Crosstalk between Immune Checkpoint Modulators, Metabolic Reprogramming and Cellular Plasticity in Triple-Negative Breast Cancer

 ,
, 


Abstract
1. Introduction
2. Metabolic Reprogramming in TNBC
2.1. Glucose Metabolism and Glycolysis
2.2. Amino Acid Metabolism
2.3. Fatty Acid Metabolism
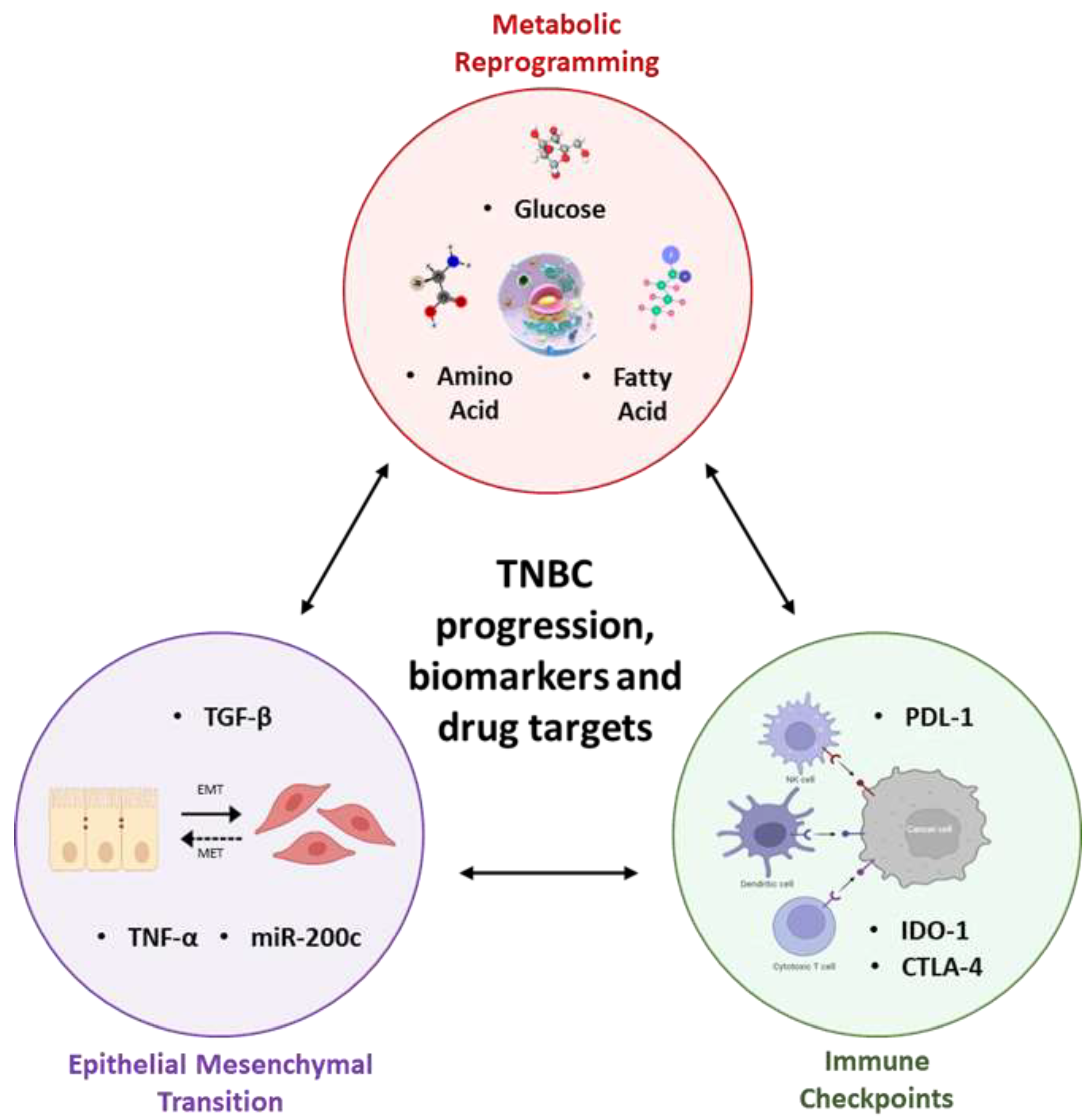
3. Synergy between Metabolic Reprogramming, Immune Checkpoints, and Epithelial-Mesenchymal Transition Lays the Foundation to TNBC Progression
4. Clinicopathological Overview
5. Future Direction
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.-Y.; Park, S.; Kwon, Y. Recent therapeutic trends and promising targets in triple negative breast cancer. Pharmacol. Ther. 2019, 199, 30–57. [Google Scholar] [CrossRef] [PubMed]
- Prithviraj, P.; Anaka, M.; Thompson, E.W.; Sharma, R.; Walkiewicz, M.; Tutuka, C.S.A.; Behren, A.; Kannourakis, G.; Jayachandran, A. Aberrant pregnancy-associated plasma protein-A expression in breast cancers prognosticates clinical outcomes. Sci. Rep. 2020, 10, 13779. [Google Scholar] [CrossRef] [PubMed]
- Denkert, C.; Liedtke, C.; Tutt, A.; von Minckwitz, G. Molecular alterations in triple-negative breast cancer—The road to new treatment strategies. Lancet 2017, 389, 2430–2442. [Google Scholar] [CrossRef]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-Negative Breast Cancer: Clinical Features and Patterns of Recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef]
- Sharma, P. Biology and Management of Patients With Triple-Negative Breast Cancer. Oncologist 2016, 21, 1050–1062. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Fang, C.; Xu, X.; Li, A.; Cai, Q.; Long, X. Androgen Receptor, EGFR, and BRCA1 as Biomarkers in Triple-Negative Breast Cancer: A Meta-Analysis. BioMed Res. Int. 2015, 2015, 357485. [Google Scholar] [CrossRef]
- Gluz, O.; Liedtke, C.; Gottschalk, N.; Pusztai, L.; Nitz, U.; Harbeck, N. Triple-negative breast cancer—Current status and future directions. Ann. Oncol. 2009, 20, 1913–1927. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef]
- Robson, M.E.; Tung, N.; Conte, P.; Im, S.-A.; Senkus, E.; Xu, B.; Masuda, N.; Delaloge, S.; Li, W.; Armstrong, A.; et al. OlympiAD final overall survival and tolerability results: Olaparib versus chemotherapy treatment of physician’s choice in patients with a germline BRCA mutation and HER2-negative metastatic breast cancer. Ann. Oncol. 2019, 30, 558–566. [Google Scholar] [CrossRef]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Emens, L.A.; Molinero, L.; Loi, S.; Rugo, H.S.; Schneeweiss, A.; Diéras, V.; Iwata, H.; Barrios, C.H.; Nechaeva, M.; Nguyen-Duc, A.; et al. Atezolizumab and nab-Paclitaxel in Advanced Triple-Negative Breast Cancer: Biomarker Evaluation of the IMpas-sion130 Study. J. Natl. Cancer Inst. 2021, 113, 1005–1016. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Ohshima, K.; Morii, E. Metabolic Reprogramming of Cancer Cells during Tumor Progression and Metastasis. Metabolites 2021, 11, 28. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Boedtkjer, E.; Pedersen, S.F. The Acidic Tumor Microenvironment as a Driver of Cancer. Annu. Rev. Physiol. 2020, 82, 103–126. [Google Scholar] [CrossRef]
- Jayachandran, A.; Lo, P.-H.; Chueh, A.C.; Prithviraj, P.; Molania, R.; Davalos-Salas, M.; Anaka, M.; Walkiewicz, M.; Cebon, J.; Behren, A. Transketolase-like 1 ectopic expression is associated with DNA hypomethylation and induces the Warburg effect in melanoma cells. BMC Cancer 2016, 16, 134. [Google Scholar] [CrossRef]
- Sun, X.; Wang, M.; Wang, M.; Yu, X.; Guo, J.; Sun, T.; Li, X.; Yao, L.; Dong, H.; Xu, Y. Metabolic Reprogramming in Triple-Negative Breast Cancer. Front. Oncol. 2020, 10, 428. [Google Scholar] [CrossRef]
- Yin, L.; Duan, J.-J.; Bian, X.-W.; Yu, S.-C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef]
- Cao, M.D.; Lamichhane, S.; Lundgren, S.; Bofin, A.; Fjøsne, H.; Giskeødegård, G.F.; Bathen, T.F. Metabolic characterization of triple negative breast cancer. BMC Cancer 2014, 14, 941. [Google Scholar] [CrossRef]
