
Synthesis and Bioactivities of Marine Pyran-Isoindolone Derivatives as Potential Antithrombotic Agents

 ,
,  ,
,
Abstract
1. Introduction
2. Results and Discussion
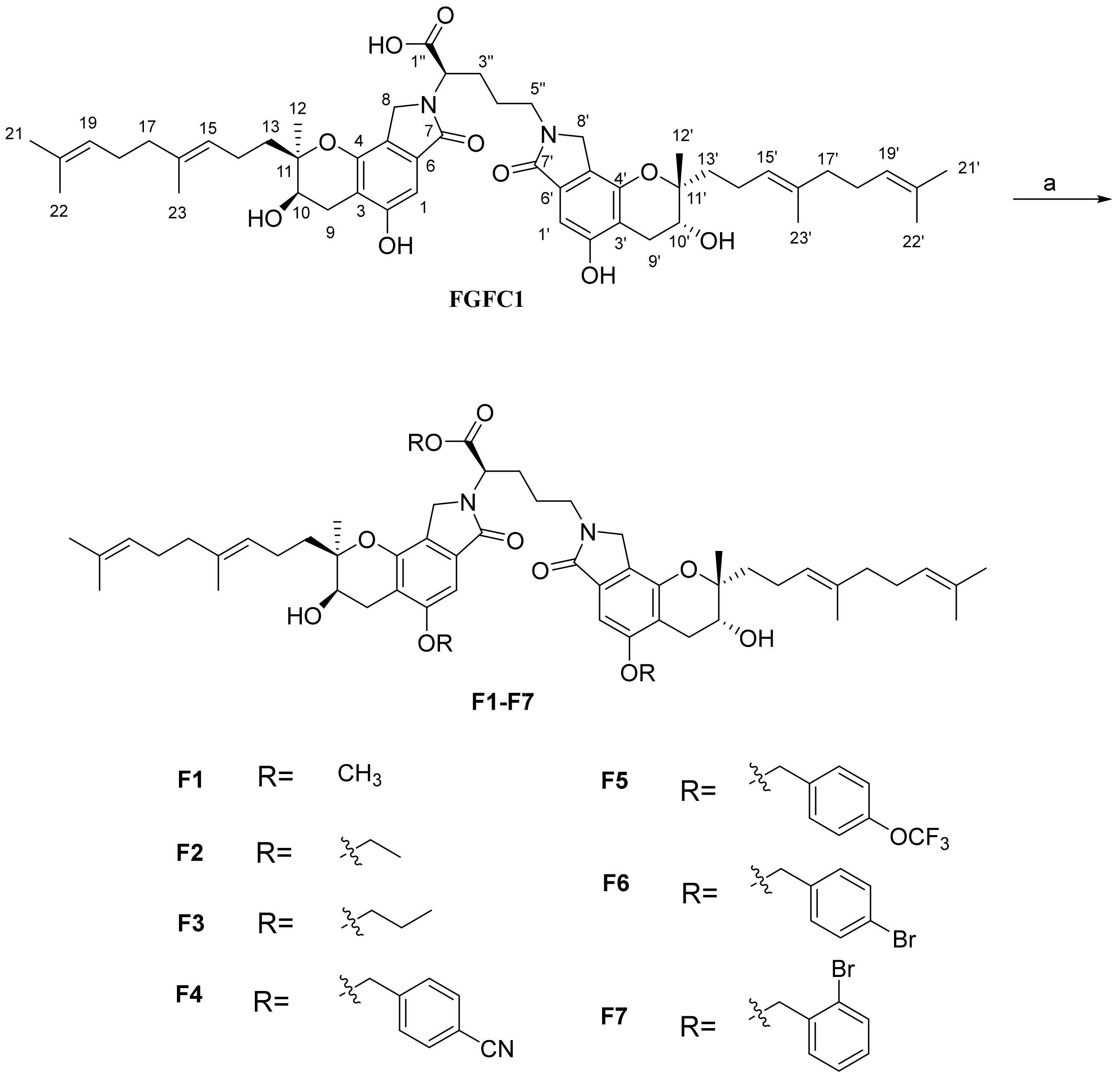
2.1. Chemistry
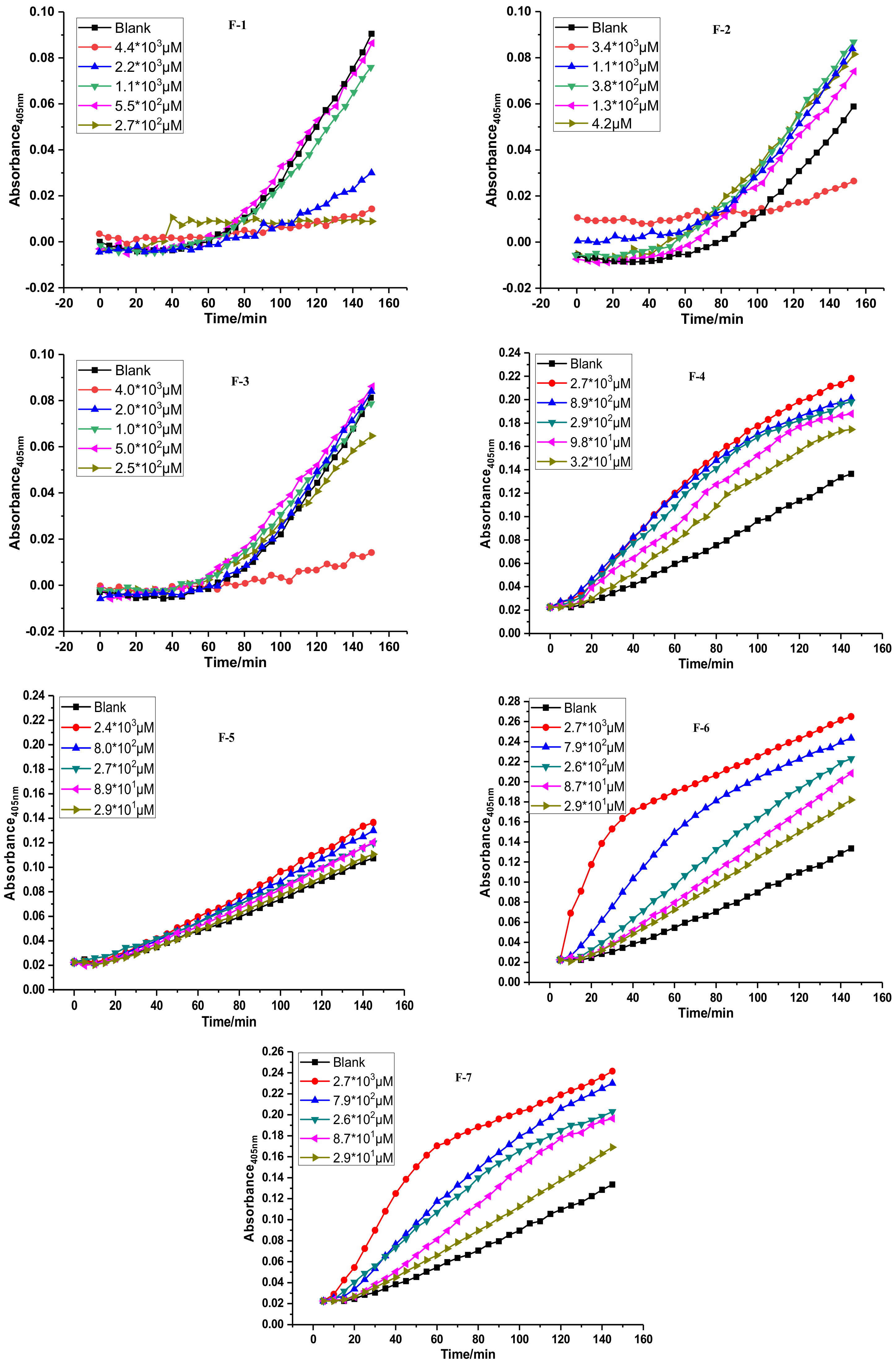
2.2. Fibrinolytic Activities of FGFC1 and F1–F7 In Vitro
2.3. Cytotoxicity, Expression of Fas/Apo-1 and IL-1 of F6 on HeLa Cells
3. Experiment
3.1. Materials
3.2. Chemistry
3.3. Fibrinolytic Activity in Vitro
3.4. Cytotoxicity, Expression of Fas/Apo-1 and IL-1 of Derivative F6 on HeLa Cells
3.4.1. Cell Lines
3.4.2. MTT Assay
3.4.3. Fas/Apo-1 Assay on HeLa Cells
3.4.4. IL-1 Assay on HeLa Cells
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yan, Y.K.; Mi, J. Noncommunicable chronic disease prevention should start from childhood. Pediatric. Investig. 2021, 5, 3–5. [Google Scholar] [CrossRef] [PubMed]
- The World Health Oragnization. Disease Burden and Mortality Estimates; The World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- Alagarsamy, K.N.; Mathan, S.; Yan, W.; Rafieerad, A.; Sekaran, S.; Manego, H.; Dhingra, S. Carbon nanomaterials for cardiovascular theranostics: Promises and challenges. Bioact. Mater. 2021, 6, 2261–2280. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.L.; Tian, Y.B.; Lv, X.; Xiao, X.; Zhan, M.M.; Cheng, K.; Li, S.Y.; Liao, C.Z. The selectivity and bioavailability improvement of novel oral anticoagulants: An overview. Eur. J. Med. Chem. 2018, 146, 299–317. [Google Scholar] [CrossRef] [PubMed]
- Zenych, A.; Fournier, L.; Chauvierre, C. Nanomedicine progress in thrombolytic therapy. Biomaterials 2020, 258, 120297. [Google Scholar] [CrossRef]
- Beaglehole, R.; Bonita, R. Global public health: A scorecard. Lancet 2008, 372, 1988–1996. [Google Scholar] [CrossRef]
- Mathers, C.D.; Dejan, L. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006, 3, 442. [Google Scholar] [CrossRef]
- Pranav, S.; Bindra, V.K. Newer antithrombotic drugs. Indian J. Crit. Care Med. 2010, 14, 188–195. [Google Scholar]
- Gemmill, J.D.; Hogg, K.J.; Dunn, F.G. Pre-dosing antibody levels and efficacy of thrombolytic drugs containing streptokinase. Heart 1994, 72, 222–225. [Google Scholar] [CrossRef]
- Verstraete, M. Thrombolytic drugs in non-coronary disorders. Drugs 1989, 38, 801–821. [Google Scholar] [CrossRef]
