
Azelaic Acid Esters as Pluripotent Immunomodulatory Molecules: Nutritional Supplements or Drugs


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemical Synthesis
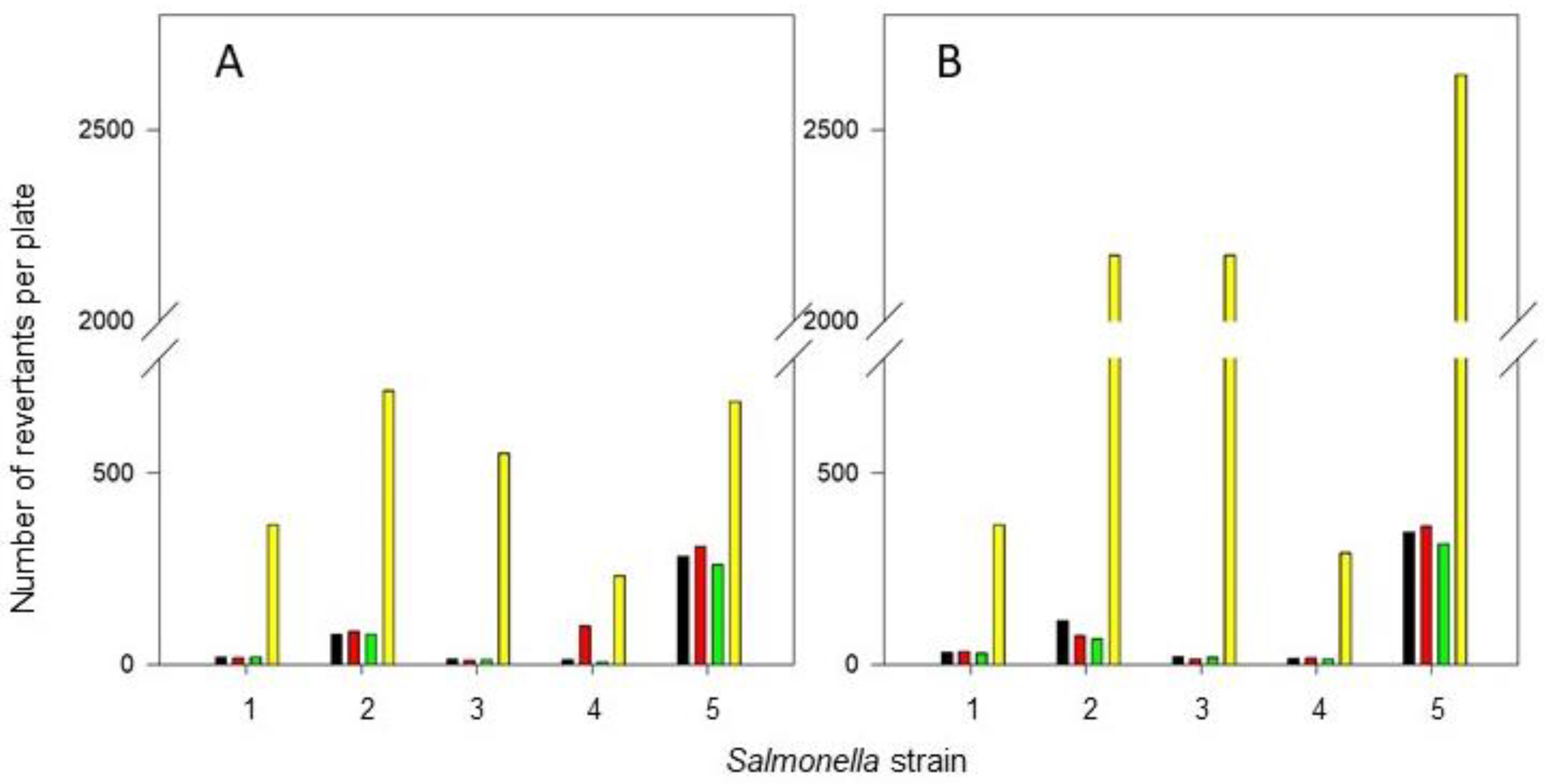
2.2. Bacterial Reverse Mutation Assay
2.3. Quantitative Liquid Chromatography (LC)-MS Method for the Analysis of DEA
2.4. Metabolism and Metabolic Profile of DEA in Rat, Dog, Monkey and Human Primary Hepatocytes
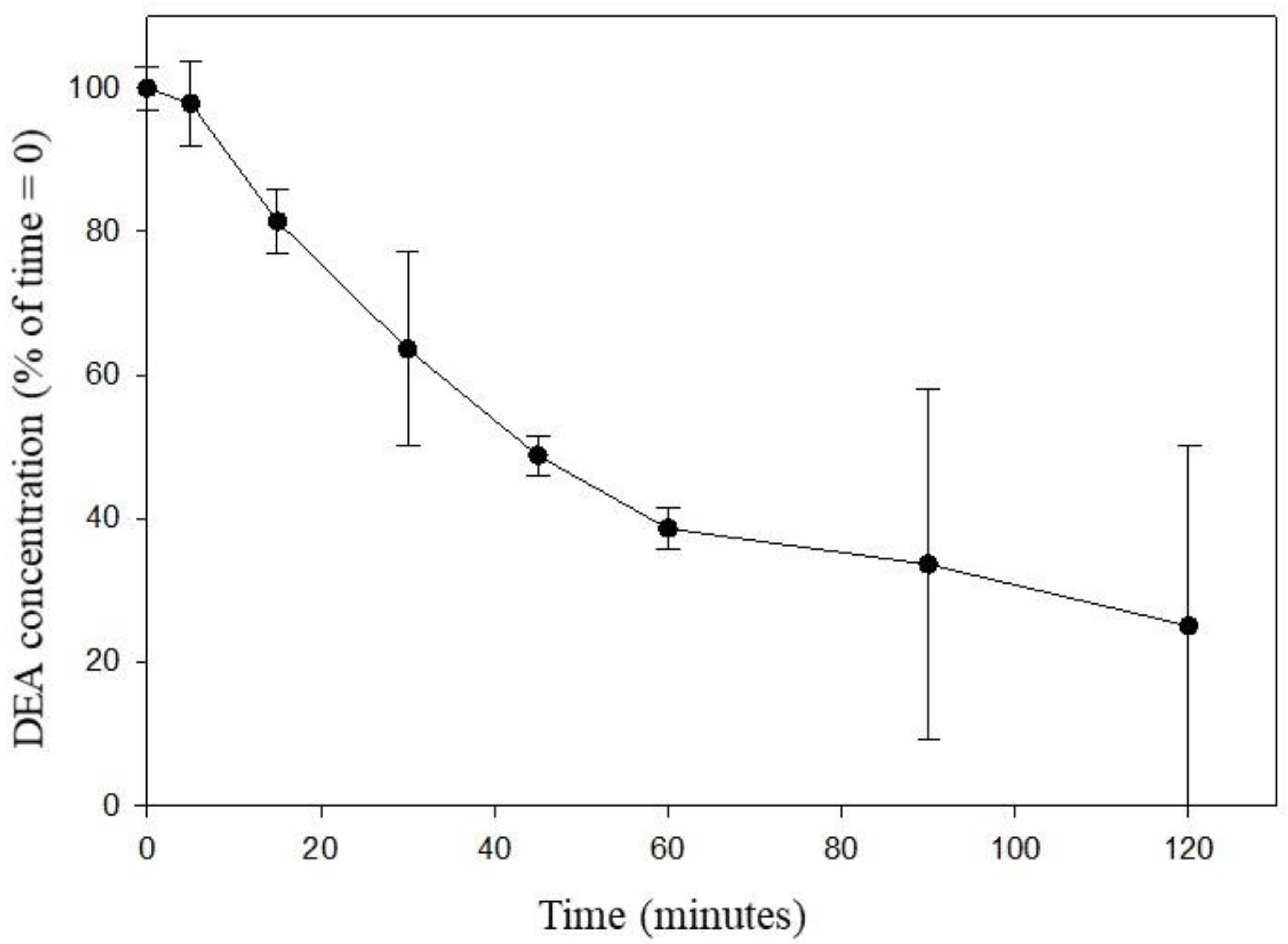
2.5. Metabolic Stability of DEA in Human Saliva
2.6. Animal Study Ethics Statement
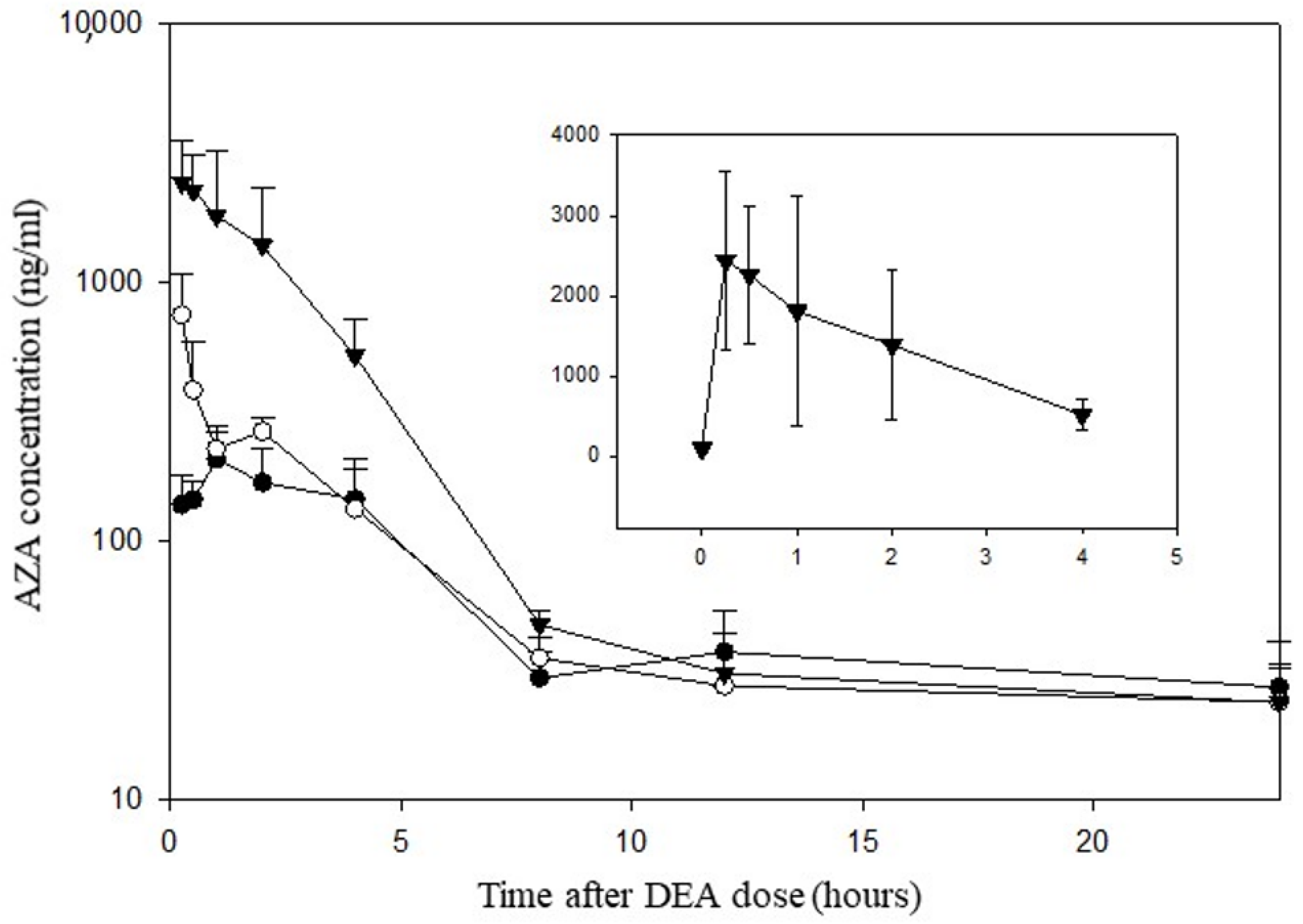
2.7. Determination of the Pharmacokinetics of DEA after an Oral Dose to Rats
2.8. Examination of Physicochemical Properties of DEA for Its Potential as a Drug
3. Results
3.1. Toxicity and Mutagenicity
3.2. Metabolism and Metabolic Profile of DEA In Vitro Using Rat, Dog, Monkey, and Human Hepatocytes
3.3. Determination of the Pharmacokinetics of DEA after an Oral Dose to Rats
3.4. Metabolism of DEA in Human Saliva
3.5. Is DEA a “Druggable” Molecule
