Subsequent Malignant Neoplasm of Bone in Children and Adolescent—Possibility of Multimodal Treatment


Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Treatment
2.2.1. First Malignant Neoplasm
2.2.2. Subsequent Malignant Neoplasm
2.3. Assessment of Response and Toxicity
2.4. Statistical Methods
3. Results
3.1. First Diagnosis and Treatment
3.2. Subsequent Malignant Bone Neoplasm Diagnosis and Treatment
3.3. Toxicity
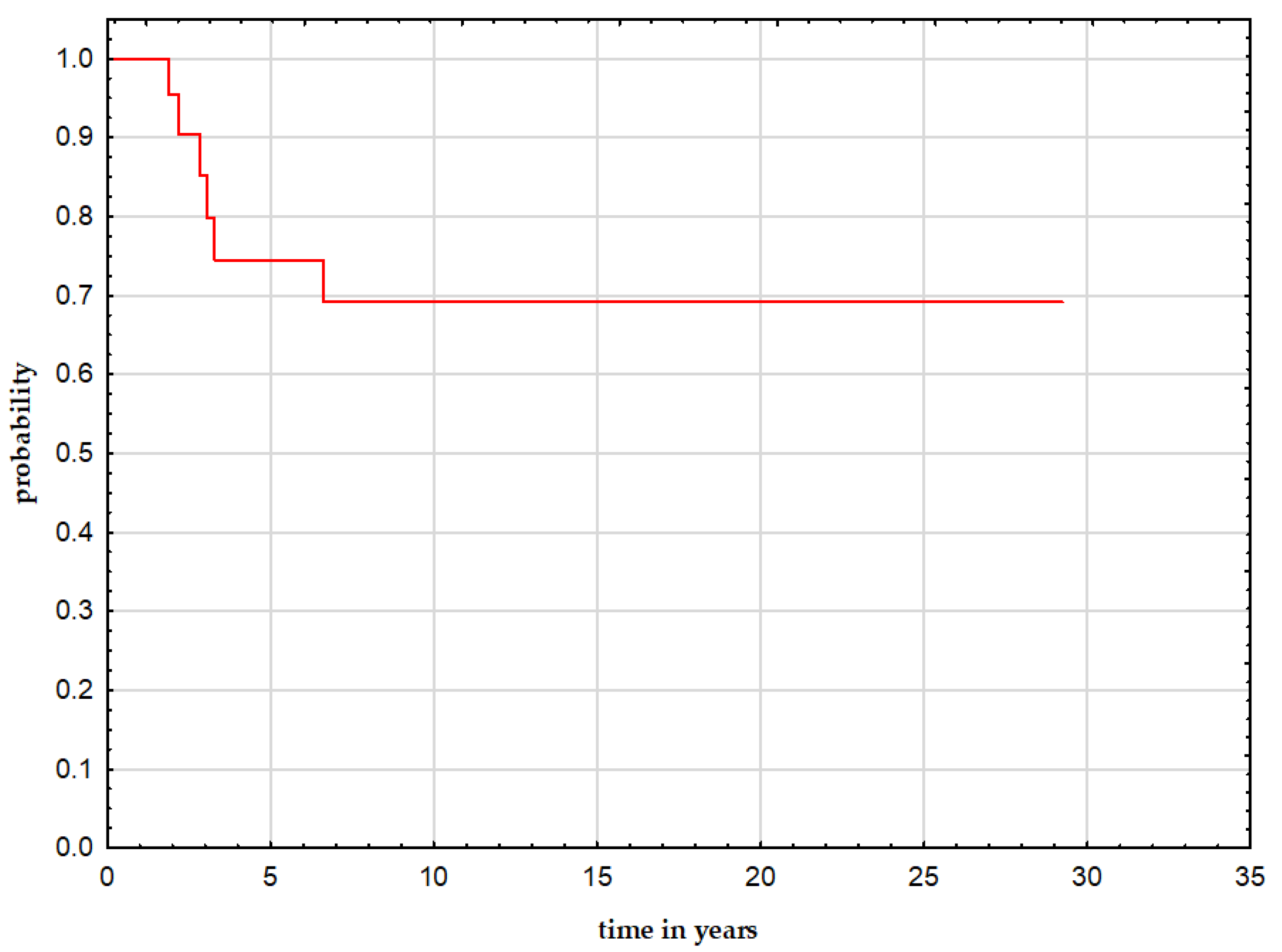
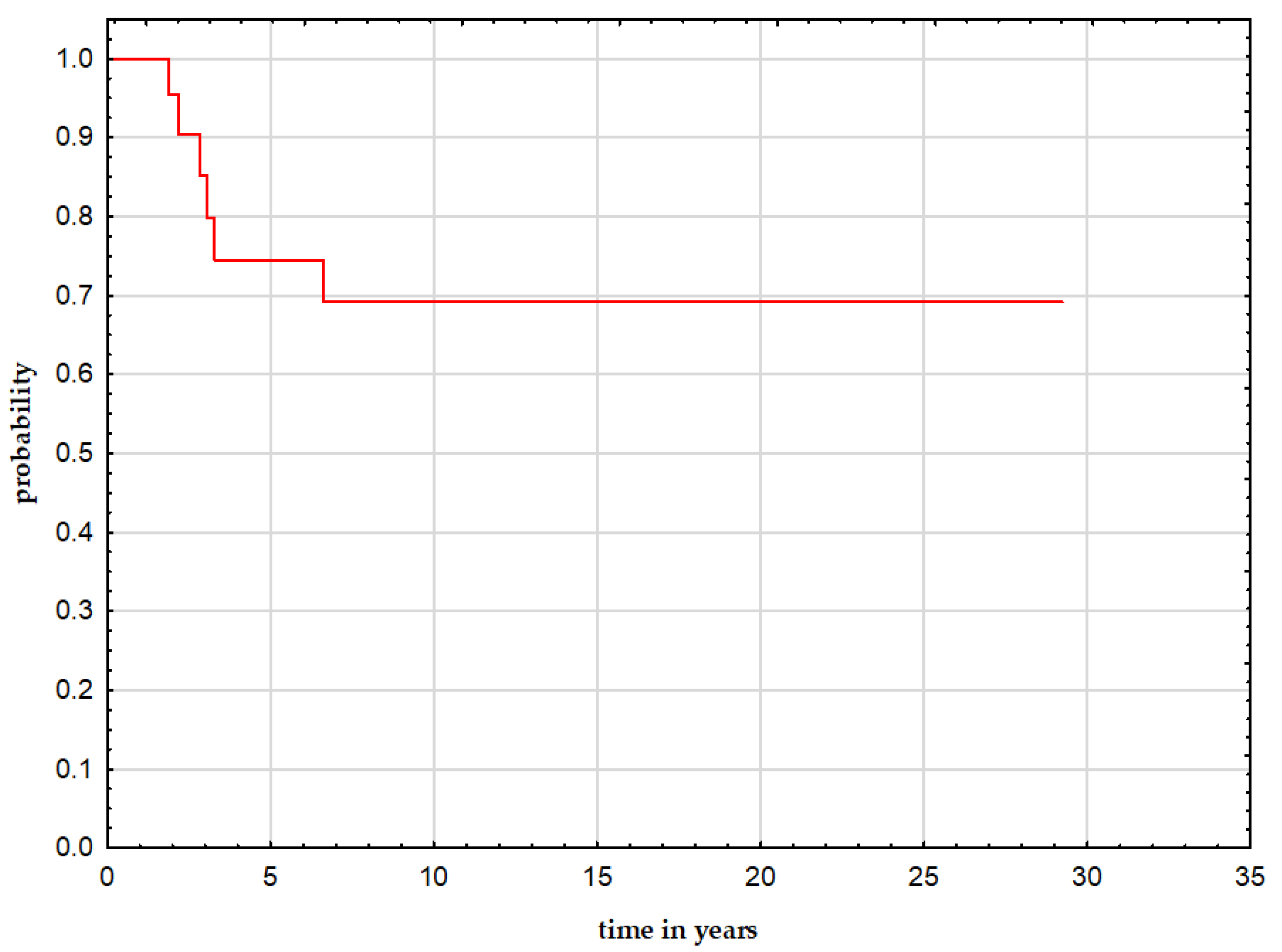
3.4. Follow-Up and Outcome
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Adamson, P.C. Improving the outcome for children with cancer: Development of targeted new agents. CA Cancer J. Clin. 2015, 65, 212–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landier, W.; Armenian, S.; Bhatia, S. Late effects of childhood cancer and its treatment. Pediatr. Clin. North Am. 2015, 62, 275–300. [Google Scholar] [CrossRef] [PubMed]
- Langer, T.; Grabow, D.; Steinmann, D.; Wörmann, B.; Calaminus, G. Late Effects and Long-Term Follow-Up after Cancer in Childhood. Oncol. Res. Treat. 2017, 40, 746–750. [Google Scholar] [CrossRef] [PubMed]
- Inskip, P.D.; Curtis, R.E. New malignancies following childhood cancer in the United States, 1973–2002. Int. J. Cancer 2007, 121, 2233–2240. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, B.V.R.; Petersen, I.A.; Arndt, C.A.; Sim, F.H. Ewing’s sarcoma and the development of secondary malignancies. Clin. Orthop. Relat. Res. 2003, 415, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Meadows, A.T.; Friedman, D.L.; Neglia, J.P.; Mertens, A.C.; Donaldson, S.S.; Stovall, M.; Hammond, S.; Yasui, Y.; Inskip, P.D. Second neoplasms in survivors of childhood cancer: Findings from the Childhood Cancer Survivor Study cohort. J. Clin. Oncol. 2009, 27, 2356–2362. [Google Scholar] [CrossRef] [PubMed]
