Targeted Isolation of Rubrolides from the New Zealand Marine Tunicate Synoicum kuranui

 , ,
, , 
Abstract
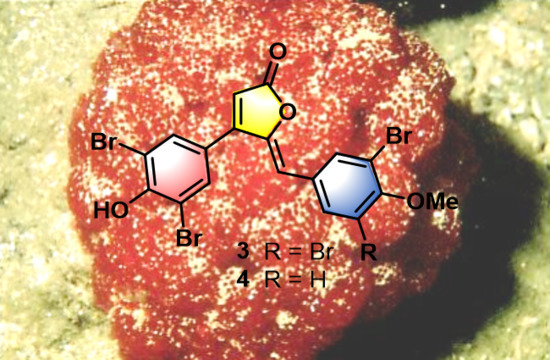
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General Procedures
3.2. Collection of Synoicum kuranui
3.3. Extraction and Isolation
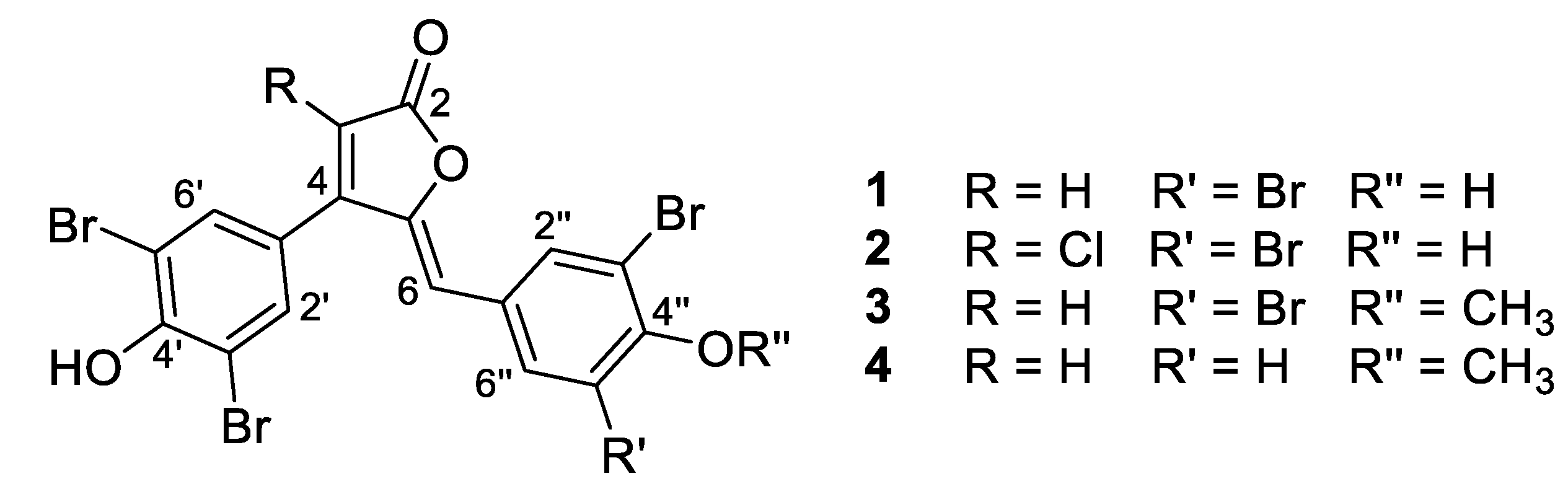
- Rubrolide A (1) yellow film; HRESIMS m/z 590.7075 [M − H]− (calcd. for C17H7Br4O4, 590.7083); all NMR data matches those previously reported [22].
- Rubrolide B (2) yellow film; HRESIMS m/z 624.6694 [M − H]− (calcd. for C17H6Br4ClO4, 624.6694); all NMR data matches those previously reported [22].
- Rubrolide T (3) yellow film; UV (MeOH/H2O) λmax 230, 250, 257 (sh), 340 nm; 13C and 1H NMR data, Table 1 and Table 2 respectively; HRESIMS m/z 604.7254 [M − H]− (calcd. for C18H9Br4O4, 604.7240); HRESIMS/MS (50 eV) m/z (% relative intensity) 589.6992 (5.4), 545.7086 (3.1), 510.7802 (20.8), 482.7853 (14.5), 454.7901 (5.0), 402.8585 (8.3), 287.8473 (18.8), 272.8544 (75), 78.9188 (100).