4. Discussion
4.1. The Levels of AZA and DEA in Human Body and in the Food
4.2. Oral Safety of AZA and Related Azelates
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Piccinin, E.; Cariello, M.; De Santis, S.; Ducheix, S.; Sabba, C.; Ntambi, J.M.; Moschetta, A. Role of oleic acid in the gut-liver axis: From diet to the regulation of its synthesis via stearoyl-CoA desaturase 1 (SCD1). Nutrients 2019, 11, 2283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everts, S. Vegetative warfare. Chem. Eng. News 2011, 89, 53–55. [Google Scholar] [CrossRef]
- Jung, H.W.; Tschaplinski, T.J.; Wang, L.; Glazebrook, J.; Greenberg, J.T. Priming in systemic plant immunity. Science 2009, 324, 89–91. [Google Scholar] [CrossRef] [PubMed]
- Mastrofrancesco, A.; Ottaviani, M.; Aspite, N.; Cardinali, G.; Izzo, E.; Graupe, K.; Zouboulis, C.C.; Camera, E.; Picardo, M. Azelaic acid modulates the inflammatory response in normal human keratinocytes through PPAR gamma activation. Exp. Dermatol. 2010, 19, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Dorschner, R.A.; Williams, M.R.; Gallo, R.L. Rosacea, the face of innate immunity. Br. J. Dermatol. 2014, 171, 1282–1284. [Google Scholar] [CrossRef] [Green Version]
- Izbicka, E.; Streeper, R.; Louden, C. Adaptive membrane fluidity modulation: A feedback regulated homeostatic system and target for pharmacological intervention. In Vivo 2021, 35, 3073–3095. [Google Scholar] [CrossRef] [PubMed]
- Izbicka, E.; Streeper, R. Adaptive membrane fluidity modulation: A feedback regulated homeostatic system hiding in plain sight. In Vivo 2021, 35, 2991–3000. [Google Scholar] [CrossRef] [PubMed]
- Saerens, S.M.; Delvaux, F.; Verstrepen, K.J.; Van Dijck, P.; Thevelein, J.M.; Delvaux, F.R. Parameters affecting ethyl ester produc-tion by Saccharomyces cerevisiae during fermentation. Appl. Environ. Microbiol. 2008, 74, 454–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostelenos, G.; Kiritsakis, A. Olive Tree History and Evolution. In Olives and Olive Oil as Functional Foods; Kiritsakis, A., Shahidi, F., Eds.; John Wiley & Sons Ltd.: Oxford, UK, 2017; pp. 1–12. [Google Scholar]
- Rahmani, M. Food hazards and quality control in table olive processing with a special reference to functional compounds. In Olives and Olive Oil as Functional Foods; Kiritsakis, A., Shahidi, F., Eds.; John Wiley & Sons Ltd.: Oxford, UK, 2017; pp. 347–352. [Google Scholar]
- Yamabe, S.; Kaneko, K.; Inoue, H.; Takita, T. Maturation of fermented rice-koji miso can be monitored by an increase in fatty acid ethyl ester. Biosci. Biotechnol. Biochem. 2004, 68, 250–252. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Chung, H. Components in commercial douchi a Chinese fermented black bean product by supercritical fluid ex-traction. J. Food Sci. Nutr. 2008, 13, 12–17. [Google Scholar]
- Yu, J. Determination of Binary Acids Diethyl Esters in Fermented Soya Beans-Flavour Liquor by Gas Chromatography. 2001. Available online: http://en.cnki.com.cn/Article_en/CJFDTOTAL-NJKJ200106033.htm (accessed on 10 May 2019).