- Anderson, J.L.; Karagounis, L.A.; Califf, R.M. Meta-analysis of five reported studies on the relation of early coronary patency grades with mortality and outcomes after acute myocardial infarction. Am. J. Cardiol. 1996, 78, 1–8. [Google Scholar] [CrossRef]
- Peter, S.; Frans, V.W. Drug variables in thrombolytic therapy. J. Vasc. Interv. Radiol. 1995, 6, 62–65. [Google Scholar]
- Verstraete, M. Third-generation thrombolytic drugs. Am. J. Med. 2000, 109, 52–58. [Google Scholar] [CrossRef]
- Marc, V.; Désiré, C. Pharmacology of thrombolytic drugs. J. Am. Coll. Cardiol. 1986, 8, 33–40. [Google Scholar]
- Felicita, A.; Biasucci, L.M.; Silvano, B.; Giovanna, C.; Antonio, G.R. A randomized study to investigate the hemostatic effect of tenecteplase (TNK-t-PA) versus alteplase (rt-PA) and streptokinase (SK) in acute myocardial infarction. J. Am. Coll. Cardiol. 2002, 39, 279. [Google Scholar]
- Thomas, O.M.; Paul, D.; Jack, C.; Barbara, Q.; Kenric, A. Bleeding complications associated with the use of rt-PA versus r-PA for peripheral arterial and venous thromboembolic occlusions. Tech. Vasc. Interv. Radiol. 2001, 4, 92–98. [Google Scholar]
- Adnan, I.; Qureshi, M.D.; Amir, M.; Siddiqui, M.D.; Fareed, K.; Suri, M.D.; Stanley, H.; Kim, M.D.; Zulfiqar, A.M.D.; Abutaher, M.; et al. Aggressive mechanical clot disruption and low-dose Intra-arterial third-generation thrombolytic agent for ischemic stroke: A prospective study. Neurosurgery 2002, 51, 1319–1329. [Google Scholar]
- Chan, N.C.; Eikelboom, J.W.; Weitz, J.I. Evolving treatments for arterial and venous thrombosis: Role of the direct oral anticoagulants. Circ. Res. 2016, 118, 1409–1424. [Google Scholar] [CrossRef]
- Johannes, R.; Katus, H.A. New antithrombotic drugs on the horizon. Expert Opin. Investig. Drugs 2003, 12, 781–797. [Google Scholar]
- Manckoundia, P.; Rosay, C.; Menu, D.; Nuss, V.; Mihai, A.M.; Vovelle, J.; Nuemi, G.; Athis, P.; Putot, A.; Barben, J. The prescription of vitamin K antagonists in a very old population: A cross-sectional study of 8696 a mbulatory subjects aged over 85 years. Int. J. Environ. Res. Public Health 2020, 17, 6685. [Google Scholar] [CrossRef]
- Josefina, C.H. Rivaroxaban versus vitamin K antagonist in antiphospholipid syndrome. Ann. Intern. Med. 2020, 73, 505–506. [Google Scholar]