- E/Z–Rubrolide U (4) yellow film; UV (MeOH/H2O) λmax 232, 254, 361 nm; 13C and 1H NMR data, Table 1 and Table 2 respectively; HRESIMS m/z 526.8143 [M − H]− (calcd. for C18H10Br3O4, 526.8135); HRESIMS/MS (50 eV) m/z (% relative intensity) 511.7898 (3.4), 483.7952 (3.0), 467.8005 (3.3), 432.8721 (12.5), 404.8767 (5.0), 375.8743 (20.0), 295.9485 (22.5), 272.8561 (50), 78.9194 (100).
3.4. LC– MS2 Analysis and Molecular Networking
3.5. Antibacterial Bioassay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotech. 2016, 34, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Fox Ramos, A.E.; Evanno, L.; Poupon, E.; Champy, P.; Beniddir, M.A. Natural products targeting strategies involving molecular networking: different manners, one goal. Nat. Prod. Rep. 2019, 36, 960–980. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yu, H.-B.; Zhang, Y.; Leao, T.; Glukhov, E.; Pierce, M.L.; Zhang, C.; Kim, H.; Mao, H.H.; Fang, F.; et al. Pagoamide A, a Cyclic Depsipeptide Isolated from a Cultured Marine Chlorophyte, Derbesia sp., Using MS/MS-Based Molecular Networking. J. Nat. Prod. 2020, 83, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Bonneau, N.; Chen, G.; Lachkar, D.; Boufridi, A.; Gallard, J.F.; Retailleau, P.; Petek, S.; Debitus, C.; Evanno, L.; Beniddir, M.A.; et al. An Unprecedented Blue Chromophore Found in Nature using a “Chemistry First” and Molecular Networking Approach: Discovery of Dactylocyanines A–H. Chem. Eur. J. 2017, 23, 14454–14461. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Wang, X.; Sims, J.; Wang, B.; Pandey, P.; Welsh, C.L.; Stone, R.P.; Avery, M.A.; Doerksen, R.J.; Ferreira, D.; et al. Computationally Assisted Discovery and Assignment of a Highly Strained and PANC-1 Selective Alkaloid from Alaska’s Deep Ocean. J. Am. Chem. Soc. 2019, 141, 4338–4344. [Google Scholar] [CrossRef] [PubMed]
- Khushi, S.; Salim, A.A.; Elbanna, A.H.; Nahar, L.; Bernhardt, P.V.; Capon, R.J. Dysidealactams and Dysidealactones: Sesquiterpene Glycinyl-Lactams, Imides, and Lactones from a Dysidea sp. Marine Sponge Collected in Southern Australia. J. Nat. Prod. 2020, 83, 1577–1584. [Google Scholar] [CrossRef] [PubMed]
- Antunes, E.M.; Copp, B.R.; Davies-Coleman, M.T.; Samaai, T. Pyrroloiminoquinone and related metabolites from marine sponges. Nat. Prod. Rep. 2005, 22, 62–72. [Google Scholar] [CrossRef]
- Kalinski, J.C.J.; Waterworth, S.C.; Siwe Noundou, X.; Jiwaji, M.; Parker-Nance, S.; Krause, R.W.M.; McPhail, K.L.; Dorrington, R.A. Molecular Networking Reveals Two Distinct Chemotypes in Pyrroloiminoquinone-Producing Tsitsikamma favus Sponges. Mar. Drugs 2019, 17, 60. [Google Scholar] [CrossRef]
- Li, F.; Janussen, D.; Peifer, C.; Pérez-Victoria, I.; Tasdemir, D. Targeted Isolation of Tsitsikammamines from the Antarctic Deep-Sea Sponge Latrunculia biformis by Molecular Networking and Anticancer Activity. Mar. Drugs 2018, 16, 268. [Google Scholar] [CrossRef]