- Turcotte, L.M.; Liu, Q.; Yasui, Y.; Henderson, T.O.; Gibson, T.M.; Leisenring, W.; Arnold, M.A.; Howell, R.M.; Green, D.M.; Armstrong, G.T.; et al. Chemotherapy and Risk of Subsequent Malignant Neoplasms in the Childhood Cancer Survivor Study Cohort. J. Clin. Oncol. 2019, 37, 3310–3319. [Google Scholar] [CrossRef] [PubMed]
- Koshy, M.; Paulino, A.C.; Mai, W.Y.; Teh, B.S. Radiation-induced osteosarcomas in the pediatric population. Int. J. Radiat. Oncol. Biol. Phys. 2005, 63, 1169–1174. [Google Scholar] [CrossRef] [PubMed]
- Kuttesch, J.F.; Wexler, L.H.; Marcus, R.B. Second malignancies after Ewing’s sarcoma: Radiation dose-dependency of secondary sarcomas. J. Clin. Oncol. 1996, 14, 2818–2825. [Google Scholar] [CrossRef] [PubMed]
- Longhi, A.; Barbieri, E.; Fabbri, N.; Macchiagodena, M.; Favale, L.; Lippo, C.; Salducca, N.; Bacci, G. Radiation-induced osteosarcoma arising 20 years after the treatment of Ewing’s sarcoma. Tumori 2003, 89, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, F.; Rubino, C.; Guerin, S. Risk of a second malignant neoplasm after cancer in childhood treated with radiotherapy: Correlation with the integral dose restricted to the irradiated fields. Int. J. Radiat. Oncol. Biol. Phys. 2008, 70, 908–915. [Google Scholar] [CrossRef] [PubMed]
- Sultan, I.; Rihani, R.; Hazin, R.; Rodriguez-Galindo, C. Second malignancies in patients with Ewing sarcoma family of tumors: A population-based study. Acta Oncol. 2010, 49, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Tahasildar, N.; Goni, V.; Bhagwat, K.; Tripathy, S.K.; Panda, B.B. Ewing’s sarcoma as second malignancy following a short latency in unilateral retinoblastoma. J. Orthop. Traumatol. 