- Lanning, N.J.; Castle, J.P.; Singh, S.J.; Leon, A.N.; Tovar, E.A.; Sanghera, A.; MacKeigan, J.P.; Filipp, F.V.; Graveel, C.R. Metabolic profiling of triple-negative breast cancer cells reveals metabolic vulnerabilities. Cancer Metab. 2017, 5, 6. [Google Scholar] [CrossRef]
- Pelicano, H.; Zhang, W.; Liu, J.; Hammoudi, N.; Dai, J.; Xu, R.-H.; Pusztai, L.; Huang, P. Mitochondrial dysfunction in some triple-negative breast cancer cell lines: Role of mTOR pathway and therapeutic potential. Breast Cancer Res. 2014, 16, 434. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Jung, W.H.; Koo, J.S. Metabolism-related proteins are differentially expressed according to the molecular subtype of in-vasive breast cancer defined by surrogate immunohistochemistry. Pathobiology 2013, 80, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Xie, R.; Cao, Y.; Tang, J.; Men, Y.; Peng, H.; Yang, W. Simvastatin induced ferroptosis for triple-negative breast cancer therapy. J. Nanobiotechnol. 2021, 19, 311. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Park, I.A.; Heo, S.-H.; Kim, Y.-A.; Gong, G.; Lee, H.J. Association between p53 Expression and Amount of Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancer. J. Pathol. Transl. Med. 2019, 53, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Mardis, E.R.; Wilson, R.K.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Wang, E.; Sorolla, A.; Cunningham, P.T.; Bogdawa, H.M.; Beck, S.; Golden, E.; Dewhurst, R.; Florez, L.; Cruickshank, M.N.; Hoffmann, K.; et al. Tumor penetrating peptides inhibiting MYC as a potent targeted therapeutic strategy for triple-negative breast cancers. Oncogene 2018, 38, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sun, X.-X.; Qian, D.Z.; Dai, M.-S. Molecular Crosstalk Between MYC and HIF in Cancer. Front. Cell Dev. Biol. 2020, 8, 590576. [Google Scholar] [CrossRef]
- Shin, E.; Koo, J.S. Glucose Metabolism and Glucose Transporters in Breast Cancer. Front. Cell Dev. Biol. 2021, 9, 728759. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic Glycolysis: Meeting the Metabolic Requirements of Cell Proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef]
- Shen, L.; O’Shea, J.M.; Kaadige, M.R.; Cunha, S.; Wilde, B.R.; Cohen, A.L.; Welm, A.L.; Ayer, D.E. Metabolic reprogramming in triple-negative breast cancer through Myc suppression of TXNIP. Proc. Natl. Acad. Sci. USA 2015, 112, 5425–5430. [Google Scholar] [CrossRef]
- Ghergurovich, J.M.; Lang, J.D.; Levin, M.K.; Briones, N.; Facista, S.J.; Mueller, C.; Cowan, A.J.; McBride, M.J.; Rodriguez, E.S.R.; Killian, A.; et al. Local production of lactate, ribose phosphate, and amino acids within human triple-negative breast cancer. Med 2021, 2, 736–754. [Google Scholar] [CrossRef]
- Kim, S.; Kim, D.H.; Jung, W.-H.; Koo, J.S. Metabolic phenotypes in triple-negative breast cancer. Tumor Biol. 2013, 34, 1699–1712. [Google Scholar] [CrossRef]
- Costa, R.L.B.; Han, H.S.; Gradishar, W.J. Targeting the PI3K/AKT/mTOR pathway in triple-negative breast cancer: A review. Breast Cancer Res. Treat. 2018, 169, 397–406. [Google Scholar] [CrossRef]
- Avanzato, D.; Pupo, E.; Ducano, N.; Isella, C.; Bertalot, G.; Luise, C.; Pece, S.; Bruna, A.; Rueda, O.M.; Caldas, C.; et al. High USP6NL Levels in Breast Cancer Sustain Chronic AKT Phosphorylation and GLUT1 Stability Fueling Aerobic Glycolysis. Cancer Res. 2018, 78, 3432–3444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, H.; Yu, W.; Qiao, F.; Su, X.; Xu, H. Downregulation of hexokinase 2 improves radiosensitivity of breast cancer. Transl. Cancer Res. 2019, 8, 290–297. [Google Scholar] [CrossRef]
- Ding, L.; Gu, H.; Xiong, X.; Ao, H.; Cao, J.; Lin, W.; Yu, M.; Lin, J.; Cui, Q. MicroRNAs Involved in Carcinogenesis, Prognosis, Therapeutic Resistance and Applications in Human Triple-Negative Breast Cancer. Cells 2019, 8, 1492. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Zu, X.; Liu, K.; Bode, A.M.; Dong, Z.; Liu, Z.; Kim, D.J. Knockdown of Pyruvate Kinase M Inhibits Cell Growth and Migration by Reducing NF-kB Activity in Triple-Negative Breast Cancer Cells. Mol. Cells 2019, 42, 628–636. [Google Scholar] [PubMed]