- Du, J.; Li, Y.; Xu, J.; Huang, M.; Wang, J.; Chao, J.; Wu, J.; Sun, H.; Ding, H.; Ye, H. Characterization of key odorants in langyatai baijiu with jian flavour by sensory-directed analysis. Food Chem. 2021, 352, 129363. [Google Scholar] [CrossRef]
- Fan, H.; Fan, W. Characterization of key odorants in Chinese chixiang aroma-type liquor by gas chromatography-olfactometry, quantitative measurements, aroma recombination, and omission studies. J. Agric. Food Chem. 2015, 63, 3660–3668. [Google Scholar] [CrossRef] [PubMed]
- Nazzaro-Porro, M. Azelaic acid. J. Am. Acad. Dermatol. 1987, 17, 1033–1041. [Google Scholar] [CrossRef]
- Nazzaro-Porro, M.; Passi, S.; Zina, G.; Bernengo, A.; Breathnach, A.; Gallagher, S. Effect of azelaic acid on human malignant melanoma. Lancet 1980, 1, 1109–1111. [Google Scholar] [CrossRef]
- Breathnach, A.; Levi-Montalcini, R. The story of azelaic acid. A tribute to Marcella Nazzaro-Porro. Rend. Fis. Acc. Lincei 1995, 6, 313–320. [Google Scholar] [CrossRef]
- Searle, T.; Ali, F.R.; Al-Niaimi, F. The versatility of azelaic acid in dermatology. J. Dermatol. Treat. 2020. [CrossRef] [PubMed]
- Wang, Z.; Xiang, H.; Dong, P.; Zhang, T.; Lu, C.; Jin, T. Pegylated azelaic acid: Synthesis, tyrosinase inhibitory activity, antibacterial activity and cytotoxic studies. J. Mol. Struct. 2021, 1234, 129234. [Google Scholar] [CrossRef]
- Streeper, R.T.; Izbicka, E. Azelaic Acid Esters in the Treatment of Insulin Resistance. U.S. Patent US 10,251,857 B2, 9 April 2019. [Google Scholar]
- Streeper, R.T.; Izbicka, E. Azelaic Acid Esters in the Treatment of Insulin Resistance. U.S. Patent US 11,026,912 B2, 8 June 2021. [Google Scholar]
- Streeper, R.T.; Izbicka, E. Azelaic Acid Esters in the Treatment of Dyslipidemia and Associated Conditions. U.S. Patent Application US/2021/0251939 A1, 19 August 2021. [Google Scholar]
- Streeper, R.T.; Louden, C.; Izbicka, E. Oral azelaic acid ester decreases markers of insulin resistance in overweight human male subjects. In Vivo 2020, 34, 1173–1186. [Google Scholar] [CrossRef]
- Maron, D.M.; Ames, B.N. Revised methods for the Salmonella mutagenicity test. Mutat. Res. 1983, 113, 173–215. [Google Scholar] [CrossRef]
- Bayliss, M.K.; Bell, J.A.; Jenner, W.N.; Park, G.R.; Wilson, K. Utility of hepatocytes to model species differences in the metabo-lism of loxtidine and to predict pharmacokinetic parameters in rat, dog and man. Xenobiotica 1999, 29, 253–268. [Google Scholar] [CrossRef]
- Tee, L.B.; Seddon, T.; Boobis, A.R.; Davies, D.S. Drug metabolising activity of freshly isolated human hepatocytes. Br. J. Clin. Pharmacol. 1985, 19, 279–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esperon-Rojas, A.A.; Torres-Palacios, C.; Santos-Luna, D.; Baeza-Jimenez, R.; Cano-Sarmiento, C.; Garcia, H.S. A specific thin layer chromatography method for the identification and separation of medium chain acylglycerols. J. Oleo Sci. 2018, 67, 1397–1403. [Google Scholar] [CrossRef] [PubMed]