- Kapur, N.K.; Shenoy, C.; Yunis, A.A.; Mohammad, N.N.; Wilson, S.; Paruchuri, V.; Mackey, E.E.; Qiao, X.; Shah, A.; Esposito, M.L.; et al. Distinct effects of unfractionated heparin versus bivalirudin on circulating angiogenic peptides. PLoS ONE 2012, 7, e34344. [Google Scholar] [CrossRef] [PubMed]
- Rupprecht, H.J.; Blank, R. Clinical pharmacology of direct and indirect factor Xa inhibitors. Drugs 2010, 70, 2153–2170. [Google Scholar] [CrossRef]
- Cragg, G.M.; Newman, D.J.; Snader, K.M. Natural Products in Drug Discovery and Development. J. Nat. Prod. 1997, 60, 52–60. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M.; Snader, K.M. Natural Products as Sources of New Drugs over the Period, 1981–2002. J. Nat. Prod. 2003, 66, 1022–1037. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M.; Snader, K.M. Natural Products as Sources of New Drugs over the Last 25 Years. J. Nat. Prod. 2007, 70, 461–477. [Google Scholar] [CrossRef]
- Wang, J.W.; Li, X.Y.; Zhang, C.Y. Recent Advances on Bioactivity of Seaweed Polysaccharides. Med. Res. 2019, 3, 200003–200007. [Google Scholar] [CrossRef]
- Li, B.L.; Wu, W.H. Review on Adaptation between Biomaterials Function of Chitosan and Its Structure. Med. Res. 2019, 3, 190013–190017. [Google Scholar] [CrossRef]
- Wang, L.L.; Xie, J.Y.; Wu, W.H.; Li, B.L.; Ou, J. Excellent Microbial Cultivation for Astaxanthin Production and Its Extraction by Rhodotorula benthica. Med. Res. 2018, 2, 180015–180018. [Google Scholar]
- Alves, K.M.A.; Cardoso, F.J.B.; Honorio, K.M.; Molfetta, F.A. Design of inhibitors for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) enzyme of leishmania Mexicana. Med. Chem. 2020, 16, 784–795. [Google Scholar] [CrossRef]
- De Sousa Luis, J.A.; da Silva Costa, N.A.; Luis, C.C.S.; Lira, B.F.; Athayde-Filho, P.F.; de Souza Lima, T.K.; da Camara Rocha, J.; Scotti, L.; Scotti, M.T. Synthesis of new cyclic lmides derived from safrole, structure- and ligand-based approaches to evaluate potential new multitarget agents against species of leishmania. Med. Chem. 2020, 16, 39–51. [Google Scholar] [CrossRef]
- Wang, G.; Wu, W.H.; Zhu, Q.G.; Fu, S.Q.; Wang, X.Y.; Hong, S.T.; Guo, R.H.; Bao, B. Identification and fibrinolytic evaluation of an isoindolone derivative isolated from a rare marine fungus Stachybotrys longispora FG216. Chin. J. Chem. 2015, 33, 1089–1095. [Google Scholar] [CrossRef]