- Romero-Cordoba, S.L.; Rodriguez-Cuevas, S.; Bautista-Pina, V.; Maffuz-Aziz, A.; D’Ippolito, E.; Cosentino, G.; Baroni, S.; Iorio, M.V.; Hidalgo-Miranda, A. Loss of function of miR-342-3p results in MCT1 over-expression and contributes to oncogenic metabolic reprogramming in triple negative breast cancer. Sci. Rep. 2018, 8, 12252. [Google Scholar] [CrossRef]
- Du, Y.; Wei, N.; Ma, R.; Jiang, S.; Song, D. A miR-210-3p regulon that controls the Warburg effect by modulating HIF-1α and p53 activity in triple-negative breast cancer. Cell Death Dis. 2020, 11, 731. [Google Scholar] [CrossRef]
- Wang, Z.; Jiang, Q.; Dong, C. Metabolic reprogramming in triple-negative breast cancer. Cancer Biol. Med. 2020, 17, 44–59. [Google Scholar] [CrossRef] [PubMed]
- Owens, K.M.; Kulawiec, M.; Desouki, M.M.; Vanniarajan, A.; Singh, K.K. Impaired OXPHOS Complex III in Breast Cancer. PLoS ONE 2011, 6, e23846. [Google Scholar] [CrossRef] [PubMed]
- Lunetti, P.; Di Giacomo, M.; Vergara, D.; De Domenico, S.; Maffia, M.; Zara, V.; Capobianco, L.; Ferramosca, A. Metabolic reprogramming in breast cancer results in distinct mitochondrial bioenergetics between luminal and basal subtypes. FEBS J. 2018, 286, 688–709. [Google Scholar] [CrossRef]
- Evans, K.W.; Yuca, E.; Scott, S.S.; Zhao, M.; Arango, N.P.; Pico, C.X.C.; Saridogan, T.; Shariati, M.; Class, C.A.; Bristow, C.A.; et al. Oxidative Phosphorylation Is a Metabolic Vulnerability in Chemotherapy-Resistant Triple-Negative Breast Cancer. Cancer Res. 2021, 81, 5572–5581. [Google Scholar] [CrossRef] [PubMed]
- Zacksenhaus, E.; Shrestha, M.; Liu, J.C.; Vorobieva, I.; Chung, P.E.; Ju, Y.; Nir, U.; Jiang, Z. Mitochondrial OXPHOS Induced by RB1 Deficiency in Breast Cancer: Implications for Anabolic Metabolism, Stemness, and Metastasis. Trends Cancer 2017, 3, 768–779. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Jones, R.; Liu, J.C.; Deng, T.; Robinson, T.; Chung, P.E.; Wang, S.; Herschkowitz, J.I.; Egan, S.E.; Perou, C.M.; et al. RB1 and p53 at the crossroad of EMT and triple-negative breast cancer. Cell Cycle 2011, 10, 1563–1570. [Google Scholar] [CrossRef] [PubMed]
- Koit, A.; Timohhina, N.; Truu, L.; Chekulayev, V.; Gudlawar, S.; Shevchuk, I.; Lepik, K.; Mallo, L.; Kutner, R.; Valvere, V.; et al. Metabolic and OXPHOS Activities Quantified by Temporal ex vivo Analysis Display Patient-Specific Metabolic Vul-nerabilities in Human Breast Cancers. Front. Oncol. 2020, 10, 1053. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.M.; Cotzia, P.; Fratamico, R.; Mikkilineni, L.; Chen, J.; Colombo, D.; Mollaee, M.; Whitaker-Menezes, D.; Domingo-Vidal, M.; Lin, Z.; et al. MCT1 in Invasive Ductal Carcinoma: Monocarboxylate Metabolism and Aggressive Breast Cancer. Front. Cell Dev. Biol. 2017, 5, 27. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, C.; Sousa, B.; Albergaria, A.; Paredes, J.; Dufloth, R.; Vieira, D.; Schmitt, F.; Baltazar, F. GLUT1 and CAIX expression profiles in breast cancer correlate with adverse prognostic factors and MCT1 overexpression. Histol. Histopathol. 2011, 26. [Google Scholar]
- Wei, Z.; Liu, X.; Cheng, C.; Yu, W.; Yi, P. Metabolism of Amino Acids in Cancer. Front. Cell Dev. Biol. 2021, 8, 603837. [Google Scholar] [CrossRef]
- Bhutia, Y.D.; Ganapathy, V. Glutamine transporters in mammalian cells and their functions in physiology and cancer. Biochim. Biophys. Acta 2015, 1863, 2531–2539. [Google Scholar] [CrossRef] [PubMed]
- Lampa, M.; Arlt, H.; He, T.; Ospina, B.; Reeves, J.; Zhang, B.; Murtie, J.; Deng, G.; Barberis, C.; Hoffmann, D.; et al. Glutaminase is essential for the growth of triple-negative breast cancer cells with a deregulated glutamine metabolism pathway and its suppression synergizes with mTOR inhibition. PLoS ONE 2017, 12, e0185092. [Google Scholar] [CrossRef]
- Ansari, R.E.; McIntyre, A.; Craze, M.L.; Ellis, I.O.; Rakha, E.A.; Green, A.R. Altered glutamine metabolism in breast cancer; subtype dependencies and alternative adaptations. Histopathology 2017, 72, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, L.A.; Holton, T.; Yuneva, M.; Louie, R.J.; Padró, M.; Daemen, A.; Hu, M.; Chan, D.A.; Ethier, S.P.; van ‘t Veer, L.J.; et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor ther-apeutic target. Cancer Cell 2013, 24, 450–465. [Google Scholar] [CrossRef] [PubMed]
