Genomics and Immunomics in the Treatment of Urothelial Carcinoma



 ,
,  and
and 
Abstract
:1. Introduction
Overview of Genomic Landscape of Urothelial Carcinoma
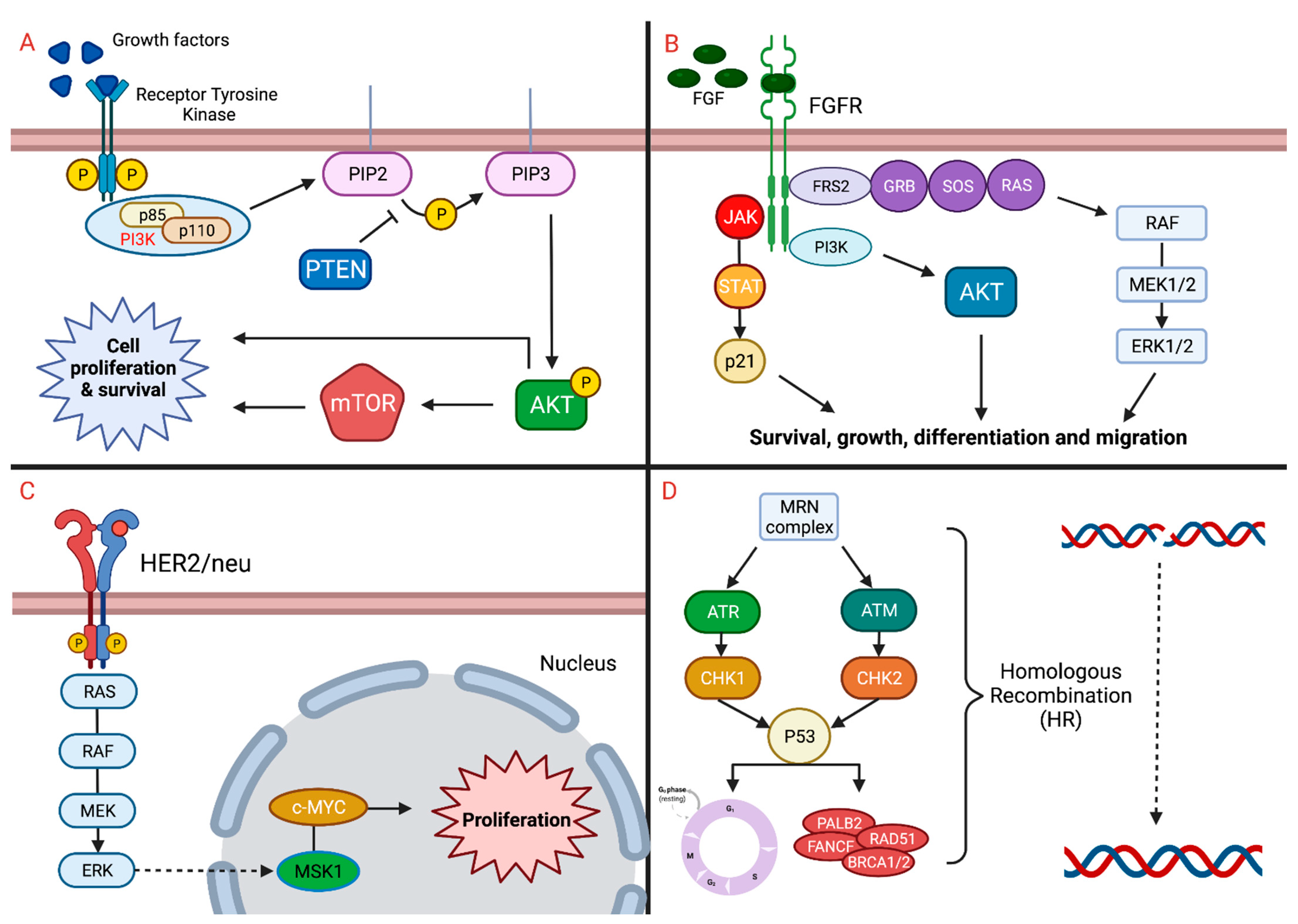
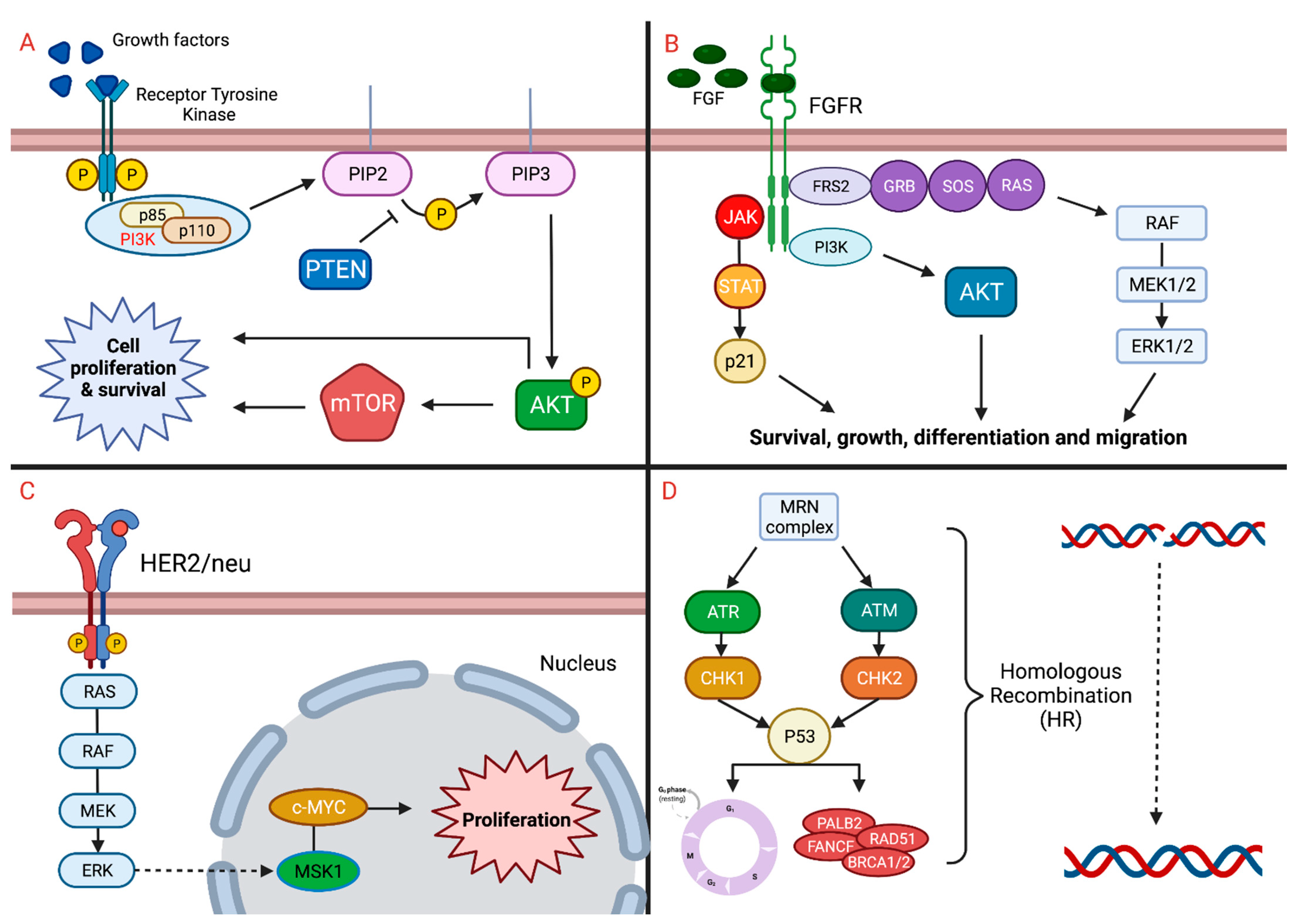
2. Implicated Pathways
2.1. FGFR
2.2. FGFR Trials and Outcomes
2.3. mTOR
2.4. PI3K/AKT/mTOR Trials and Outcomes
2.5. HER2
2.6. HER2 Trials and Outcomes
2.7. DDR Trials and Outcomes
2.8. DDR Trials and Outcomes
2.9. Antibody-Drug Conjugate
3. Immunotherapy in Urothelial Carcinoma
3.1. Immune Checkpoint Inhibitors
3.2. Hyperprogressive Disease
4. Future Directions and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Mollica, V.; Rizzo, A.; Montironi, R.; Cheng, L.; Giunchi, F.; Schiavina, R.; Santoni, M.; Fiorentino, M.; Lopez-Beltran, A.; Brunocilla, E.; et al. Current Strategies and Novel Therapeutic Approaches for Metastatic Urothelial Carcinoma. Cancers 2020, 12, 1449. [Google Scholar] [CrossRef] [PubMed]
- Glaser, A.; Fantini, D.; Shilatifard, A.; Schaeffer, E.M.; Meeks, J.J. The evolving genomic landscape of urothelial carcinoma. Nat. Rev. Urol. 2017, 14, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Sabour, L.; Sabour, M.; Ghorbian, S. Clinical Applications of Next-Generation Sequencing in Cancer Diagnosis. Pathol. Oncol. Res. 2016, 23, 225–234. [Google Scholar] [CrossRef]
- Kato, S.; Kim, K.H.; Lim, H.J.; Boichard, A.; Nikanjam, M.; Weihe, E.; Kuo, D.J.; Eskander, R.N.; Goodman, A.; Galanina, N.; et al. Real-world data from a molecular tumor board demonstrates improved outcomes with a precision N-of-One strategy. Nat. Commun. 2020, 11, 4965. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2017, 171, 540–556.e25. [Google Scholar] [CrossRef] [Green Version]
- Kamoun, A.; de Reyniès, A.; Allory, Y.; Sjödahl, G.; Robertson, A.G.; Seiler, R.; Hoadley, K.A.; Groeneveld, C.S.; Al-Ahmadie, H.; Choi, W.; et al. A Consensus Molecular Classification of Muscle-invasive Bladder Cancer. Eur. Urol. 2020, 77, 420–433. [Google Scholar] [CrossRef]
- Rizzo, A.; Mollica, V.; Cimadamore, A.; Santoni, M.; Scarpelli, M.; Schiavina, R.; Cheng, L.; Lopez-Beltran, A.; Brunocilla, E.; Montironi, R.; et al. TNM staging towards a personalized approach in metastatic urothelial carcinoma: What will the future be like?—A narrative review. Transl. Androl. Urol. 2021, 10, 1541–1552. [Google Scholar] [CrossRef]
- Mollica, V.; Maggio, I.; Lopez-Beltran, A.; Montironi, R.; Cimadamore, A.; Cheng, L.; Rizzo, A.; Giunchi, F.; Schiavina, R.; Fiorentino, M.; et al. Combination therapy in advanced urothelial cancer: The role of PARP, HER-2 and mTOR inhibitors. Expert Rev. Anticancer Ther. 2020, 20, 755–763. [Google Scholar] [CrossRef]
- Mateo, J.; Lord, C.J.; Serra, V.; Tutt, A.; Balmaña, J.; Castroviejo-Bermejo, M.; Cruz, C.; Oaknin, A.; Kaye, S.B.; de Bono, J.S. A decade of clinical development of PARP inhibitors in perspective. Ann. Oncol. 2019, 30, 1437–1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adashek, J.J.; Jain, R.K.; Zhang, J. Clinical Development of PARP Inhibitors in Treating Metastatic Castration-Resistant Prostate Cancer. Cells 2019, 8, 860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, K.L.; Kiyotani, K.; Tamura, K.; Antic, T.; Jang, M.; Montoya, M.; Campanile, A.; Yew, P.Y.; Ganshert, C.; Fujioka, T.; et al. Whole-Exome Sequencing of Muscle-Invasive Bladder Cancer Identifies Recurrent Mutations of UNC5C and Prognostic Importance of DNA Repair Gene Mutations on Survival. Clin. Cancer Res. 2014, 20, 6605–6617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meeks, J.J.; Al-Ahmadie, H.; Faltas, B.M.; Taylor, J.A., 3rd; Flaig, T.W.; DeGraff, D.J.; Christensen, E.; Woolbright, B.L.; McConkey, D.J.; Dyrskjøt, L. Genomic heterogeneity in bladder cancer: Challenges and possible solutions to improve outcomes. Nat. Rev. Urol. 2020, 17, 259–270. [Google Scholar] [CrossRef]
- Günes, C.; Wezel, F.; Southgate, J.; Bolenz, C. Implications of TERT promoter mutations and telomerase activity in urothelial carcinogenesis. Nat. Rev. Urol. 2018, 15, 386–393. [Google Scholar] [CrossRef] [Green Version]
- De Kouchkovsky, I.; Zhang, L.; Philip, E.J.; Wright, F.; Kim, D.M.; Natesan, D.; Kwon, D.; Ho, H.; Ho, S.; Chan, E.; et al. TERT promoter mutations and other prognostic factors in patients with advanced urothelial carcinoma treated with an immune checkpoint inhibitor. J. Immunother Cancer 2021, 9, e002127. [Google Scholar] [CrossRef]
- Xie, Y.; Su, N.; Yang, J.; Tan, Q.; Huang, S.; Jin, M.; Ni, Z.; Zhang, B.; Zhang, D.; Luo, F.; et al. FGF/FGFR signaling in health and disease. Signal Transduct. Target. Ther. 2020, 5, 181. [Google Scholar] [CrossRef]