- Yan, T.; Wu, W.H.; Su, T.W.; Chen, J.J.; Zhu, Q.G.; Zhang, C.Y.; Wang, X.Y.; Bao, B. Effects of a novel marine natural product: Pyrano indolone alkaloid fibrinolytic compound on thrombolysis and hemorrhagic activities in vitro and in vivo. Arch. Pharm. Res. 2014, 38, 1530–1540. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.H.; Duan, D.; Hong, S.T.; Zhou, Y.; Wang, F.; Wang, S.J.; Wu, W.H.; Bao, B. A marine fibrinolytic compound FGFC1 stimulating enzymatic kinetic parameters of a reciprocal activation system based on a single chain urokinase-type plasminogen activator and plasminogen. Process Biochem. 2018, 68, 190–196. [Google Scholar] [CrossRef]
- Wang, M.X.; He, H.; Na, K.; Cai, M.H.; Zhou, X.S.; Zhao, W.J.; Zhang, Y.X. Designing novel glucose/ornithine replenishment strategies by biosynthetic and bioprocess analysis to improve fibrinolytic FGFC1 production by the marine fungus Stachybotrys longispora. Process Biochem. 2015, 50, 2012–2018. [Google Scholar] [CrossRef]
- Su, T.W.; Wu, W.H.; Yan, T.; Zhang, C.Y.; Zhu, Q.G.; Bao, B. Pharmacokinetics and tissue distribution of a novel marine fibrinolytic compound in Wistar rat following intravenous administrations. J. Chromatogr. B 2013, 942, 77–82. [Google Scholar] [CrossRef]
- Ma, Z.B.; Guo, R.H.; Elango, J.; Bao, B.; Wu, W.H. Evaluation of marine diindolinonepyrane in vitro and in vivo: Permeability characterization in Caco-2 cells monolayer and pharmacokinetic properties in Beagle dogs. Mar. Drugs 2019, 17, 651. [Google Scholar] [CrossRef]
- Wang, Y.N.; Wang, J.M.; Fu, Z.; Sheng, R.L.; Wu, W.H.; Fan, J.T.; Guo, R.H. Syntheses and evaluation of daphnetin derivatives as novel G protein-coupled receptor inhibitors and activators. Bioorg. Chem. 2020, 104, 104342–104350. [Google Scholar] [CrossRef]
- Wang, J.M.; Bian, C.H.; Wang, Y.N.; Shen, Q.; Bao, B.; Fan, J.T.; Zuo, A.X.; Wu, W.H.; Guo, R.H. Syntheses and bioactivities of Songorine derivatives as novel G protein-coupled receptor antagonists. Bioorg. Med. Chem. 2019, 27, 1903–1910. [Google Scholar] [CrossRef]
- Florea, A.M.; Büsselberg, D. Cisplatin as an anti-tumor drug: Cellular mechanisms of activity, drug resistance and induced side effects. Cancers 2011, 3, 1351–1371. [Google Scholar] [CrossRef]
- Jane, A.P. Cell sensitivity assays: The MTT assay. Methods Mol. Med. 1999, 80, 165–169. [Google Scholar]