- Montero, J.C.; Esparís-Ogando, A.; Re-Louhau, M.F.; Seoane, S.; Abad, M.; Calero, R.; Ocaña, A.; Pandiella, A. Active kinase profiling, genetic and pharmacological data define mTOR as an important common target in tri-ple-negative breast cancer. Oncogene 2014, 33, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Combs, J.A.; DeNicola, G.M. The Non-Essential Amino Acid Cysteine Becomes Essential for Tumor Proliferation and Survival. Cancers 2019, 11, 678. [Google Scholar] [CrossRef]
- Chen, M.-S.; Wang, S.-F.; Hsu, C.-Y.; Yin, P.-H.; Yeh, T.-S.; Lee, H.-C.; Tseng, L.-M. CHAC1 degradation of glutathione enhances cystine-starvation-induced necroptosis and ferroptosis in human triple negative breast cancer cells via the GCN2-eIF2α-ATF4 pathway. Oncotarget 2017, 8, 114588–114602. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Ding, C.-K.; Wu, J.; Sjol, J.; Wardell, S.; Spasojevic, I.; George, D.; McDonnell, D.P.; Hsu, D.S.; Chang, J.T.; et al. Cystine addiction of triple-negative breast cancer associated with EMT augmented death signaling. Oncogene 2016, 36, 4235–4242. [Google Scholar] [CrossRef]
- Li, M.; Bu, X.; Cai, B.; Liang, P.; Li, K.; Qu, X.; Shen, L. Biological role of metabolic reprogramming of cancer cells during epithelial-mesenchymal transition (Review). Oncol. Rep. 2018, 41, 727–741. [Google Scholar] [CrossRef]
- Kim, S.K.; Jung, W.H.; Koo, J.S. Differential Expression of Enzymes Associated with Serine/Glycine Metabolism in Different Breast Cancer Subtypes. PLoS ONE 2014, 9, e101004. [Google Scholar] [CrossRef] [PubMed]
- Chisari, A.; Golán, I.; Campisano, S.; Gélabert, C.; Moustakas, A.; Sancho, P.; Caja, L. Glucose and Amino Acid Metabolic Dependencies Linked to Stemness and Metastasis in Different Aggressive Cancer Types. Front. Pharmacol. 2021, 12, 723798. [Google Scholar] [CrossRef] [PubMed]
- D’Amato, N.C.; Rogers, T.J.; Gordon, M.A.; Greene, L.I.; Cochrane, D.R.; Spoelstra, N.S.; Nemkov, T.G.; D’Alessandro, A.; Hansen, K.C.; Richer, J.K. A TDO2-AhR Signaling Axis Facilitates Anoikis Resistance and Metastasis in Triple-Negative Breast Cancer. Cancer Res. 2015, 75, 4651–4664. [Google Scholar] [CrossRef] [PubMed]
- Greene, L.I.; Bruno, T.C.; Christenson, J.L.; D’Alessandro, A.; Culp-Hill, R.; Torkko, K.; Borges, V.F.; Slansky, J.E.; Richer, J.K. A Role for Tryptophan-2,3-dioxygenase in CD8 T-cell Suppression and Evidence of Tryptophan Catabolism in Breast Cancer Patient Plasma. Mol. Cancer Res. 2019, 17, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Monaco, M.E. Fatty acid metabolism in breast cancer subtypes. Oncotarget 2017, 8, 29487–29500. [Google Scholar] [CrossRef]
- Ma, Y.; Temkin, S.M.; Hawkridge, A.M.; Guo, C.; Wang, W.; Wang, X.-Y.; Fang, X. Fatty acid oxidation: An emerging facet of metabolic transformation in cancer. Cancer Lett. 2018, 435, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular Fatty Acid Metabolism and Cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef]
- Giró-Perafita, A.; Palomeras, S.; Lum, D.H.; Blancafort, A.; Viñas, G.; Oliveras, G.; Pérez-Bueno, F.; Sarrats, A.; Welm, A.L.; Puig, T. Preclinical Evaluation of Fatty Acid Synthase and EGFR Inhibition in Triple-Negative Breast Cancer. Clin. Cancer Res. 2016, 22, 4687–4697. [Google Scholar] [CrossRef]
- Camarda, R.; Zhou, A.Y.; Kohnz, R.A.; Balakrishnan, S.; Mahieu, C.; Anderton, B.; Eyob, H.; Kajimura, S.; Tward, A.; Krings, G.; et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat. Med. 2016, 22, 427–432. [Google Scholar] [CrossRef]
- Kuemmerle, N.B.; Rysman, E.; Lombardo, P.S.; Flanagan, A.J.; Lipe, B.C.; Wells, W.A.; Pettus, J.R.; Froehlich, H.M.; Memoli, V.A.; Morganelli, P.M.; et al. Lipoprotein Lipase Links Dietary Fat to Solid Tumor Cell Proliferation. Mol. Cancer Ther. 2011, 10, 427–436. [Google Scholar] [CrossRef]
- Wang, B.; Yan, N.; Wu, D.; Dou, Y.; Hu, X.; Chen, C. Combination inhibition of triple-negative breast cancer cell growth with CD36 siRNA-loaded DNA nanoprism and genistein. Nanotechnology 2021, 32, 395101. [Google Scholar] [CrossRef]