- Wiedlocha, A.; Haugsten, E.M.; Zakrzewska, M. Roles of the FGF-FGFR Signaling System in Cancer Development and Inflammation. Cells 2021, 10, 2231. [Google Scholar] [CrossRef]
- Helsten, T.; Elkin, S.; Arthur, E.; Tomson, B.N.; Carter, J.; Kurzrock, R. The FGFR Landscape in Cancer: Analysis of 4,853 Tumors by Next-Generation Sequencing. Clin. Cancer Res. 2015, 22, 259–267. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, H.; Smith, M.; Francavilla, C. Fibroblast Growth Factor Receptors (FGFRs) and Noncanonical Partners in Cancer Signaling. Cells 2021, 10, 1201. [Google Scholar] [CrossRef]
- Katoh, M. Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat. Rev. Clin. Oncol. 2018, 16, 105–122. [Google Scholar] [CrossRef]
- Touat, M.; Ileana, E.; Postel-Vinay, S.; André, F.; Soria, J.-C. Targeting FGFR Signaling in Cancer. Clin. Cancer Res. 2015, 21, 2684–2694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stecca, C.; Abdeljalil, O.; Sridhar, S.S. Metastatic Urothelial Cancer: A rapidly changing treatment landscape. Ther. Adv. Med Oncol. 2021, 13, 17588359211047352. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Patel, V.; Galsky, M.D. Urothelial carcinoma: The development of FGFR inhibitors in combination with immune checkpoint inhibitors. Expert Rev. Anticancer Ther. 2020, 20, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Adashek, J.J.; Kato, S.; Lippman, S.M.; Kurzrock, R. The paradox of cancer genes in non-malignant conditions: Implications for precision medicine. Genome Med. 2020, 12, 16. [Google Scholar] [CrossRef] [Green Version]
- Hubert, P.; Selmani, Z.; Loriot, Y.; Thiery-Vuillemin, A. FGFR alterations in urothelial carcinoma: Picking the right target. Bull. Cancer 2021, 108, 566–570. [Google Scholar] [CrossRef]
- Garje, R.; An, J.; Obeidat, M.; Kumar, K.; Yasin, H.A.; Zakharia, Y. Fibroblast Growth Factor Receptor (FGFR) Inhibitors in Urothelial Cancer. Oncologist 2020, 25, e1711–e1719. [Google Scholar] [CrossRef]
- Roskoski, R.J. The role of fibroblast growth factor receptor (FGFR) protein-tyrosine kinase inhibitors in the treatment of cancers including those of the urinary bladder. Pharmacol. Res. 2019, 151, 104567. [Google Scholar] [CrossRef]
- Mazzola, C.; Siddiqui, K.M.; Billia, M.; Chin, J. Dovitinib: Rationale, preclinical and early clinical data in urothelial carcinoma of the bladder. Expert Opin. Investig. Drugs 2014, 23, 1553–1562. [Google Scholar] [CrossRef]
- Pal, S.K.; Bajorin, D.; Dizman, N.; Hoffman-Censits, J.; Quinn, D.I.; Petrylak, D.P.; Galsky, M.D.; Vaishampayan, U.; De Giorgi, U.; Gupta, S.; et al. Infigratinib in upper tract urothelial carcinoma versus urothelial carcinoma of the bladder and its association with comprehensive genomic profiling and/or cell-free DNA results. Cancer 2020, 126, 2597–2606. [Google Scholar] [CrossRef]
- Krook, M.A.; Reeser, J.W.; Ernst, G.; Barker, H.; Wilberding, M.; Li, G.; Chen, H.-Z.; Roychowdhury, S. Fibroblast growth factor receptors in cancer: Genetic alterations, diagnostics, therapeutic targets and mechanisms of resistance. Br. J. Cancer 2020, 124, 880–892. [Google Scholar] [CrossRef] [PubMed]
- Roubal, K.; Myint, Z.W.; Kolesar, J.M. Erdafitinib: A novel therapy for FGFR-mutated urothelial cancer. Am. J. Health. Pharm. 2020, 77, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Loriot, Y.; Necchi, A.; Park, S.H.; Garcia-Donas, J.; Huddart, R.; Burgess, E.; Fleming, M.; Rezazadeh, A.; Mellado, B.; Varlamov, S.; et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2019, 381, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Siefker-Radtke, A.O.; Necchi, A.; Park, S.H.; García-Donas, J.; Huddart, R.A.; Burgess, E.F.; Fleming, M.T.; Kalebasty, A.R.; Mellado, B.; Varlamov, S.; et al. Efficacy and safety of erdafitinib in patients with locally advanced or metastatic urothelial carcinoma: Long-term follow-up of a phase 2 study. Lancet Oncol. 2022, 23, 248–258. [Google Scholar] [CrossRef]
- Casadei, C.; Dizman, N.; Schepisi, G.; Cursano, M.C.; Basso, U.; Santini, D.; Pal, S.K.; De Giorgi, U. Targeted therapies for advanced bladder cancer: New strategies with FGFR inhibitors. Ther. Adv. Med. Oncol. 2019, 11, 1758835919890285. [Google Scholar] [CrossRef]
- Pal, S.K.; Rosenberg, J.E.; Hoffman-Censits, J.H.; Berger, R.; Quinn, D.I.; Galsky, M.D.; Wolf, J.; Dittrich, C.; Keam, B.; Delord, J.-P.; et al. Efficacy of BGJ398, a Fibroblast Growth Factor Receptor 1–3 Inhibitor, in Patients with Previously Treated Advanced Urothelial Carcinoma with FGFR3 Alterations. Cancer Discov. 2018, 8, 812–821. [Google Scholar] [CrossRef] [Green Version]