2011, 12, 167–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, M.E.; Vogel, V.; Ng, A.; Foxhall, L.; Goodwin, P.; Travis, L.B. Second malignant neoplasms: Assessment and strategies for risk reduction. J. Clin. Oncol. 2012, 30, 3734–3745. [Google Scholar] [CrossRef] [PubMed]
- Caruso, J.; Shulman, D.S.; DuBois, S.G. Second malignancies in patients treated for Ewing sarcoma: A systematic review. Pediatr. Blood Cancer 2019, 66, e27938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, G.E.; Beach, B.; Gastier-Foster, J.M.; Murata-Collins, J.L.; Rowland, J.M.; O’Donnell, R.J.; Goldsby, R.E. Ewing sarcoma as a second malignant neoplasm after acute lymphoblastic leukemia. Pediatr. Blood Cancer 2005, 45, 57–59. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Pts no. | GD | Age (y) FMN | Type of Disease | Treatment |
|---|---|---|---|---|
| 1 | M | 3.3 | ALL | CHT |
| 2 | F | 5.5 | ALL | CHT, RT, alHSCT |
| 3 | M | 5.6 | ALL | CHT, RT |
| 4 | F | 7.4 | ES | CHT, S, RT |
| 5 | F | 15.4 | ES | CHT, S, RT |
| 6 | M | 5.2 | ES | CHT, S, RT |
| 7 | F | 12.1 | ES | CHT, S, RT |
| 8 | F | 7.9 | ES | CHT, S, RT |
| 9 | F | 7.3 | FA | CHT, S, RT |
| 10 | F | 3.0 | GCT | CHT, S |
| 11 | M | 0.1 | GCT | CHT, S, aHSCT |
| 12 | M | 3.3 | GCT | CHT, S |
| 13 | F | 7.5 | CNS tu | CHT, S, RT |
| 14 | M | 10.2 | MM | CHT, S |
| 15 | M | 1.6 | NBL | CHT, S |
| 16 | M | 0.5 | RBL | CHT, S |
| 17 | F | 0.2 | RBL | CHT, S |
| 18 | F | 0.1 | RBL | CHT, S |
| 19 | F | 3.2 | RBL | CHT, S |
| 20 | M | 1.9 | RMS | CHT, S, RT |
| 21 | M | 6.0 | RMS | CHT, S, RT |
| 22 | F | 4.7 | RMS | CHT, S |
| 23 | M | 1.6 | Wilms | CHT, S, RT |
| 24 | M | 7.8 | Wilms | CHT, S, RT |