- Ehmsen, S.; Pedersen, M.H.; Wang, G.; Terp, M.G.; Arslanagic, A.; Hood, B.L.; Conrads, T.P.; Leth-Larsen, R.; Ditzel, H.J. Increased Cholesterol Biosynthesis Is a Key Characteristic of Breast Cancer Stem Cells Influencing Patient Outcome. Cell Rep. 2019, 27, 3927–3938.e6. [Google Scholar] [CrossRef]
- Pacheco-Torres, J.; Penet, M.-F.; Mironchik, B.; Krishnamachary, B.; Bhujwalla, Z.M. The PD-L1 metabolic interactome intersects with choline metabolism and inflammation. Cancer Metab. 2021, 9, 10. [Google Scholar] [CrossRef]
- Bhatia, S.; Wang, P.; Toh, A.; Thompson, E.W. New Insights Into the Role of Phenotypic Plasticity and EMT in Driving Cancer Progression. Front. Mol. Biosci. 2020, 7, 71. [Google Scholar] [CrossRef]
- Xia, L.; Oyang, L.; Lin, J.; Tan, S.; Han, Y.; Wu, N.; Yi, P.; Tang, L.; Pan, Q.; Rao, S.; et al. The cancer metabolic reprogramming and immune response. Mol. Cancer 2021, 20, 28. [Google Scholar] [CrossRef] [PubMed]
- Naik, A.; Decock, J. Lactate Metabolism and Immune Modulation in Breast Cancer: A Focused Review on Triple Negative Breast Tumors. Front. Oncol. 2020, 10, 598626. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Tanikawa, T.; Kryczek, I.; Xia, H.; Li, G.; Wu, K.; Wei, S.; Zhao, L.; Vatan, L.; Wen, B.; et al. Aerobic Glycolysis Controls Myeloid-Derived Suppressor Cells and Tumor Immunity via a Specific CEBPB Isoform in Triple-Negative Breast Cancer. Cell Metab. 2018, 28, 87–103.e6. [Google Scholar] [CrossRef]
- Byun, J.-K.; Park, M.; Lee, S.; Yun, J.W.; Lee, J.; Kim, J.S.; Cho, S.J.; Jeon, H.-J.; Lee, I.-K.; Choi, Y.-K.; et al. Inhibition of Glutamine Utilization Synergizes with Immune Checkpoint Inhibitor to Promote Antitumor Immunity. Mol. Cell 2020, 80, 592–606.e8. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Lin, H.; Yuan, L.; Li, B. Combination therapy with L-arginine and α-PD-L1 antibody boosts immune response against osteosarcoma in immuno-competent mice. Cancer Biol. Ther. 2017, 18, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yue, C.; Herrmann, A.; Song, J.; Egelston, C.; Wang, T.; Zhang, Z.; Li, W.; Lee, H.; Aftabizadeh, M.; et al. STAT3 Activation-Induced Fatty Acid Oxidation in CD8+ T Effector Cells Is Critical for Obesity-Promoted Breast Tumor Growth. Cell Metab. 2019, 31, 148–161.e5. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.; Al-Khadairi, G.; Decock, J. Immune Checkpoint Inhibitors in Triple Negative Breast Cancer Treatment: Promising Future Prospects. Front. Oncol. 2021, 10, 600573. [Google Scholar] [CrossRef]
- Qin, Y.; Vasilatos, S.N.; Chen, L.; Wu, H.; Cao, Z.; Fu, Y.; Huang, M.; Vlad, A.M.; Lu, B.; Oesterreich, S.; et al. Inhibition of histone lysine-specific demethylase 1 elicits breast tumor immunity and enhances antitumor efficacy of immune checkpoint blockade. Oncogene 2018, 38, 390–405. [Google Scholar] [CrossRef]
- Racioppi, L.; Nelson, E.R.; Huang, W.; Mukherjee, D.; Lawrence, S.A.; Lento, W.; Masci, A.M.; Jiao, Y.; Park, S.; York, B.; et al. CaMKK2 in myeloid cells is a key regulator of the immune-suppressive microenvironment in breast cancer. Nat. Commun. 2019, 10, 2450. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Xu, J.; Xie, Z.; Huang, H.; Li, N.; Wei, X.; Li, T.; Yang, H.; Li, S.; Qin, X.; et al. Light-responsive hyaluronic acid nanomicelles co-loaded with an IDO inhibitor focus targeted photoimmunotherapy against “immune cold” cancer. Biomater. Sci. 2021, 9, 8019–8031. [Google Scholar] [CrossRef] [PubMed]
- Jayachandran, A.; Dhungel, B.; Steel, J.C. Epithelial-to-mesenchymal plasticity of cancer stem cells: Therapeutic targets in hepa-tocellular carcinoma. J. Hematol. Oncol. 2016, 9, 74. [Google Scholar] [CrossRef]
- Shrestha, R.; Prithviraj, P.; Bridle, K.; Crawford, D.; Jayachandran, A. Combined Inhibition of TGF-β1-Induced EMT and PD-L1 Silencing Re-Sensitizes Hepatocellular Carcinoma to Sorafenib Treatment. J. Clin. Med. 2021, 10, 1889. [Google Scholar] [CrossRef] [PubMed]
- Bakir, B.; Chiarella, A.M.; Pitarresi, J.R.; Rustgi, A.K. EMT, MET, Plasticity, and Tumor Metastasis. Trends Cell Biol. 2020, 30, 764–776. [Google Scholar] [CrossRef]
