell, F.E., Jr.; Lee, K.L.; Mark, D.B. Multivariable prognostic models: Issues in developing models, evaluating assumptions and adequacy, and measuring and reducing errors. Stat. Med. 1996, 15, 361–387. [Google Scholar] [CrossRef]
- Zhi, S.; Yang, B.; Zhou, S.; Tan, J.; Zhong, G.; Han, F. Immune-Related LncRNAs to Construct a Prognosis Risk-Assessment Model for Gastric Cancer. Curr. Oncol. 2022, 29, 4923–4935. [Google Scholar] [CrossRef]
- Xiao, B.; Liu, L.; Li, A.; Xiang, C.; Wang, P.; Li, H.; Xiao, T. Identification and Verification of Immune-Related Gene Prognostic Signature Based on ssGSEA for Osteosarcoma. Front. Oncol. 2020, 10, 607622. [Google Scholar] [CrossRef]
- Geeleher, P.; Cox, N.; Huang, R.S. pRRophetic: An R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS ONE 2014, 9, e107468. [Google Scholar] [CrossRef]
- Geeleher, P.; Cox, N.J.; Huang, R.S. Clinical drug response can be predicted using baseline gene expression levels and in vitro drug sensitivity in cell lines. Genome Biol. 2014, 15, R47. [Google Scholar] [CrossRef]
- Jeggari, A.; Marks, D.S.; Larsson, E. miRcode: A map of putative microRNA target sites in the long non-coding transcriptome. Bioinformatics 2012, 28, 2062–2063. [Google Scholar] [CrossRef]
- von Mering, C.; Huynen, M.; Jaeggi, D.; Schmidt, S.; Bork, P.; Snel, B. STRING: A database of predicted functional associations between proteins. Nucleic Acids Res. 2003, 31, 258–261. [Google Scholar] [CrossRef]
- Ahmadi, M.; Pashangzadeh, S.; Mousavi, P.; Saffarzadeh, N.; Habibi, M.A.; Hajiesmaeili, F.; Rezaei, N. ACE2 correlates with immune infiltrates in colon adenocarcinoma: Implication for COVID-19. Int. Immunopharmacol. 2021, 95, 107568. [Google Scholar] [CrossRef]
- Wilson, B.G.; Roberts, C.W. SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 2011, 11, 481–492. [Google Scholar] [CrossRef]
- Chen, C.; Enomoto, A. Complex roles of the actin-binding protein Girdin/GIV in DNA damage-induced apoptosis of cancer cells. Cancer Sci. 2020, 111, 4303–4317. [Google Scholar] [CrossRef]
- Qin, L.; Chen, C.; Chen, L.; Xue, R.; Ou-Yang, M.; Zhou, C.; Zhao, S.; He, Z.; Xia, Y.; He, J.; et al. Worldwide malaria incidence and cancer mortality are inversely associated. Infect. Agents Cancer 2017, 12, 14. [Google Scholar] [CrossRef]
- Chen, L.; He, Z.; Qin, L.; Li, Q.; Shi, X.; Zhao, S.; Chen, L.; Zhong, N.; Chen, X. Antitumor effect of malaria parasite infection in a murine Lewis lung cancer model through induction of innate and adaptive immunity. PLoS ONE 2011, 6, e24407. [Google Scholar] [CrossRef]
- Chu, J.H.; Gao, Z.H.; Qu, X.J. Down-regulation of sphingosine kinase 2 (SphK2) increases the effects of all-trans-retinoic acid (ATRA) on colon cancer cells. Biomed. Pharmacother. 2014, 68, 1089–1097. [Google Scholar] [CrossRef]
- Wang, G.; Huang, Y.; Wu, Z.; Zhao, C.; Cong, H.; Ju, S.; Wang, X. KRAS-mutant colon cancer cells respond to combined treatment of ABT263 and axitinib. Biosci. Rep. 2019, 39, BSR20181786. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Q.; Chen, L.; Yang, H.S. Inhibition of p70S6K1 Activation by Pdcd4 Overcomes the Resistance to an IGF-1R/IR Inhibitor in Colon Carcinoma Cells. Mol. Cancer Ther. 2015, 14, 799–809. [Google Scholar] [CrossRef]