- Moriguchi, I.; Hirono, S.; Liu, Q.; Nakagome, I.; Matsushita, Y. Simple method of calculating octanol/water partition coefficient. Chem. Pharm. Bull. 1992, 40, 127–130. [Google Scholar] [CrossRef] [Green Version]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Escobar, P.A.; Kemper, R.A.; Tarca, J.; Nicolette, J.; Kenyon, M.; Glowienke, S. Bacterial mutagenicity screening in the pharmaceutical industry. Mutat. Res. 2013, 752, 99–118. [Google Scholar] [CrossRef]
- Dash, R.; Veeravalli, V.; Thomas, J.; Rosenfeld, C.; Mehta, N.; Srinivas, N. Whole blood or plasma: What is the ideal matrix for pharmacokinetic-driven drug candidate selection? Future Med. Chem. 2020, 13, 157–171. [Google Scholar] [CrossRef]
- Lindqvist, L.; Augustinsson, K. Esterases in human saliva. Enzyme 1975, 20, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Perez-Jimenez, M.; Munoz-Gonzales, C.; Pozo-Bayon, M.A. Understanding human salivary esterases activity and its varia-tion under wine consumption conditions. R. Soc. Chem. 2020, 10, 24352–24361. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioa-vailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Australian Government Department of Health. Nonanedioic acid (Azelaic Acid). Evaluation Statement. 2021. Available online: https://www.industrialchemicals.gov.au/sites/default/files/2021-09/EVA00010%20-%20Evaluation%20Statement%20-2014%20September%202021%20%5B702%20KB%5D.pdf (accessed on 20 September 2021).
- Leeming, J.P.; Holland, K.T.; Bojar, R.A. The in vitro antimicrobial effect of azelaic acid. Br. J. Derm. 1986, 115, 551–556. [Google Scholar] [CrossRef]
- Charnock, C.; Brudeli, B.; Klaveness, J. Evaluation of the antibacterial efficacy of diesters of azelaic acid. Eur. J. Pharm. Sci. 2004, 21, 589–596. [Google Scholar] [CrossRef]
- Goncharov, N.V.; Belinskaia, D.A.; Shmurak, V.I.; Terpilowski, M.A.; Jenkins, R.O.; Avdonin, P.V. Serum albumin binding and esterase activity: Mechanistic interactions with organophosphates. Molecules 2017, 22, 1201. [Google Scholar] [CrossRef] [Green Version]
- Muthulakshmi, S.; Saravanan, R. Efficacy of azelaic acid on hepatic key enzymes of carbohydrate metabolism in high fat diet induced type 2 diabetic mice. Biochimie 2013, 95, 1239–1244. [Google Scholar] [CrossRef]
- Thach, T.T.; Hong, Y.J.; Lee, S.; Lee, S.J. Molecular determinants of the olfactory receptor Olfr544 activation by azelaic acid. Biochem. Biophys. Res. Commun. 2017, 485, 241–248. [Google Scholar] [CrossRef]
- Thach, T.T.; Wu, C.; Hwang, K.Y.; Lee, S.J. Azelaic Acid Induces Mitochondrial Biogenesis in Skeletal Muscle by Activation of Olfactory Receptor 544. Front. Physiol. 2020, 11, 329. [Google Scholar] [CrossRef] [Green Version]
- Munoz-Gonzalez, C.; Perez-Jimenez, M.; Pozo-Bayon, M.A. Oral persistence of esters is affected by wine matrix composition. Food Res. Int. 2020, 135, 109286. [Google Scholar] [CrossRef]
- Perez-Jimenez, M.; Esteban-Fernandez, A.; Munoz-Gonzalez, C.; Pozo-Bayon, M.A. Interactions among odorants, phenolic compounds, and oral components and their effects on wine aroma volatility. Molecules 2020, 25, 1701. [Google Scholar] [CrossRef] [Green Version]