- Soundararajan, R.; Fradette, J.J.; Konen, J.M.; Moulder, S.; Zhang, X.; Gibbons, D.L.; Varadarajan, N.; Wistuba, I.I.; Tripathy, D.; Bernatchez, C.; et al. Targeting the Interplay between Epithelial-to-Mesenchymal-Transition and the Immune System for Effective Immunotherapy. Cancers 2019, 11, 714. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Prithviraj, P.; Shrestha, R.; Sharma, R.; Anaka, M.; Bridle, K.; Kannourakis, G.; Crawford, D.; Jayachandran, A. Prognostic Role of Immune Checkpoint Regulators in Cholangiocarcinoma: A Pilot Study. J. Clin. Med. 2021, 10, 2191. [Google Scholar] [CrossRef]
- Woods, K.; Pasam, A.; Jayachandran, A.; Andrews, M.C.; Cebon, J. Effects of Epithelial to Mesenchymal Transition on T Cell Targeting of Melanoma Cells. Front. Oncol. 2014, 4, 367. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Rashidian, M.; Eaton, E.N.; Reinhardt, F.; Thiru, P.; Zagorulya, M.; Nepal, S.; Banaz, T.; Martner, A.; Spranger, S.; et al. Direct and Indirect Regulators of Epithelial-Mesenchymal Transition-Mediated Immunosuppression in Breast Car-cinomas. Cancer Discov. 2021, 11, 1286–1305. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.D.; Sitter, B.; Bathen, T.F.; Bofin, A.; Lønning, P.E.; Lundgren, S.; Gribbestad, I.S. Predicting long-term survival and treatment response in breast cancer patients receiving neoadjuvant chemotherapy by MR metabolic profiling. NMR Biomed. 2011, 25, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Baek, H.-M.; Chen, J.-H.; Nie, K.; Yu, H.J.; Bahri, S.; Mehta, R.S.; Nalcioglu, O.; Su, M.-Y. Predicting Pathologic Response to Neoadjuvant Chemotherapy in Breast Cancer by Using MR Imaging and Quantitative1H MR Spectroscopy. Radiology 2009, 251, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; Whitaker-Menezes, D.; Dasgupta, A.; Philp, N.J.; Lin, Z.; Gandara, R.; Sneddon, S.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Using the “reverse Warburg effect” to identify high-risk breast cancer patients: Stromal MCT4 predicts poor clinical outcome in triple-negative breast cancers. Cell Cycle 2012, 11, 1108–1117. [Google Scholar] [CrossRef]
- Huang, X.; Xie, X.; Wang, H.; Xiao, X.; Yang, L.; Tian, Z.; Guo, X.; Zhang, L.; Tang, H.; Xie, X. PDL1 And LDHA act as ceRNAs in triple negative breast cancer by regulating miR-34a. J. Exp. Clin. Cancer Res. 2017, 36, 129. [Google Scholar] [CrossRef]
- Huang, X.; Li, X.; Xie, X.; Ye, F.; Chen, B.; Song, C.; Tang, H.; Xie, X. High expressions of LDHA and AMPK as prognostic biomarkers for breast cancer. Breast 2016, 30, 39–46. [Google Scholar] [CrossRef]
- McCleland, M.L.; Adler, A.S.; Shang, Y.; Hunsaker, T.; Truong, T.; Peterson, D.; Torres, E.; Li, L.; Haley, B.; Stephan, J.-P.; et al. An Integrated Genomic Screen Identifies LDHB as an Essential Gene for Triple-Negative Breast Cancer. Cancer Res. 2012, 72, 5812–5823. [Google Scholar] [CrossRef]
- Asleh, K.; Negri, G.L.; Miko, S.E.S.; Colborne, S.; Hughes, C.S.; Wang, X.Q.; Gao, D.; Gilks, C.B.; Chia, S.K.L.; Nielsen, T.O.; et al. Proteomic analysis of archival breast cancer clinical specimens identifies biological subtypes with distinct survival outcomes. Nat. Commun. 2022, 13, 896. [Google Scholar] [CrossRef]
- Pinheiro, C.; Albergaria, A.; Paredes, J.; Sousa, B.; Dufloth, R.; Vieira, D.; Schmitt, F.; Baltazar, F. Monocarboxylate transporter 1 is up-regulated in basal-like breast carcinoma. Histopathology 2010, 56, 860–867. [Google Scholar] [CrossRef]
- Davis, S.; Scott, C.; Oetjen, J.; Charles, P.D.; Kessler, B.M.; Ansorge, O.; Fischer, R. Deep topographic proteomics of a human brain tumour. bioRxiv 2022. [Google Scholar] [CrossRef]
- Seydel, C. Single-cell metabolomics hits its stride. Nat. Methods 2021, 18, 1452–1456. [Google Scholar] [CrossRef] [PubMed]
- Mund, A.; Brunner, A.-D.; Mann, M. Unbiased spatial proteomics with single-cell resolution in tissues. Mol. Cell 2022, 82, 2335–2349. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Gene/Protein/Pathway | Status in TNBC | Study Model | Clinicopathological Evaluation | Reference |
|---|---|---|---|---|
| Oxidative phosphorylation | Glycolysis dominates over oxidative phosphorylation | 12 TNBC patients, 12–14-week-old male C57BL/6 mice | Not evaluated | [34] |