- Bian, Z.; Fan, R. A Novel Cuproptosis-Related Prognostic Gene Signature and Validation of Differential Expression in Clear Cell Renal Cell Carcinoma. Genes 2022, 13, 851. [Google Scholar] [CrossRef]
- Li, H.; Liu, L.; Huang, T.; Jin, M.; Zheng, Z.; Zhang, H.; Ye, M.; Liu, K. Establishment of a novel ferroptosis-related lncRNA pair prognostic model in colon adenocarcinoma. Aging 2021, 13, 23072–23095. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, R. LINC01140 regulates osteosarcoma proliferation and invasion by targeting the miR-139-5p/HOXA9 axis. Biochem. Biophys. Rep. 2022, 31, 101301. [Google Scholar] [CrossRef]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Su, Z.; Lu, S.; Fu, W.; Liu, Z.; Jiang, X.; Tai, S. LncRNA HOXA-AS2 and its molecular mechanisms in human cancer. Clin. Chim. Acta Int. J. Clin. Chem. 2018, 485, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Lu, Z.; Li, L.; Ma, M.; Long, F.; Wu, R.; Huang, L.; Chou, J.; Yang, K.; Zhang, Y.; et al. Identification and Validation of Ferroptosis-Related LncRNA Signatures as a Novel Prognostic Model for Colon Cancer. Front. Immunol. 2021, 12, 783362. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Jiang, C.; Zhu, Z.; Han, G.; Xu, N.; Ye, J.; Wang, R. Long non-coding RNA LINC00511 facilitates colon cancer development through regulating microRNA-625-5p to target WEE1. Cell Death Discov. 2022, 8, 233. [Google Scholar] [CrossRef]
- Samuels, T.L.; Zimmermann, M.T.; Zeighami, A.; Demos, W.; Southwood, J.E.; Blumin, J.H.; Bock, J.M. RNA Sequencing Reveals Cancer-Associated Changes in Laryngeal Cells Exposed to Non-Acid Pepsin. Laryngoscope 2021, 131, 121–129. [Google Scholar] [CrossRef]
- Addeo, A.; Friedlaender, A.; Banna, G.L.; Weiss, G.J. TMB or not TMB as a biomarker: That is the question. Crit. Rev. Oncol./Hematol. 2021, 163, 103374. [Google Scholar] [CrossRef]
- Liu, L.; Bai, X.; Wang, J.; Tang, X.R.; Wu, D.H.; Du, S.S.; Du, X.J.; Zhang, Y.W.; Zhu, H.B.; Fang, Y.; et al. Combination of TMB and CNA Stratifies Prognostic and Predictive Responses to Immunotherapy Across Metastatic Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 7413–7423. [Google Scholar] [CrossRef]
- Lv, J.; Zhu, Y.; Ji, A.; Zhang, Q.; Liao, G. Mining TCGA database for tumor mutation burden and their clinical significance in bladder cancer. Biosci. Rep. 2020, 40, BSR20194337. [Google Scholar] [CrossRef]
- Pai, S.G.; Carneiro, B.A.; Chae, Y.K.; Costa, R.L.; Kalyan, A.; Shah, H.A.; Helenowski, I.; Rademaker, A.W.; Mahalingam, D.; Giles, F.J. Correlation of tumor mutational burden and treatment outcomes in patients with colorectal cancer. J. Gastrointest. Oncol. 2017, 8, 858–866. [Google Scholar] [CrossRef]
- Lee, D.W.; Han, S.W. Tumor Mutation Burden and Prognosis in Patients with Colorectal Cancer Treated with Adjuvant Fluoropyrimidine and Oxaliplatin. Clin. Cancer Res. 2019, 25, 6141–6147. [Google Scholar] [CrossRef]
- Bravaccini, S.; Bronte, G. TMB in NSCLC: A Broken Dream? Int. J. Mol. Sci. 2021, 22, 6536. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| lncRNA Name | Coefficient | HR | HR.95L | HR.95H | p Value |
|---|---|---|---|---|---|
| AP003119.3 | 2.443247512 | 4.291960 | 1.570556 | 11.728919 | 0.004510 |