- Bracegirdle, J.; Gordon, D.P.; Harvey, J.E.; Keyzers, R.A. Kinase-Inhibitory Nucleoside Derivatives from the Pacific Bryozoan Nelliella nelliiformis. J. Nat. Prod. 2020, 83, 547–551. [Google Scholar] [CrossRef]
- Taufa, T.; Singh, A.J.; Harland, C.R.; Patel, V.; Jones, B.; Halafihi, T.; Miller, J.H.; Keyzers, R.A.; Northcote, P.T. Zampanolides B–E from the Marine Sponge Cacospongia mycofijiensis: Potent Cytotoxic Macrolides with Microtubule-Stabilizing Activity. J. Nat. Prod. 2018, 81, 2539–2544. [Google Scholar] [CrossRef] [PubMed]
- Taufa, T.; Gordon, R.M.A.; Hashmi, M.A.; Hira, K.; Miller, J.H.; Lein, M.; Fromont, J.; Northcote, P.T.; Keyzers, R.A. Pyrroloquinoline derivatives from a Tongan specimen of the marine sponge Strongylodesma tongaensis. Tetrahedron Lett. 2019, 60, 1825–1829. [Google Scholar] [CrossRef]
- Woolner, V.H.; Gordon, R.M.A.; Miller, J.H.; Lein, M.; Northcote, P.T.; Keyzers, R.A. Halogenated Meroditerpenoids from a South Pacific Collection of the Red Alga Callophycus serratus. J. Nat. Prod. 2018, 81, 2446–2454. [Google Scholar] [CrossRef] [PubMed]
- Bracegirdle, J.; Robertson, L.P.; Hume, P.A.; Page, M.J.; Sharrock, A.V.; Ackerley, D.F.; Carroll, A.R.; Keyzers, R.A. Lamellarin Sulfates from the Pacific Tunicate Didemnum ternerratum. J. Nat. Prod. 2019, 82, 2000–2008. [Google Scholar] [CrossRef]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2020, 37, 175–223. [Google Scholar] [CrossRef]
- World Register of Marine Species 2020. Available online: http://www.marinespecies.org/ (accessed on 26 June 2020).
- Diyabalanage, T.; Amsler, C.D.; McClintock, J.B.; Baker, B.J. Palmerolide A, a Cytotoxic Macrolide from the Antarctic Tunicate Synoicum adareanum. J. Am. Chem. Soc. 2006, 128, 5630–5631. [Google Scholar] [CrossRef]
- Tadesse, M.; Strøm, M.B.; Svenson, J.; Jaspars, M.; Milne, B.F.; Tørfoss, V.; Andersen, J.H.; Stensvåg, K.; Haug, T. Synoxazolidinones A and B: Novel Bioactive Alkaloids from the Ascidian Synoicum pulmonaria. Org. Lett. 2010, 12, 4752–4755. [Google Scholar] [CrossRef]
- Tadesse, M.; Svenson, J.; Jaspars, M.; Strøm, M.B.; Abdelrahman, M.H.; Andersen, J.H.; Hansen, E.; Kristiansen, P.E.; Stensvåg, K.; Haug, T. Synoxazolidinone C; a bicyclic member of the synoxazolidinone family with antibacterial and anticancer activities. Tetrahedron Lett. 2011, 52, 1804–1806. [Google Scholar] [CrossRef]
- Tadesse, M.; Svenson, J.; Sepčić, K.; Trembleau, L.; Engqvist, M.; Andersen, J.H.; Jaspars, M.; Stensvåg, K.; Haug, T. Isolation and Synthesis of Pulmonarins A and B, Acetylcholinesterase Inhibitors from the Colonial Ascidian Synoicum pulmonaria. J. Nat. Prod. 2014, 77, 364–369. [Google Scholar] [CrossRef]
- Shen, G.Q.; Baker, B.J. Biosynthetic studies of eudistomin H in the tunicate Eudistoma olivaceum. Tetrahedron Lett. 1994, 35, 4923–4926. [Google Scholar] [CrossRef]