- Passi, S.P.M.; De Luca, C.; Nazzaro-Porro, M. Mechanism of azelaic acid action in acne. G. Ital. Derm. Venereol. 1989, 124, 455–463. [Google Scholar]
- Bertuzzi, A.; Gandolfi, A.; Salinari, S.; Mingrone, G.; Arcieri-Mastromattei, E.; Finotti, E.; Greco, A.V. Pharmacokinetic analysis of azelaic acid disodium salt. A proposed substrate for total parenteral nutrition. Clin. Pharm. 1991, 20, 411–419. [Google Scholar] [CrossRef]
- Fiume, M.M.; Eldreth, H.B.; Bergfeld, W.F.; Belsito, D.V.; Hill, R.A.; Klaassen, C.D.; Liebler, D.; Marks, J.G.; Shank, R.C.; Slaga, T.J.; et al. Final Report of the Cosmetic Ingredient Review Expert Panel on the Safety Assessment of Dicarboxylic Acids, Salts, and Esters. Int. J. Toxicol. 2012, 31 (Suppl. SI), 5S–76S. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T.; Tanaka, N.; Krausz, K.W.; Manna, S.K.; Kang, D.W.; Anderson, E.R. Metabolomics identifies an inflammatory cascade involved in dioxin- and diet-induced steatohepatitis. Cell Metab. 2012, 16, 634–644. [Google Scholar] [CrossRef] [Green Version]
- Grego, A.V.; Mingrone, G. Dicarboxylic acids, an alternate fuel substrate in parenteral nutrition: An update. Clin. Nutr. 1995, 14, 143–148. [Google Scholar] [CrossRef]
- Passi, S.; Picardo, M.; Mingrone, G.; Breathnach, A.S.; Nazzaro-Porro, M. Azelaic acid-biochemistry and metabolism. Acta Derm. Venereol. 1989, 143, 8–13. [Google Scholar] [CrossRef]
- Alhomsi, K.; Cluette-Brown, J.E.; Laposata, M. Fatty acid ethyl esters in human mononuclear cells: Production by endogenous synthesis greatly exceeds the uptake of preformed ethyl esters. Alcohol. Clin. Exp. Res. 2006, 30, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, G.F.; Meier-Augenstein, W.; Stockler, S.; Surtees, R.; Rating, D.; Nyhan, W.L. Physiology and pathophysiology of organic acids in cerebrospinal fluid. J. Inherit. Metab. Dis. 1993, 16, 648–669. [Google Scholar] [CrossRef]
- Sugimoto, M.; Saruta, J.; Matsuki, C.; To, M.; Onuma, H.; Kaneko, M.; Soga, T.; Tomita, M.; Tsukinoki, K. Physiological and environmental parameters associated with mass spectrometry-based salivary metabolomic profiles. Metabolomics 2013, 9, 454–463. [Google Scholar] [CrossRef]
- Smilowitz, J.T.; O’Sullivan, A.; Barile, D.; German, J.B.; Lonnerdal, B.; Slupsky, C.M. The human milk metabolome reveals diverse oligosaccharide profiles. J. Nutr. 2013, 143, 1709–1718. [Google Scholar] [CrossRef] [Green Version]
- Gunderson, E.P.; Hurston, S.R.; Ning, X.; Lo, J.C.; Crites, Y.; Walton, D.; Dewey, K.G.; Azevedo, R.A.; Young, S.; Fox, G.; et al. Study of Women IF and Type 2 Diabetes After GDMPI: Lactation and progression to type 2 diabetes mellitus after gestational diabetes mellitus: A prospective cohort study. Ann. Intern. Med. 2015, 163, 889–898. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Lai, M.; Piro, A.L.; Alexeeff, S.E.; Allalou, A.; Rost, H.L.; Dai, F.F.; Wheeler, M.B.; Gunderson, E.P. Intensive lactation among women with recent gestational diabetes significantly alters the early postpartum circulating lipid profile: The swift study. BMC Med. 2021, 19, 241. [Google Scholar] [CrossRef]