- Manikandan, A.; Arumugam, S. Molecular explorations of substituted 2-(4-phenylquinolin-2-yl) phenols as phosphoinositide 3-kinase inhibitors and anticancer agents. Cancer Chemother. Pharmacol. 2017, 79, 389–397. [Google Scholar]
- Park, I.J.; Kim, M.J.; Ock, J.P.; Myoung, G.P.; Joohun, H. Cryptotanshinone sensitizes DU145 prostate cancer cells to Fas (APO1/CD95)-mediated apoptosis through Bcl-2 and MAPK regulation. Cancer Lett. 2010, 298, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Laurie, B.O.S.; Zhang, W.; James, C.C.; Laura, S.A.; Sybil, M.S.; Toshiyoshi, F.; Jack, A.R.; Albert, B.D.; Zhang, W.W.; Ewa, K.; et al. Wild-type human p53 and a temperature-sensitive mutant induce Fas/APO-1 expression. Mol. Cell. Biol. 1995, 15, 3032–3040. [Google Scholar]
- Mark, P.B.; Tanya, M.G.; Yury, V.G.; David, W. Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor–induced cell death. Cell 1996, 85, 803–815. [Google Scholar]
- Philip, L.S.; Jeffrey, T.L.; John, C.L. A modified assay for interleukin-1 (IL-1). J. Immunol. Methods 1985, 85, 85–94. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
 | ||
| Compounds | R | EC50/μM |
| FGFC1 | H | 115.0 |
| F1 | CH3 | 59.7 |
| F2 |  | 87.1 |
| F3 |  | 66.6 |
| F4 |  | 82.8 |
| F5 |  | 133.3 |
| F6 |  | 42.3 |
| F7 |  | 119.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Chen, H.; Sheng, R.; Fu, Z.; Fan, J.; Wu, W.; Tu, Q.; Guo, R. Synthesis and Bioactivities of Marine Pyran-Isoindolone Derivatives as Potential Antithrombotic Agents. Mar. Drugs 2021, 19, 218. https://doi.org/10.3390/md19040218
Wang Y, Chen H, Sheng R, Fu Z, Fan J, Wu W, Tu Q, Guo R. Synthesis and Bioactivities of Marine Pyran-Isoindolone Derivatives as Potential Antithrombotic Agents. Marine Drugs. 2021; 19(4):218. https://doi.org/10.3390/md19040218
Chicago/Turabian StyleWang, Yinan, Hui Chen, Ruilong Sheng, Zhe Fu, Junting Fan, Wenhui Wu, Qidong Tu, and Ruihua Guo. 2021. "Synthesis and Bioactivities of Marine Pyran-Isoindolone Derivatives as Potential Antithrombotic Agents" Marine Drugs 19, no. 4: 218. https://doi.org/10.3390/md19040218
APA StyleWang, Y., Chen, H., Sheng, R., Fu, Z., Fan, J., Wu, W., Tu, Q., & Guo, R. (2021). Synthesis and Bioactivities of Marine Pyran-Isoindolone Derivatives as Potential Antithrombotic Agents. Marine Drugs, 19(4), 218. https://doi.org/10.3390/md19040218