- Miao, S.; Andersen, R.J. Rubrolides A-H, metabolites of the colonial tunicate Ritterella rubra. J. Org. Chem. 1991, 56, 6275–6280. [Google Scholar] [CrossRef]
- Sikorska, J.; Parker-Nance, S.; Davies-Coleman, M.T.; Vining, O.B.; Sikora, A.E.; McPhail, K.L. Antimicrobial Rubrolides from a South African Species of Synoicum Tunicate. J. Nat. Prod. 2012, 75, 1824–1827. [Google Scholar] [CrossRef] [PubMed]
- Ortega, M.J.; Zubía, E.; Ocaña, J.M.; Naranjo, S.; Salvá, J. New Rubrolides from the Ascidian Synoicum blochmanni. Tetrahedron 2000, 56, 3963–3967. [Google Scholar] [CrossRef]
- Pearce, A.N.; Chia, E.W.; Berridge, M.V.; Maas, E.W.; Page, M.J.; Webb, V.L.; Harper, J.L.; Copp, B.R. E/Z-Rubrolide O, an Anti-inflammatory Halogenated Furanone from the New Zealand Ascidian Synoicum. sp. J. Nat. Prod. 2007, 70, 111–113. [Google Scholar] [CrossRef]
- Smitha, D.; Kumar, M.M.K.; Ramana, H.; Rao, D.V. Rubrolide R: A new furanone metabolite from the ascidian Synoicum of the Indian Ocean. Nat. Prod. Res. 2014, 28, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Kim, H.; Nam, S.J.; Rho, B.J.; Kang, H. Antibacterial Butenolides from the Korean Tunicate Pseudodistoma antinboja. J. Nat. Prod. 2012, 75, 2049–2054. [Google Scholar] [CrossRef]
- Zhu, T.; Chen, Z.; Liu, P.; Wang, Y.; Xin, Z.; Zhu, W. New rubrolides from the marine-derived fungus Aspergillus terreus OUCMDZ-1925. J. Antibiot. 2013, 67, 315. [Google Scholar] [CrossRef] [PubMed]
- Brewin, B.J. Ascidians of New Zealand. Part 5: Ascidians from the East Coast of Great Barrier Island. Trans. R. Soc. N.Z. 1950, 78, 354–362. [Google Scholar]
- Page, M.J.; Kelly, M. Awesome Ascidians—A Guide to the Sea Squirts of New Zealand 2016. Available online: https://niwa.co.nz/coasts-and-oceans/marine-identification-guides-and-fact-sheets/seasquirt-id-guide (accessed on 26 June 2020).
- Marinlit 2019. Available online: http://pubs.rsc.org/marinlit/ (accessed on 26 June 2020).
- Wang, W.; Kim, H.; Patil, R.S.; Giri, A.G.; Won, D.H.; Hahn, D.; Sung, Y.; Lee, J.; Choi, H.; Nam, S.J.; et al. Cadiolides J–M, antibacterial polyphenyl butenolides from the Korean tunicate Pseudodistoma antinboja. Bioorg. Med. Chem. Lett. 2017, 27, 574–577. [Google Scholar] [CrossRef]
- Kiriyama, N.; Nitta, K.; Sakaguchi, Y.; Taguchi, Y.; Yamamoto, Y. Studies on the Metabolic Products of Aspergillus terreus. III. Metabolites of the Strain IFO 8835. (1). Chem. Pharm. Bull. 1977, 25, 2593–2601. [Google Scholar] [CrossRef]
- Nitta, K.; Fujita, N.; Yoshimura, T.; Arai, K.; Yamamoto, Y. Metabolic Products of Aspergillus terreus. IX. Biosynthesis of Butyrolactone Derivatives isolated from Strains IFO 8835 and 4100. Chem. Pharm. Bull. 1983, 31, 1528–1533. [Google Scholar] [CrossRef]
- Miao, S. Novel Secondary Metabolites from Selected Marine Invertebrates. Ph.D. Thesis, University of British Columbia, Vancouver, BC, Canada, 1985. [Google Scholar]