| Complex III component absent and Complex V was reduced by 90% in MDA-MB-231 | several breast cancer cell lines | Not evaluated | [44] | |
| Lower levels of Complex I subunit, higher levels of Complex IV in MDA-MB31 | Cancer cell lines MCF-7, MDA-MB-231, T47D, and MDA-MB-435 | Not evaluated | [45] | |
| High OXPHOS signature, with overexpression of Complex I | 43 TNBC biopsy samples | Correlation with relapse and poor survival | [46] | |
| GLUT1 | 65.2% overexpression | 132 TNBC patients | Not evaluated | [30,35] |
| PI3K/Akt pathways | overexpression of Akt and p-p44/42 MAPK proteins in samples bearing PIK3CA mutation; elevated GTPase-activating protein USP6NL | 97 TNBC and 36 non-TNBC tumour samples; TNBC cell lines HCC70 and HCC1187, human mammary epithelial cell line HMEC and non-small cell lung carcinoma cell line H1299 | Not evaluated | [37,38] |
| Hexokinase 2 (HK2) | HK2 suppression improves radiosensitivity | TNBC cell line MDA-MB-231; 6-week-old female BALB/c mice with MDA-MB-231 injections. | Not evaluated | [38] |
| LDHA and PDL-1 | PDL-1 elevated in TNBC cell lines (especially HCC38 and MDA-MB-231 cells) and in 17 of 20 patients. LDHA elevated in TNBC cell lines compared to MCF-10A and in 18 of 20 patients. | human mammary epithelial (HME) cell line MCF-10A and TNBC cell lines MDA-MB-453, MDA-MB-468, MDA-MB-231, BT-549, HCC38 and 4 T1; 20 TNBC tissues and normal adjacent tissues; tissue microarrays (TMAs) of 554 cases of breast cancer tissues | PDL-1 and LDHA are linked with worse OS and DFS | [50] |
| MCT1 | Elevated expression in TNBC as compared with ER+ and/or PR+ and HER-2+ (p < 0.001). | TMAs of 257 cases of breast cancer tissues | Increased MCT1 predictive of recurrence. However, correlation with OS is not significant. | [50] |
| MCT4 and CD147 | Enhanced expression; correlating with poor prognosis and metastases | 249 FFPE breast cancer samples | Not evaluated | [51] |
| Gene/Protein/Pathway | Status in TNBC | Study Model | Clinicopathological Evaluation | Ref. |
|---|---|---|---|---|
| Glutaminase, ASCT2 or SLC1A5, LAT1 or SLC7A5 (all up) | Low glutamine levels in conjunction with high glutamate levels, demonstrating elevated glutaminolysis | TNBC cell lines MDA-MB-231 and SUM-159PT, and basal cell lines HCC38, HCC70, MDA-MB-468 and MDA-MB-157; ER-positive (SKBR3) and luminal cell lines MDA-MB-453 and MCF7 | Not evaluated | [54,55] |
| Glutamine sensitivity | Out of cell lines analysed, 11 TNBC (3 basal, 8 claudin low) identified as ‘glutamine auxotrophs’. | Metabolic profiles of 46 independently-derived breast cell lines | Not evaluated | [56] |
| RTK signalling pathways, mTOR activation | Most frequently activated: EGFR, 75% of patients, HER2 33% of patients, HER4 25% of patients, PDGFRβ; 71% of patients, Akt (88% of patients), Erk1/2; 53% of patients. | 26 patient derived TNBC tumours; HBL100, HS578T and MDA-MB-231 cell lines; TNBC mice | In vivo analysis of BEZ235 combinations with taxotere or carboplatin—delay in tumour growth observed. | [57] |
| Cystine susceptibility | Most essential amino acid for TNBC cell growth—Starvation led to necroptosis and ferroptosis in MDA-MB-231, Hs 578T, and HCC 1937. Positively correlates with CHAC1 expression. | Cell lines: MDA-MB-231, Hs 578T, HCC 1937, MCF-7 | Not evaluated | [59] |
| EMT strongly correlates with cystine dependence. | luminal (BT474, ZR751, MCF7) and basal (MDA-MB-157, BT20, MDA-MB-231) subtypes | Not evaluated | [60] | |
| Serine/glycine metabolising enzymes | High expression of PHGDH and PSPH in TNBC cell lines; elevated PHGDH, PSPH, and SHMT1 in epithelial component of TNBC cancer. | Cell lines: MCF-7, MDA-MB-361, MDA-MB-453, MDA-MB-435S, MDA-MB-231, and MDA-MB-468; tissue samples from 709 patients with invasive ductal carcinoma | Not evaluated | [62] |
| Tryptophan catabolism and AhR signalling | Tryptophan 2,3-dioxygenase (TDO2), kynureninase (KYNU) upregulated in suspension as compared to attached cultures of SUM159PT, MDA-MB-231, BT549. TDO2 inhibition decreased metastasis in vivo. | Cell lines: SUM159PT, MDA-MB-231, BT549, MCF7 and T47D. Tail-vein injection of MDA-MB-231 cells into NOD.CB17-Prkdcscid/J (NOD/SCID) mice. primary TNBC patient samples. Gene expression microarray from Curtis et al. dataset (n = 1998). | Above-median TDO2 expression had about 3 years shorter OS as compared with below-median. expression (p = 0.0002). Above-median IDO1 expression had a shorter OS than with reduced IDO1, but difference was less than TDO2 expression. | [64] |