- Necchi, A.; Pouessel, D.; Leibowitz-Amit, R.; Flechon, A.; Gupta, S.; Barthelemy, P.; Maio, M.; Zhu, X.; Asatiani, E.; Serbest, G.; et al. Interim results of fight-201, a phase II, open-label, multicenter study of INCB054828 in patients (pts) with metastatic or surgically unresectable urothelial carcinoma (UC) harboring fibroblast growth factor (FGF)/FGF receptor (FGFR) genetic alterations (GA). Ann. Oncolo. 2018, 29, viii319–viii320. [Google Scholar]
- Bellmunt, J.; Lalani, A.-K.A.; Jacobus, S.; Wankowicz, S.; Polacek, L.; Takeda, D.Y.; Harshman, L.C.; Wagle, N.; Moreno, I.; Lundgren, K.; et al. Everolimus and pazopanib (E/P) benefit genomically selected patients with metastatic urothelial carcinoma. Br. J. Cancer 2018, 119, 707–712. [Google Scholar] [CrossRef]
- Powles, T.; Huddart, R.A.; Elliott, T.; Sarker, S.J.; Ackerman, C.; Jones, R.; Hussain, S.; Crabb, S.; Jagdev, S.; Chester, J.; et al. Phase III, Double-Blind, Randomized Trial That Compared Maintenance Lapatinib Versus Placebo After First-Line Chemotherapy in Patients With Human Epidermal Growth Factor Receptor 1/2-Positive Metastatic Bladder Cancer. J. Clin. Oncol. 2017, 35, 48–55. [Google Scholar] [CrossRef]
- Hussain, M.H.; MacVicar, G.R.; Petrylak, D.P.; Dunn, R.L.; Vaishampayan, U.; Lara, P.N.J.; Chatta, G.S.; Nanus, D.M.; Glode, L.M.; Trump, D.L.; et al. Trastuzumab, paclitaxel, carboplatin, and gemcitabine in advanced human epidermal growth factor receptor-2/neu-positive urothelial carcinoma: Results of a multicenter phase II National Cancer Institute trial. J. Clin. Oncol. 2007, 25, 2218–2224. [Google Scholar] [CrossRef]
- Hainsworth, J.D.; Meric-Bernstam, F.; Swanton, C.; Hurwitz, H.; Spigel, D.R.; Sweeney, C.; Burris, H.A.; Bose, R.; Yoo, B.; Stein, A.; et al. Targeted Therapy for Advanced Solid Tumors on the Basis of Molecular Profiles: Results From MyPathway, an Open-Label, Phase IIa Multiple Basket Study. J. Clin. Oncol. 2018, 36, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Tagawa, S.T.; Balar, A.V.; Petrylak, D.P.; Kalebasty, A.R.; Loriot, Y.; Fléchon, A.; Jain, R.K.; Agarwal, N.; Bupathi, M.; Barthelemy, P.; et al. TROPHY-U-01: A Phase II Open-Label Study of Sacituzumab Govitecan in Patients With Metastatic Urothelial Carcinoma Progressing After Platinum-Based Chemotherapy and Checkpoint Inhibitors. J. Clin. Oncol. 2021, 39, 2474–2485. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Rosenberg, J.E.; Sonpavde, G.P.; Loriot, Y.; Durán, I.; Lee, J.-L.; Matsubara, N.; Vulsteke, C.; Castellano, D.; Wu, C.; et al. Enfortumab Vedotin in Previously Treated Advanced Urothelial Carcinoma. N. Engl. J. Med. 2021, 384, 1125–1135. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://oncologypro.esmo.org/meeting-resources/esmo-congress/erdafitinib-erda-or-erda-plus-cetrelimab-cet-for-patients-with-metastatic-or-locally-advanced-urothelial-carcinoma-muc-and-fibroblast-growth (accessed on 12 March 2022).
- Gao, Q.; Wang, Z.-C.; Duan, M.; Lin, Y.-H.; Zhou, X.; Worthley, D.L.; Wang, X.-Y.; Niu, G.; Xia, Y.; Deng, M.; et al. Cell Culture System for Analysis of Genetic Heterogeneity Within Hepatocellular Carcinomas and Response to Pharmacologic Agents. Gastroenterology 2017, 152, 232–242.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansel, D.E.; Platt, E.; Orloff, M.; Harwalker, J.; Sethu, S.; Hicks, J.L.; De Marzo, A.; Steinle, R.E.; Hsi, E.D.; Theodorescu, D.; et al. Mammalian Target of Rapamycin (mTOR) Regulates Cellular Proliferation and Tumor Growth in Urothelial Carcinoma. Am. J. Pathol. 2010, 176, 3062–3072. [Google Scholar] [CrossRef]
- Sun, C.-H.; Chang, Y.-H.; Pan, C.-C. Activation of the PI3K/Akt/mTOR pathway correlates with tumour progression and reduced survival in patients with urothelial carcinoma of the urinary bladder. Histopathology 2011, 58, 1054–1063. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [Green Version]
- Madka, V.; Mohammed, A.; Li, Q.; Zhang, Y.; Biddick, L.; Patlolla, J.M.; Lightfoot, S.; Towner, R.A.; Wu, X.-R.; Steele, V.E.; et al. Targeting mTOR and p53 Signaling Inhibits Muscle Invasive Bladder Cancer In Vivo. Cancer Prev. Res. 2015, 9, 53–62. [Google Scholar] [CrossRef] [Green Version]
- Makhlin, I.; Zhang, J.; Long, C.J.; Devarajan, K.; Zhou, Y.; Klein-Szanto, A.J.; Huang, M.; Chernoff, J.; Boorjian, S.A. The mTOR pathway affects proliferation and chemosensitivity of urothelial carcinoma cells and is upregulated in a subset of human bladder cancers. Br. J. Urol. 2010, 108, E84–E90. [Google Scholar] [CrossRef] [Green Version]