| Pts no. | Age (y) SMN | Type of Disease | Site of SMN | Meta of SMN | Treatment | CHT Modif. | Toxicity (Grade 3–4) | ADM Discont. | Best Resp. | TMN | Outcome Follow-Up (y) from dgn of SMN |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 15.0 | Osteosa | Lower limb | no | CHT, S | yes | blood, renal | no | CR | na | AWD (13.9) |
| 2 | 11.6 | ES | Axial | no | CHT, S, RT | yes | blood | no | CR | na | AWD (1.9) |
| 3 | 14.0 | ES | Axial | no | CHT, S, RT | yes | blood | yes | CR | na | AWD (10.8) |
| 4 | 12.5 | UPS | Lower limb | no | CHT, S | yes | blood | yes | CR | na | AWD (11.0) |
| 5 | 20.3 | CHS | Axial | no | CHT, S | yes | blood | yes | CR | na | AWD (23.6) |
| 6 | 15.2 | CHS | Lower limb | yes | CHT, S | yes | blood | yes | CR | na | AWD (17.6) |
| 7 | 16.1 | Osteosa | Lower limb | yes | CHT, S | yes | blood | yes | CR | Pleom.sa | DOD (2.8) |
| 8 | 11.8 | Pleom.sa | Lower limb | no | CHT, S | yes | blood | no | CR | na | AWD (1.9) |
| 9 | 15.6 | CHS | Axial | no | CHT, S | yes | blood | no | CR | na | AWD (17.0) |
| 10 | 16.1 | CHS | Lower limb | no | CHT, S | yes | renal | yes | CR | AML | DOD (6.6) |
| 11 | 14.6 | Osteosa | Lower limb | no | CHT, S | yes | renal | no | CR | na | AWD (0.7) |
| 12 | 13.2 | Osteosa | Axial | no | CHT, S | no | na | no | CR | na | AWD (8.4) |
| 13 | 9.9 | Osteosa | Axial | yes | CHT, S | no | na | no | PD | na | DOD (3.3) |
| 14 | 20.3 | Osteosa | Axial | no | CHT, S | no | na | no | CR | na | AWD (12.2) |
| 15 | 5.9 | Osteosa | Upper limb | no | CHT, S | no | na | no | CR | na | AWD (16.2) |
| 16 | 5.9 | Osteosa | Lower limb | yes | CHT, S | no | na | no | PD | na | DOD (2.2) |
| 17 | 11.6 | Osteosa | Lower limb | yes | CHT, S | no | na | no | PD | na | DOD (3.1) |