- Bellina, F.; Anselmi, C.; Martina, F.; Rossi, R. Mucochloric Acid: A Useful Synthon for the Selective Synthesis of 4-Aryl-3-chloro-2(5H)-furanones, (Z)-4-Aryl-5-[1-(aryl)methylidene]-3-chloro-2(5H)-furanones and 3,4-Diaryl-2(5H)-furanones. Eur. J. Org. Chem. 2003, 2003, 2290–2302. [Google Scholar] [CrossRef]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef]
- Pierens, G.K.; Carroll, A.R.; Davis, R.A.; Palframan, M.E.; Quinn, R.J. Determination of Analyte Concentration Using the Residual Solvent Resonance in 1H NMR Spectroscopy. J. Nat. Prod. 2008, 71, 810–813. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Wiegand, I.; Hilpert, K.; Hancock, R.E.W. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carbon | 3 | Z-4 | E-4 |
|---|---|---|---|
| 2 | 168.5, C | 168.6, C | 168.1, C |
| 3 | 105.7, CH | 105.0, CH | 110.3, CH |
| 4 | 156.7, C | 156.8, C | 154.7, C |
| 5 | 148.8, C | 146.7, C | 147.6, C |
| 6 | 108.2, CH | 110.0, CH | 113.8, CH |
| 1’ | 129.9, C | 131.6, C | nd |
| 2’/6’ | 131.9, CH | 131.5, CH | 132.0, CH |
| 3’/5’ | 115.8, C | 115.4, C | 114.3, C |
| 4’ | 164.9, C | 164.6, C | 164.0, C |
| 1’’ | 133.0, C | 127.4, C | 126.5, C |
| 2’’ | 134.2, CH | 131.1, CH | 130.7, CH |
| 3’’ | 117.7, C | 112.6, CH | 111.9, CH |
| 4’’ | 153.1, C | 155.3, C | 155.4, C |
| 5’’ | 117.7, C | 110.7, C | 110.3, C |
| 6’’ | 134.2, CH | 134.1, CH | 134.1, CH |
| CH3O–4’’ | 60.6, CH3 | 56.4, CH3 | 56.2, CH3 |
| Proton | 3 | Z-4 | E-4 |
|---|---|---|---|
| 3 | 6.21, s | 6.13, s | 6.18, s |
| 6 | 6.44, s | 6.37, s | 6.88, s |
| 2’/6’ | 7.59, s | 7.56, s | 7.04, s |
| 2’’ | 8.15, s | 7.81, dd (8.7, 2.2) | 7.05, dd (8.7, 2.2) |
| 3’’ | 7.19, d (8.7) | 6.87, d (8.7) | |
| 6’’ | 8.15, s | 8.11, d (2.2) | 7.40, d (2.2) |
| CH3O–4’’ | 3.83, s | 3.90, s | 3.79, s |
| Compound | MIC (µg mL−1) | MIC (µM) |
|---|---|---|
| 1 | 2 | 3.36 |
| 2 | 0.5 | 0.79 |
| 3 | ≤0.25 | ≤0.41 |
| 4 | 0.5 | 0.94 |
| Tetracycline | 2 | 4.5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bracegirdle, J.; Stevenson, L.J.; Page, M.J.; Owen, J.G.; Keyzers, R.A. Targeted Isolation of Rubrolides from the New Zealand Marine Tunicate Synoicum kuranui. Mar. Drugs 2020, 18, 337. https://doi.org/10.3390/md18070337
Bracegirdle J, Stevenson LJ, Page MJ, Owen JG, Keyzers RA. Targeted Isolation of Rubrolides from the New Zealand Marine Tunicate Synoicum kuranui. Marine Drugs. 2020; 18(7):337. https://doi.org/10.3390/md18070337
Chicago/Turabian StyleBracegirdle, Joe, Luke J. Stevenson, Michael J. Page, Jeremy G. Owen, and Robert A. Keyzers. 2020. "Targeted Isolation of Rubrolides from the New Zealand Marine Tunicate Synoicum kuranui" Marine Drugs 18, no. 7: 337. https://doi.org/10.3390/md18070337
APA StyleBracegirdle, J., Stevenson, L. J., Page, M. J., Owen, J. G., & Keyzers, R. A. (2020). Targeted Isolation of Rubrolides from the New Zealand Marine Tunicate Synoicum kuranui. Marine Drugs, 18(7), 337. https://doi.org/10.3390/md18070337