- Milowsky, M.I.; Iyer, G.; Regazzi, A.M.; Al-Ahmadie, H.; Gerst, S.R.; Ostrovnaya, I.; Gellert, L.L.; Kaplan, R.; Garcia-Grossman, I.R.; Pendse, D.; et al. Phase II study of everolimus in metastatic urothelial cancer. Br. J. Urol. 2013, 112, 462–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seront, E.; Pinto, A.; Bouzin, C.; Bertrand, L.; Machiels, J.-P.; Feron, O. PTEN deficiency is associated with reduced sensitivity to mTOR inhibitor in human bladder cancer through the unhampered feedback loop driving PI3K/Akt activation. Br. J. Cancer 2013, 109, 1586–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powles, T.; Carroll, D.; Chowdhury, S.; Gravis, G.; Joly, F.; Carles, J.; Fléchon, A.; Maroto, P.; Petrylak, D.; Rolland, F.; et al. An adaptive, biomarker-directed platform study of durvalumab in combination with targeted therapies in advanced urothelial cancer. Nat. Med. 2021, 27, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Mooso, B.A.; Vinall, R.L.; Mudryj, M.; Yap, S.A.; White, R.W.D.; Ghosh, P.M. The Role of EGFR Family Inhibitors in Muscle Invasive Bladder Cancer: A Review of Clinical Data and Molecular Evidence. J. Urol. 2015, 193, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Ménard, S.; Pupa, S.M.; Campiglio, M.; Tagliabue, E. Biologic and therapeutic role of HER2 in cancer. Oncogene 2003, 22, 6570–6578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, M.; Schwaederle, M.; Arguello, D.; Millis, S.Z.; Gatalica, Z.; Kurzrock, R. HER2 expression status in diverse cancers: Review of results from 37,992 patients. Cancer Metastasis Rev. 2015, 34, 157–164. [Google Scholar] [CrossRef] [Green Version]
- Chow, N.H.; Chan, S.H.; Tzai, T.S.; Ho, C.L.; Liu, H.S. Expression profiles of ErbB family receptors and prognosis in primary transitional cell carcinoma of the urinary bladder. Clin. Cancer Res. 2001, 7, 1957–1962. [Google Scholar]
- Krüger, S.; Weitsch, G.; Büttner, H.; Matthiensen, A.; Böhmer, T.; Marquardt, T.; Sayk, F.; Feller, A.C.; Böhle, A. HER2 overexpression in muscle-invasive urothelial carcinoma of the bladder: Prognostic implications. Int. J. Cancer 2002, 102, 514–518. [Google Scholar] [CrossRef]
- Rizzo, A.; Mollica, V.; Giunchi, F.; Dall’Olio, F.G.; Rosellini, M.; Marchetti, A.; Franceschini, T.; Schiavina, R.; Brunocilla, E.; Fiorentino, M.; et al. Impact of HER2 assessment by CISH in urothelial carcinoma: A retrospective single-center experience. Pathol. Res. Pract. 2021, 220, 153410. [Google Scholar] [CrossRef]
- Volotnikova, V.A.; El’Shanskaia, M.P. Morpho-histochemical study of the processes of healing of tuberculosis during treatment with streptomycin and hyaluronidase. Probl. Tuberk. 1975, 58–64. [Google Scholar]
- Junttila, T.T.; Akita, R.W.; Parsons, K.; Fields, C.; Lewis Phillips, G.D.; Friedman, L.S.; Sampath, D.; Sliwkowski, M.X. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell. 2009, 15, 429–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oudard, S.; Culine, S.; Vano, Y.A.; Goldwasser, F.; Théodore, C.; Nguyen, T.; Voog, E.; Banu, E.; Vieillefond, A.; Priou, F.; et al. Multicentre randomised phase II trial of gemcitabine+platinum, with or without trastuzumab, in advanced or metastatic urothelial carcinoma overexpressing Her2. Eur. J. Cancer 2014, 51, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Galsky, M.D.; Del Conte, G.; Foti, S.; Yu, E.Y.; Machiels, J.-P.H.; Doger, B.; Necchi, A.; De Braud, F.G.; Hamilton, E.P.; Hennequin, A.; et al. Primary analysis from DS8201-A-U105: A phase 1b, two-part, open-label study of trastuzumab deruxtecan (T-DXd) with nivolumab (nivo) in patients (pts) with HER2-expressing urothelial carcinoma (UC). J. Clin. Oncol. 2022, 40, 438. [Google Scholar] [CrossRef]
- Choudhury, N.J.; Campanile, A.; Antic, T.; Yap, K.L.; Fitzpatrick, C.A.; Wade, J.L., 3rd; Karrison, T.; Stadler, W.M.; Nakamura, Y.; O’Donnell, P.H. Afatinib Activity in Platinum-Refractory Metastatic Urothelial Carcinoma in Patients with ERBB Alterations. J. Clin. Oncol. 2016, 34, 2165–2171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweis, R.F.; Heiss, B.; Segal, J.; Ritterhouse, L.; Kadri, S.; Churpek, J.E.; Allen, K.; Conway, D.; Marinier, C.; Smith, N.D.; et al. Clinical Activity of Olaparib in Urothelial Bladder Cancer With DNA Damage Response Gene Mutations. JCO Precis. Oncol. 2018, 1–7. [Google Scholar] [CrossRef]
- Grivas, P.; Loriot, Y.; Morales-Barrera, R.; Teo, M.Y.; Zakharia, Y.; Feyerabend, S.; Vogelzang, N.J.; Grande, E.; Adra, N.; Alva, A.; et al. Efficacy and safety of rucaparib in previously treated, locally advanced or metastatic urothelial carcinoma from a phase 2, open-label trial (ATLAS). BMC Cancer 2021, 21, 593. [Google Scholar] [CrossRef]