| 18 | 11.2 | Osteosa | Lower limb | no | CHT, S | no | na | no | CR | Thyroid tu | AWD (29.2) |
| 19 | 6.4 | ES | Lower limb | no | CHT, S | no | na | no | CR | na | AWD (15.0) |
| 20 | 8.8 | Osteosa | Lower limb | no | CHT, S | yes | renal | no | CR | na | AWD (0.6) |
| 21 | 12.0 | Osteosa | Axial | yes | CHT, S | yes | blood, renal | no | PD | na | DOD (1.9) |
| 22 | 9.1 | CHS | Lower limb | no | CHT, S | no | na | no | CR | na | AWD (2.5) |
| 23 | 17.9 | Osteosa | Axial | yes | CHT, S | yes | blood | yes | CR | na | AWD (16.2) |
| 24 | 23.4 | Pleom.sa | Axial | no | CHT, S | no | na | no | CR | na | AWD (11.9) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raciborska, A.; Bilska, K.; Koziński, T.; Rodriguez-Galindo, C. Subsequent Malignant Neoplasm of Bone in Children and Adolescent—Possibility of Multimodal Treatment. Curr. Oncol. 2022, 29, 1001-1007. https://doi.org/10.3390/curroncol29020085
Raciborska A, Bilska K, Koziński T, Rodriguez-Galindo C. Subsequent Malignant Neoplasm of Bone in Children and Adolescent—Possibility of Multimodal Treatment. Current Oncology. 2022; 29(2):1001-1007. https://doi.org/10.3390/curroncol29020085
Chicago/Turabian StyleRaciborska, Anna, Katarzyna Bilska, Tomasz Koziński, and Carlos Rodriguez-Galindo. 2022. "Subsequent Malignant Neoplasm of Bone in Children and Adolescent—Possibility of Multimodal Treatment" Current Oncology 29, no. 2: 1001-1007. https://doi.org/10.3390/curroncol29020085
APA StyleRaciborska, A., Bilska, K., Koziński, T., & Rodriguez-Galindo, C. (2022). Subsequent Malignant Neoplasm of Bone in Children and Adolescent—Possibility of Multimodal Treatment. Current Oncology, 29(2), 1001-1007. https://doi.org/10.3390/curroncol29020085