| Kynurenine, tryptophan 2,3-dioxygenase (TDO) | Induces CD8 T cell death and reduces its viability. | Cell lines: BT549 cells. Pre-surgical breast cancer patient plasma (n = 77) and tumour-free donors (n = 40). | Not evaluated | [65] |
| Gene/Protein/Pathway | Status in TNBC | Study Model | Clinicopathological Evaluation | Ref. |
|---|---|---|---|---|
| FAS and FASN enzyme | Expression of FASN in TNBC cell lines, sensitivity to FASN inhibitors especially in doxorubicin resistant models. | TNBC cell lines MDA-MB-231, MDA-MB-468, MDA-MB-157, HCC1806, Du4475, and BT549, Doxorubicin-resistant cells MDA-MB-231 (231DXR) and HCC1806 (HCCDXR); paraffin-embedded biopsy samples of 29 TNBC patients; orthoxenografts of NRG (NOD-Rag1<null> IL2rg<null>) mice. | Combination treatment with doxorubicin, C75, cetuximab and epigallocatechin gallate (EGCG) showed a strong synergistic effect in chemo-sensitive and chemo-resistant cells. | [69] |
| MYC-dependent FAO dysregulation | FAO upregulated in TNBC mice model that correlates with high MYC expression, FAS is preferntial through ACC1 activity rather than ACC2, treatment with etomoxir in cells with high MYC reduced proliferation but not viability. | MMTV-rtTA/TetO-MYC mice, orthotopic allograft and xenograft in NOD/SCID female mice and WT FVB/N models; The Cancer Genome Atlas (TCGA) breast cancer dataset (771 patients); cell lines—human mammary epithelial cell line HMEC, six TN and three RP cell lines HCC1428 and T47D | Reduced ACC2, a critical FAS gene, indicative of increased FAO that leads to aggressiveness of breast tumours, is linked with worse prognosis and outcome in TNBC cohort. | [70] |
| Lipoprotein lipase (LPL) and CD36 overexpression | TNBC cells Du4475 expressed the highest levels of LPL mRNA and lowest levels of heparanase mRNA. | 45 human breast cancer cell lines (ICBP45) | Not evaluated | [71] |
| CD36 overexpression | Lower dose of genistein combined with CD36 siRNA loaded nanoparticles suppressed proliferation and promoted apoptosis. | Cell lines: MDA-MB-231 | Not evaluated | [72] |
| Cholesterol biosynthesis | Increase in expression of cholesterol biosynthesis associated proteins. | Cell lines MDA-MB-468 and MDA-MB-231; cohort of 615 basal-like breast cancer patients | Cholesterol biosynthesis-associated proteins significantly correlated with high gene expression and shorter relapse-free survival in the basal-like cohort. | [73] |
| Chk-α and PD-L1 | High expression and interdependence of Chk-α and PD-L1 | Cell lines MDA-MB-231, SUM-149; TCGA TARGET GTEx database | Not evaluated | [74] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poddar, A.; Rao, S.R.; Prithviraj, P.; Kannourakis, G.; Jayachandran, A. Crosstalk between Immune Checkpoint Modulators, Metabolic Reprogramming and Cellular Plasticity in Triple-Negative Breast Cancer. Curr. Oncol. 2022, 29, 6847-6863. https://doi.org/10.3390/curroncol29100540
Poddar A, Rao SR, Prithviraj P, Kannourakis G, Jayachandran A. Crosstalk between Immune Checkpoint Modulators, Metabolic Reprogramming and Cellular Plasticity in Triple-Negative Breast Cancer. Current Oncology. 2022; 29(10):6847-6863. https://doi.org/10.3390/curroncol29100540
Chicago/Turabian StylePoddar, Arpita, Sushma R. Rao, Prashanth Prithviraj, George Kannourakis, and Aparna Jayachandran. 2022. "Crosstalk between Immune Checkpoint Modulators, Metabolic Reprogramming and Cellular Plasticity in Triple-Negative Breast Cancer" Current Oncology 29, no. 10: 6847-6863. https://doi.org/10.3390/curroncol29100540
APA StylePoddar, A., Rao, S. R., Prithviraj, P., Kannourakis, G., & Jayachandran, A. (2022). Crosstalk between Immune Checkpoint Modulators, Metabolic Reprogramming and Cellular Plasticity in Triple-Negative Breast Cancer. Current Oncology, 29(10), 6847-6863. https://doi.org/10.3390/curroncol29100540