- Mouw, K.W.; Goldberg, M.S.; Konstantinopoulos, P.A.; D’Andrea, A.D. DNA Damage and Repair Biomarkers of Immunotherapy Response. Cancer Discov. 2017, 7, 675–693. [Google Scholar] [CrossRef] [Green Version]
- Teo, M.Y.; Seier, K.; Ostrovnaya, I.; Regazzi, A.M.; Kania, B.E.; Moran, M.M.; Cipolla, C.K.; Bluth, M.J.; Chaim, J.; Al-Ahmadie, H.; et al. Alterations in DNA Damage Response and Repair Genes as Potential Marker of Clinical Benefit From PD-1/PD-L1 Blockade in Advanced Urothelial Cancers. J. Clin. Oncol. 2018, 36, 1685–1694. [Google Scholar] [CrossRef]
- Crabb, S.J.; Hussain, S.A.; Soulis, E.; Hinsley, S.; Dempsey, L.; Trevethan, A.; Song, Y.P.; Barber, J.; Frew, J.A.; Gale, J.; et al. A randomized, double blind, biomarker selected, phase II clinical trial of maintenance PARP inhibition following chemotherapy for metastatic urothelial carcinoma (mUC): Final analysis of the ATLANTIS rucaparib arm. J. Clin. Oncol. 2022, 40, 436. [Google Scholar] [CrossRef]
- Rosenberg, J.E.; Park, S.H.; Dao, T.V.; Castellano, D.E.; Li, J.-R.; Mukherjee, S.; Howells, K.; Dry, H.; Lanasa, M.C.; Stewart, R.; et al. BAYOU: A phase II, randomized, multicenter, double-blind, study of durvalumab (D) in combination with olaparib (O) for the first-line treatment of platinum-ineligible patients with unresectable, stage IV urothelial carcinoma (UC). J. Clin. Oncol. 2022, 40, 437. [Google Scholar] [CrossRef]
- Grivas, P.; Pouessel, D.; Park, C.H.; Barthélémy, P.; Bupathi, M.; Petrylak, D.P.; Agarwal, N.; Flechon, A.; Ramamurthy, C.; Davis, N.B.; et al. TROPHY-U-01 Cohort 3: Sacituzumab govitecan (SG) in combination with pembrolizumab (Pembro) in patients (pts) with metastatic urothelial cancer (mUC) who progressed after platinum (PLT)-based regimens. J. Clin. Oncol. 2022, 40, 434. [Google Scholar] [CrossRef]
- Challita-Eid, P.M.; Satpayev, D.; Yang, P.; An, Z.; Morrison, K.; Shostak, Y.; Raitano, A.; Nadell, R.; Liu, W.; Lortie, D.R.; et al. Enfortumab Vedotin Antibody-Drug Conjugate Targeting Nectin-4 Is a Highly Potent Therapeutic Agent in Multiple Preclinical Cancer Models. Cancer Res. 2016, 76, 3003–3013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, E.Y.; Petrylak, D.P.; O’Donnell, P.H.; Lee, J.L.; van der Heijden, M.S.; Loriot, Y.; Stein, M.N.; Necchi, A.; Kojima, T.; Harrison, M.R.; et al. Enfortumab vedotin after PD-1 or PD-L1 inhibitors in cisplatin-ineligible patients with advanced urothelial carcinoma (EV201): A multicentre, single-arm, phase 2 trial. Lancet Oncol. 2021, 22, 872–882. [Google Scholar] [CrossRef]
- Friedlander, T.W.; Milowsky, M.I.; Bilen, M.A.; Srinivas, S.; McKay, R.R.; Flaig, T.W.; Hoimes, C.J.; Balar, A.V.; Henry, E.; Petrylak, D.P.; et al. Study EV-103: Update on durability results and long term outcome of enfortumab vedotin + pembrolizumab in first line locally advanced or metastatic urothelial carcinoma (la/mUC). J. Clin. Oncol. 2021, 39, 4528. [Google Scholar] [CrossRef]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; van der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; O’Donnell, P.H.; Balmanoukian, A.; Loriot, Y.; et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 2016, 387, 1909–1920. [Google Scholar] [CrossRef] [Green Version]
- Balar, A.V.; Galsky, M.D.; Rosenberg, J.E.; Powles, T.; Petrylak, D.P.; Bellmunt, J.; Loriot, Y.; Necchi, A.; Hoffman-Censits, J.; Perez-Gracia, J.L.; et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: A single-arm, multicentre, phase 2 trial. Lancet 2017, 389, 67–76, Erratum in Lancet 2017, 390, 848. [Google Scholar] [CrossRef] [Green Version]
- Powles, T.; Csőszi, T.; Özgüroğlu, M.; Matsubara, N.; Géczi, L.; Cheng, S.Y.-S.; Fradet, Y.; Oudard, S.; Vulsteke, C.; Barrera, R.M.; et al. Pembrolizumab alone or combined with chemotherapy versus chemotherapy as first-line therapy for advanced urothelial carcinoma (KEYNOTE-361): A randomised, open-label, phase 3 trial. Lancet Oncol. 2021, 22, 931–945. [Google Scholar] [CrossRef]
- Flaig, T.W.; Spiess, P.E.; Agarwal, N.; Bangs, R.; Boorjian, S.A.; Buyyounouski, M.K.; Chang, S.; Downs, T.M.; Efstathiou, J.A.; Friedlander, T.; et al. Bladder Cancer, Version 3.2020, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2020, 18, 329–354. [Google Scholar] [CrossRef] [Green Version]
- Bellmunt, J.; De Wit, R.; Vaughn, D.J.; Fradet, Y.; Lee, J.-L.; Fong, L.; Vogelzang, N.J.; Climent, M.A.; Petrylak, D.P.; Choueiri, T.K.; et al. Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. N. Engl. J. Med. 2017, 376, 1015–1026. [Google Scholar] [CrossRef] [Green Version]