- Abdelmagid, S.A.; Clarke, S.E.; Nielsen, D.E.; Badawi, A.; El-Sohemy, A.; Mutch, D.M.; Ma, D.W. Comprehensive profiling of plasma fatty acid concentrations in young healthy Canadian adults. PLoS ONE 2015, 10, e0116195. [Google Scholar] [CrossRef] [Green Version]
- Bourre, J.M.; Dumont, O.L.; Clement, M.E.; Durand, G.A. Endogenous synthesis cannot compensate for absence of dietary oleic acid in rats. J. Nutr. 1997, 127, 488–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourre, J.M.; Dumont, O.; Durand, G. Dose-effect of dietary oleic acid: Oleic acid is conditionally essential for some organs. Reprod. Nutr. Dev. 2004, 44, 371–380. [Google Scholar] [CrossRef] [Green Version]
- Catlin, N.R.; Collins, B.J.; Auerbach, S.S.; Ferguson, S.S.; Harnly, J.M.; Gennings, C.; Waidyanatha, S.; Rice, G.E.; Smith-Roe, S.L.; Witt, K.L.; et al. How similar is similar enough? A sufficient similarity case study with ginkgo biloba extract. Food Chem. Toxicol. 2018, 118, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Center for Drug Evaluation and Research. Application Number 21-470; Finacea Approval. Clinical Pharmacology and Bio-Pharmaceutics Review(s). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2002/21-470_Finacea_Approv.pdf (accessed on 8 July 2021).
- Palagano, R.; Valli, E.; Tura, M.; Cevoli, C.; Perez-Camino, M.D.C.; Moreda, W.; Bendini, A.; Toschi, T. Fatty acid ethyl esters in virgin olive oils: In-house validation of a revised method. Foods 2020, 9, 924. [Google Scholar] [CrossRef]
- Regulation European Commission. Opinion of the scientific panel on food additives, flavourings, processing aids and mate-rials in contact with food (AFC) on a request from the commission related to flavouring group evaluation 10: Aliphatic pri-mary and secondary saturated and unsaturated alcohols, aldehydes, acetals, carboxylic acids and esters containing an addi-tional oxygenated functional group and lactones from chemical groups 9, 13 and 30 (commission regulation (EC) no 1565/2000 of 18 July 2000). EFSA J. 2005, 246, 1–110. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Izbicka, E.; Streeper, R.T. Azelaic Acid Esters as Pluripotent Immunomodulatory Molecules: Nutritional Supplements or Drugs. Nutraceuticals 2021, 1, 42-53. https://doi.org/10.3390/nutraceuticals1010006
Izbicka E, Streeper RT. Azelaic Acid Esters as Pluripotent Immunomodulatory Molecules: Nutritional Supplements or Drugs. Nutraceuticals. 2021; 1(1):42-53. https://doi.org/10.3390/nutraceuticals1010006
Chicago/Turabian StyleIzbicka, Elzbieta, and Robert T. Streeper. 2021. "Azelaic Acid Esters as Pluripotent Immunomodulatory Molecules: Nutritional Supplements or Drugs" Nutraceuticals 1, no. 1: 42-53. https://doi.org/10.3390/nutraceuticals1010006
APA StyleIzbicka, E., & Streeper, R. T. (2021). Azelaic Acid Esters as Pluripotent Immunomodulatory Molecules: Nutritional Supplements or Drugs. Nutraceuticals, 1(1), 42-53. https://doi.org/10.3390/nutraceuticals1010006