| RNF216P1 | 1.367425711 | 3.088040 | 1.200965 | 7.940272 | 0.019283 |
| AC156455.1 | 0.733807815 | 1.681309 | 1.150079 | 2.457917 | 0.007326 |
| AL360270.1 | 1.773933174 | 2.986836 | 1.163762 | 7.665821 | 0.022887 |
| AC073896.3 | −2.343500132 | 0.356807 | 0.151339 | 0.841232 | 0.018520 |
| AC139720.2 | 1.047691955 | 1.814512 | 1.159697 | 2.839062 | 0.009090 |
| AC092614.1 | 1.213541229 | 2.343952 | 1.077011 | 5.101259 | 0.031797 |
| LINC00511 | −1.445411714 | 0.150115 | 0.062617 | 0.359881 | 0.000021 |
| LRP4-AS1 | 1.80630443 | 1.799438 | 1.119335 | 2.892770 | 0.015292 |
| AC026979.4 | −3.402432218 | 0.029192 | 0.001764 | 0.483129 | 0.013586 |
| AC103703.1 | −4.616328154 | 0.016495 | 0.000326 | 0.833521 | 0.040272 |
| AP003555.1 | 0.58763672 | 2.636331 | 1.595781 | 4.355387 | 0.000154 |
| AL513550.1 | 1.525410885 | 2.399151 | 1.194502 | 4.818681 | 0.013914 |
| LRP1-AS | −7.45422088 | 0.083526 | 0.007523 | 0.927419 | 0.043247 |
| AL512306.2 | 2.464575716 | 4.895259 | 1.643549 | 14.580379 | 0.004341 |
| Covariates | Type | 446 Patients | Validation Group | Training Group | p Value |
|---|---|---|---|---|---|
| Age | <60 years | 121 | 56 | 65 | 0.1236 |
| ≥60 years | 325 | 167 | 158 | ||
| gender | FEMALE | 212 | 101 | 111 | 0.3935 |
| MALE | 234 | 122 | 112 | ||
| stage | Stage I | 75 | 35 | 40 | 0.6808 |
| Stage II | 175 | 93 | 82 | ||
| Stage III | 124 | 64 | 60 | ||
| Stage IV | 61 | 28 | 33 | ||
| unknown | 11 | 3 | 8 | ||
| Pathologic T stage | T1 | 10 | 3 | 7 | 0.6372 |
| T2 | 76 | 38 | 38 | ||
| T3 | 303 | 153 | 150 | ||
| T4 | 56 | 29 | 27 | ||
| unknown | 1 | 0 | 1 | ||
| Pathologic M stage | M0 | 329 | 169 | 160 | 0.519 |
| M1 | 61 | 28 | 33 | ||
| unknown | 56 | 26 | 30 | ||
| Pathologic N stage | N0 | 265 | 135 | 130 | 0.8764 |
| N1 | 102 | 49 | 53 | ||
| N2 | 79 | 39 | 40 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Huang, X.; Chen, S.; Jiang, H.; Rao, H.; Lu, L.; Wen, F.; Pei, J. In Silico Identification and Validation of Cuproptosis-Related LncRNA Signature as a Novel Prognostic Model and Immune Function Analysis in Colon Adenocarcinoma. Curr. Oncol. 2022, 29, 6573-6593. https://doi.org/10.3390/curroncol29090517
Wang Y, Huang X, Chen S, Jiang H, Rao H, Lu L, Wen F, Pei J. In Silico Identification and Validation of Cuproptosis-Related LncRNA Signature as a Novel Prognostic Model and Immune Function Analysis in Colon Adenocarcinoma. Current Oncology. 2022; 29(9):6573-6593. https://doi.org/10.3390/curroncol29090517
Chicago/Turabian StyleWang, Yue, Xulong Huang, Siyu Chen, Huajuan Jiang, Huanan Rao, Lijie Lu, Feiyan Wen, and Jin Pei. 2022. "In Silico Identification and Validation of Cuproptosis-Related LncRNA Signature as a Novel Prognostic Model and Immune Function Analysis in Colon Adenocarcinoma" Current Oncology 29, no. 9: 6573-6593. https://doi.org/10.3390/curroncol29090517
APA StyleWang, Y., Huang, X., Chen, S., Jiang, H., Rao, H., Lu, L., Wen, F., & Pei, J. (2022). In Silico Identification and Validation of Cuproptosis-Related LncRNA Signature as a Novel Prognostic Model and Immune Function Analysis in Colon Adenocarcinoma. Current Oncology, 29(9), 6573-6593. https://doi.org/10.3390/curroncol29090517