- Powles, T.; Durán, I.; Van Der Heijden, M.S.; Loriot, Y.; Vogelzang, N.J.; De Giorgi, U.; Oudard, S.; Retz, M.M.; Castellano, D.; Bamias, A.; et al. Atezolizumab versus chemotherapy in patients with platinum-treated locally advanced or metastatic urothelial carcinoma (IMvigor211): A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2018, 391, 748–757. [Google Scholar] [CrossRef]
- Sharma, P.; Retz, M.; Siefker-Radtke, A.; Baron, A.; Necchi, A.; Bedke, J.; Plimack, E.R.; Vaena, D.; Grimm, M.-O.; Bracarda, S.; et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): A multicentre, single-arm, phase 2 trial. Lancet Oncol. 2017, 18, 312–322. [Google Scholar] [CrossRef]
- Grimm, M.-O.; Grün, B.; Niegisch, G.; Pichler, M.; Roghmann, F.; Schmitz-Dräger, B.; Baretton, G.B.; Schmitz, M.; Foller, S.; Leucht, K.; et al. Tailored immunotherapy approach with nivolumab in advanced transitional cell carcinoma (TITAN-TCC). J. Clin. Oncol. 2022, 40, 441. [Google Scholar] [CrossRef]
- Powles, T.; van der Heijden, M.S.; Castellano, D.; Galsky, M.D.; Loriot, Y.; Petrylak, D.P.; Ogawa, O.; Park, S.H.; Lee, J.-L.; De Giorgi, U.; et al. Durvalumab alone and durvalumab plus tremelimumab versus chemotherapy in previously untreated patients with unresectable, locally advanced or metastatic urothelial carcinoma (DANUBE): A randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 2020, 21, 1574–1588. [Google Scholar] [CrossRef]
- Rizzo, A.; Mollica, V.; Massari, F. Adjuvant immunotherapy in muscle-invasive urothelial carcinoma. Lancet Oncol. 2021, 22, e237. [Google Scholar] [CrossRef]
- Powles, T.; Park, S.H.; Voog, E.; Caserta, C.; Valderrama, B.P.; Gurney, H.; Kalofonos, H.; Radulović, S.; Demey, W.; Ullén, A.; et al. Avelumab Maintenance Therapy for Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2020, 383, 1218–1230. [Google Scholar] [CrossRef]
- Nuvola, G.; Rizzo, A.; Mollica, V.; Massari, F. The dilemma of neoadjuvant and adjuvant therapy in urothelial carcinoma: Will immunotherapy solve the problem? Immunotherapy 2022, 14, 171–174. [Google Scholar] [CrossRef]
- Powles, T.; Kockx, M.; Rodriguez-Vida, A.; Duran, I.; Crabb, S.J.; Van Der Heijden, M.S.; Szabados, B.; Pous, A.F.; Gravis, G.; Herranz, U.A.; et al. Publisher Correction: Clinical efficacy and biomarker analysis of neoadjuvant atezolizumab in operable urothelial carcinoma in the ABACUS trial. Nat. Med. 2020, 26, 983. [Google Scholar] [CrossRef]
- Necchi, A.; Anichini, A.; Raggi, D.; Briganti, A.; Massa, S.; Lucianò, R.; Colecchia, M.; Giannatempo, P.; Mortarini, R.; Bianchi, M.; et al. Pembrolizumab as Neoadjuvant Therapy Before Radical Cystectomy in Patients With Muscle-Invasive Urothelial Bladder Carcinoma (PURE-01): An Open-Label, Single-Arm, Phase II Study. J. Clin. Oncol. 2018, 36, 3353–3360. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Navai, N.; Alhalabi, O.; Siefker-Radtke, A.; Campbell, M.T.; Tidwell, R.S.; Guo, C.C.; Kamat, A.M.; Matin, S.F.; Araujo, J.C.; et al. Neoadjuvant PD-L1 plus CTLA-4 blockade in patients with cisplatin-ineligible operable high-risk urothelial carcinoma. Nat. Med. 2020, 26, 1845–1851. [Google Scholar] [CrossRef]
- Van Dijk, N.; Gil-Jimenez, A.; Silina, K.; Hendricksen, K.; Smit, L.A.; de Feijter, J.M.; van Montfoort, M.L.; van Rooijen, C.; Peters, D.; Broeks, A.; et al. Preoperative ipilimumab plus nivolumab in locoregionally advanced urothelial cancer: The NABUCCO trial. Nat Med. 2020, 26, 1839–1844. [Google Scholar] [CrossRef]
- Bajorin, D.F.; Witjes, J.A.; Gschwend, J.E.; Schenker, M.; Valderrama, B.P.; Tomita, Y.; Bamias, A.; Lebret, T.; Shariat, S.F.; Park, S.H.; et al. Adjuvant Nivolumab versus Placebo in Muscle-Invasive Urothelial Carcinoma. N. Engl. J. Med. 2021, 384, 2102–2114. [Google Scholar] [CrossRef] [PubMed]
- Mollica, V.; Rizzo, A.; Massari, F. Adjuvant Nivolumab in Muscle-Invasive Urothelial Carcinoma. N. Engl. J. Med. 2021, 385, 956–957. [Google Scholar] [PubMed]
- Sjödahl, G.; Eriksson, P.; Liedberg, F.; Höglund, M. Molecular classification of urothelial carcinoma: Global mRNA classification versus tumour-cell phenotype classification. J. Pathol. 2017, 242, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Sjödahl, G.; Lauss, M.; Lövgren, K.; Chebil, G.; Gudjonsson, S.; Veerla, S.; Patschan, O.; Aine, M.; Fernö, M.; Ringnér, M.; et al. A Molecular Taxonomy for Urothelial Carcinoma. Clin. Cancer Res. 2012, 18, 3377–3386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellmunt, J.; de Wit, R.; Fradet, Y.; Climent, M.A.; Petrylak, D.P.; Lee, J.L.; Fong, L.; Necchi, A.; Sternberg, C.N.; O’Donnell, P.H.; et al. Putative Biomarkers of Clinical Benefit With Pembrolizumab in Advanced Urothelial Cancer: Results From the KEYNOTE-045 and KEYNOTE-052 Landmark Trials. Clin. Cancer Res. 2022. Online ahead of print. [Google Scholar] [CrossRef]
- Adashek, J.J.; Subbiah, I.M.; Matos, I.; Garralda, E.; Menta, A.K.; Ganeshan, D.M.; Subbiah, V. Hyperprogression and Immunotherapy: Fact, Fiction, or Alternative Fact? Trends Cancer 2020, 6, 181–191. [Google Scholar] [CrossRef] [Green Version]
- Adashek, J.J.; Kato, S.; Ferrara, R.; Lo Russo, G.; Kurzrock, R. Hyperprogression and Immune Checkpoint Inhibitors: Hype or Progress? Oncologist 2019. [Google Scholar] [CrossRef] [Green Version]
- Hwang, I.; Park, I.; Yoon, S.K.; Lee, J.L. Hyperprogressive Disease in Patients With Urothelial Carcinoma or Renal Cell Carcinoma Treated With PD-1/PD-L1 Inhibitors. Clin. Genitour. Cancer 2020, 18, e122–e133. [Google Scholar] [CrossRef]
- Miyama, Y.; Morikawa, T.; Miyakawa, J.; Koyama, Y.; Kawai, T.; Kume, H.; Ushiku, T. Squamous differentiation is a potential biomarker predicting tumor progression in patients treated with pembrolizumab for urothelial carcinoma. Pathol. Res. Pract. 2021, 219, 153364. [Google Scholar] [CrossRef]
- Kato, S.; Goodman, A.; Walavalkar, V.; Barkauskas, D.A.; Sharabi, A.; Kurzrock, R. Hyperprogressors after Immunotherapy: Analysis of Genomic Alterations Associated with Accelerated Growth Rate. Clin. Cancer Res. 2017, 23, 4242–4250. [Google Scholar] [CrossRef] [Green Version]
- Champiat, S.; Ferrara, R.; Massard, C.; Besse, B.; Marabelle, A.; Soria, J.-C.; Ferté, C. Hyperprogressive disease: Recognizing a novel pattern to improve patient management. Nat. Rev. Clin. Oncol. 2018, 15, 748–762. [Google Scholar] [CrossRef] [PubMed]
- Adashek, J.; Janku, F.; Kurzrock, R. Signed in Blood: Circulating Tumor DNA in Cancer Diagnosis, Treatment and Screening. Cancers 2021, 13, 3600. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Assaf, Z.J.; Davarpanah, N.; Banchereau, R.; Szabados, B.E.; Yuen, K.C.; Grivas, P.; Hussain, M.; Oudard, S.; Gschwend, J.E.; et al. ctDNA guiding adjuvant immunotherapy in urothelial carcinoma. Nature 2021, 595, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Shohdy, K.S.; Villamar, D.M.; Cao, Y.; Trieu, J.; Price, K.S.; Nagy, R.; Tagawa, S.T.; Molina, A.M.; Sternberg, C.N.; Nanus, D.M.; et al. Serial ctDNA analysis predicts clinical progression in patients with advanced urothelial carcinoma. Br. J. Cancer 2022, 126, 430–439. [Google Scholar] [CrossRef]
- Hicks, J.K.; Howard, R.; Reisman, P.; Adashek, J.J.; Fields, K.K.; Gray, J.E.; McIver, B.; McKee, K.; O’Leary, M.F.; Perkins, R.M.; et al. Integrating Somatic and Germline Next-Generation Sequencing Into Routine Clinical Oncology Practice. JCO Precis. Oncol. 2021, 5, 884–895. [Google Scholar] [CrossRef]
- Pal, S.K.; Bergerot, P.; Dizman, N.; Bergerot, C.; Adashek, J.; Madison, R.; Chung, J.; Ali, S.M.; Jones, J.O.; Salgia, R. Responses to Alectinib in ALK-rearranged Papillary Renal Cell Carcinoma. Eur. Urol. 2018, 74, 124–128. [Google Scholar] [CrossRef]
- Kato, S.; Adashek, J.J.; Shaya, J.; Okamura, R.; Jimenez, R.E.; Lee, S.; Sicklick, J.K.; Kurzrock, R. Concomitant MEK and Cyclin Gene Alterations: Implications for Response to Targeted Therapeutics. Clin. Cancer Res. 2021, 27, 2792–2797. [Google Scholar] [CrossRef]
- Kato, S.; Okamura, R.; Adashek, J.J.; Khalid, N.; Lee, S.; Nguyen, V.; Sicklick, J.K.; Kurzrock, R. Targeting G1/S phase cell-cycle genomic alterations and accompanying co-alterations with individualized CDK4/6 inhibitor-based regimens. JCI Insight 2021, 6, e142547. [Google Scholar] [CrossRef]
- Sicklick, J.K.; Kato, S.; Okamura, R.; Schwaederle, M.; Hahn, M.E.; Williams, C.B.; De, P.; Krie, A.; Piccioni, D.E.; Miller, V.A.; et al. Molecular profiling of cancer patients enables personalized combination therapy: The I-PREDICT study. Nat. Med. 2019, 25, 744–750. [Google Scholar] [CrossRef]
- Szeto, C.; Kurzrock, R.; Kato, S.; Goloubev, A.; Veerapaneni, S.; Preble, A.; Reddy, S.; Adashek, J. Association of differential expression of immunoregulatory molecules and presence of targetable mutations may inform rational design of clinical trials. ESMO Open 2022, 7, 100396. [Google Scholar] [CrossRef]
- Adashek, J.J.; Subbiah, V.; Kurzrock, R. From Tissue-Agnostic to N-of-One Therapies: (R)Evolution of the Precision Paradigm. Trends Cancer 2020, 7, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Adashek, J.J.; Kato, S.; Parulkar, R.; Szeto, C.W.; Sanborn, J.Z.; Vaske, C.J.; Benz, S.; Reddy, S.K.; Kurzrock, R. Transcriptomic silencing as a potential mechanism of treatment resistance. JCI Insight 2020, 5, e134824. [Google Scholar] [CrossRef] [PubMed]
- Rodon, J.; Soria, J.-C.; Berger, R.; Miller, W.H.; Rubin, E.; Kugel, A.; Tsimberidou, A.; Saintigny, P.; Ackerstein, A.; Braña, I.; et al. Genomic and transcriptomic profiling expands precision cancer medicine: The WINTHER trial. Nat. Med. 2019, 25, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Adashek, J.J.; Goloubev, A.; Kato, S.; Kurzrock, R. Missing the target in cancer therapy. Nat. Cancer 2021, 2, 369–371. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Gene Alteration | Drug | Mechanism of Action | Number of Patients | Outcome | Reference |
|---|---|---|---|---|---|
| FGFR | Erdafitinib | Tyrosine kinase inhibitor of FGFR1–4 | 99 | ORR 40% PFS 5.5 months OS 13.8 months | [34] |
| FGFR | Pemigatinib | Tyrosine kinase inhibitor of FGFR1-3 | 140 (Interim analysis: 100) | ORR 25% | [37] |
| FGFR | Infigratinib | Tyrosine kinase inhibitor of FGFR1-3 | 67 | ORR 25% PFS 3.75 months OS 7.75 months | [36] |
| mTOR | Everolimus + pazopanib | Inhibitor of mTOR + inhibitor of VEGF | 19 | ORR 21% PFS 3.6 months OS 9.1 months | [38] |
| HER2 | Lapatinib | Tyrosine-kinase inhibitor against HER2 and EGFR | 232 | PFS 4.5 months OS 12.6 months | [39] |
| HER2 | Trastuzumab + carboplatin, paclitaxel, gemcitabine | Monoclonal antibody against HER2 | 44 | PFS 9.3 months OS 14.1 months | [40] |
| HER2 | Trastuzumab + pertuzumab | Monoclonal antibody against HER2 | 9 | ORR 33% | [41] |
| Trop2 | Sacituzumab govitecan | ADC of active metabolite of the cytotoxic agent irinotecan and transmembrane glycoprotein highly expressed on epithelial cancer cells surface | 113 | ORR 27% PFS 5.4 months OS 10.9 months | [42] |
| Nectin-4 | Enfortumab vedotin | ADC of anti-nectin-4 conjugated to monomethyl auristatin E | 608 | ORR 52% PFS 5.55 months | [43] |
| Gene Alteration | Drug | Number of Patients Planned to Accrue | Primary Outcome | NCT Number |
|---|---|---|---|---|
| FGFR aberrations | Erdafitinib | 631 | OS | NCT03390504 (THOR) |
| TSC1/TSC2 mutations | Sapanisertib | 209 | ORR | NCT03047213 |
| Unselected | Buparlisib | 19 | 2-months PFS; PFS in the expansion cohort | NCT01551030 |
| Unselected | Nivolumab + nabrapamycin | 34 | Maximum tolerated dose | NCT03190174 |
| Unselected | Nivolumab + IPI-549 | 160 | ORR | NCT03980041 (MARIO-275) |
| Unselected | Paclitaxel + sapanisertib | 52 | ORR | NCT03745911 |
| HER2 overexpressed | Trastuzumab deruxtecan + nivolumab | 99 | Part 1: dose-limiting toxicity Part 2: ORR | NCT03523572 |
| EGFR, HER2, VEGFR, FGFR1/2, MET | Afatinib Regorafenib Cabozantinib | 100 | ORR | NCT02795156 |
| ERBB1, ERBB2, ERBB3 | Afatinib | 42 | 6-months PFS | NCT02780687 |
| HER2-negative | RC48-ADC | 19 | ORR | NCT04073602 |
| HER2-positive | RC48-ADC | 60 | ORR | NCT03809013 |
| HER2-positive | PRS-343 | 85 | Incidence and severity of adverse events | NCT03330561 |
| HER2-positive | PRS-343 + atezolizumab | 45 | Incidence of dose-limiting toxicities; recommended phase 2 dose | NCT03650348 |
| DDR genes | Olaparib | 30 | ORR | NCT03448718 |
| DDR genes | Olaparib | 60 | ORR | NCT03375307 |
| ARID1A, ATM | Olaparib + AZD6738 | 68 | ORR | NCT03682289 |
| Unselected | Niraparib + cabozantinib | 20 | Maximum tolerated dose; PFS | NCT03425201 |
| Unselected | Niraparib | 58 | PFS | NCT03945084 |
| Unselected | Durvalumab + olaparib | 154 | PFS | NCT03459846 |
| Unselected | Atezolizumab + enfortumab vedotin; Atezolizumab + niraparib; Atezolizumab + Hu5F9-G4; Atezolizumab + tiragolumab; Atezolizumab + sacituzumab govitecan; Atezolizumab + tocilizumab; Atezolizumab + RO7122290 | 645 | ORR | NCT03869190 (MORPHEUS-UC) |
| BRCA1, BRCA2, PALB2, RAD51C, RAD51D | Rucaparib + lucitanib; Rucaparib + sacituzumab govitecan | 329 | Phase 1b: Safety and tolerability; Dose-limiting toxicityPhase 2: ORR | NCT03992131 (SEASTAR) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mollica, V.; Massari, F.; Rizzo, A.; Ferrara, R.; Menta, A.K.; Adashek, J.J. Genomics and Immunomics in the Treatment of Urothelial Carcinoma. Curr. Oncol. 2022, 29, 3499-3518. https://doi.org/10.3390/curroncol29050283
Mollica V, Massari F, Rizzo A, Ferrara R, Menta AK, Adashek JJ. Genomics and Immunomics in the Treatment of Urothelial Carcinoma. Current Oncology. 2022; 29(5):3499-3518. https://doi.org/10.3390/curroncol29050283
Chicago/Turabian StyleMollica, Veronica, Francesco Massari, Alessandro Rizzo, Roberto Ferrara, Arjun K. Menta, and Jacob J. Adashek. 2022. "Genomics and Immunomics in the Treatment of Urothelial Carcinoma" Current Oncology 29, no. 5: 3499-3518. https://doi.org/10.3390/curroncol29050283
APA StyleMollica, V., Massari, F., Rizzo, A., Ferrara, R., Menta, A. K., & Adashek, J. J. (2022). Genomics and Immunomics in the Treatment of Urothelial Carcinoma. Current Oncology, 29(5), 3499-3518. https://doi.org/10.3390/curroncol29050283