Astaxanthin Counteracts Excitotoxicity and Reduces the Ensuing Increases in Calcium Levels and Mitochondrial Reactive Oxygen Species Generation

 ,
, 
 ,
, 
 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
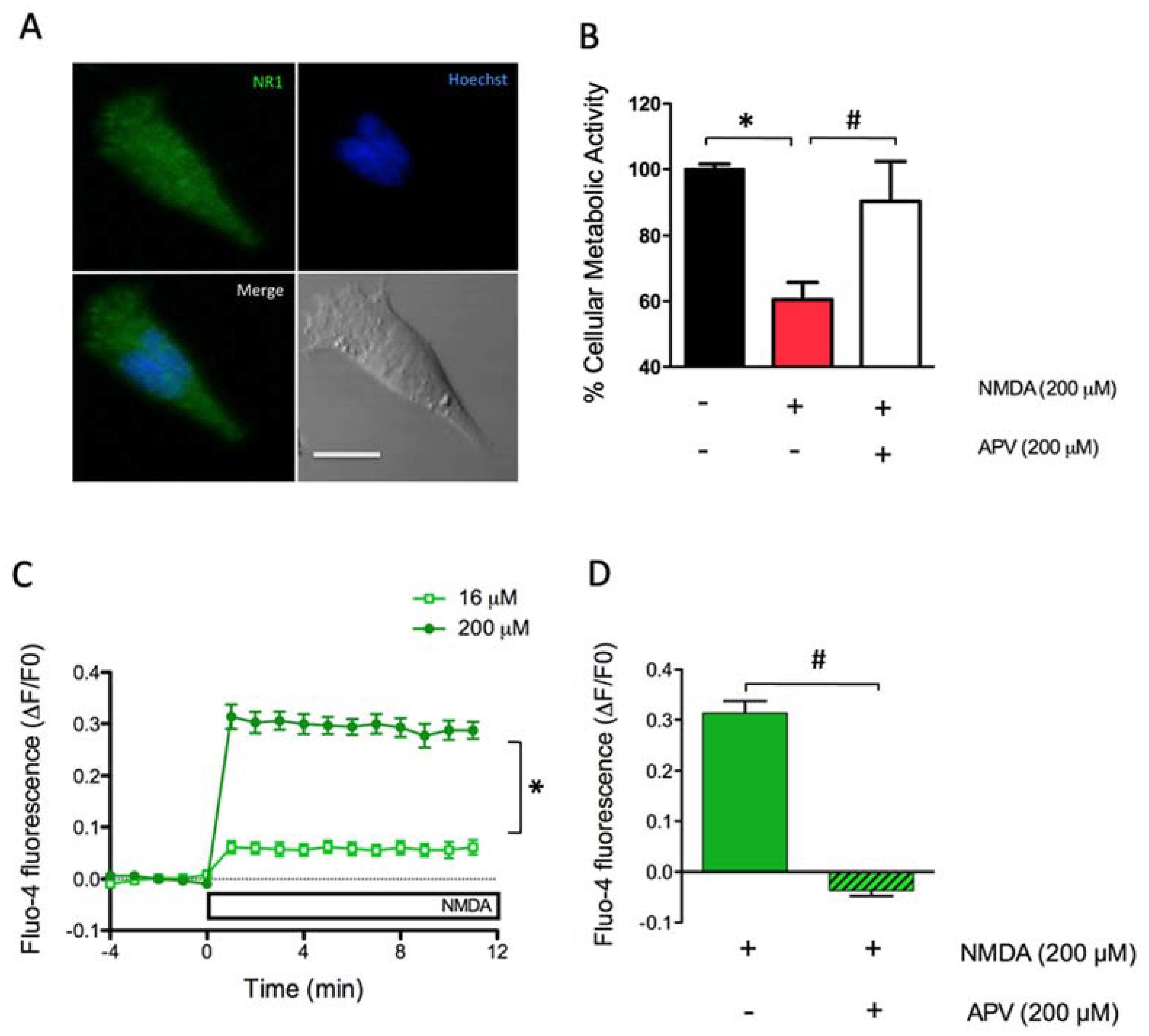
2.1. Dose-Dependent Activation of NMDAR Increases Cytoplasmic [Ca2+] and Causes Excitoxicity in SH-SY5Y Cells
2.2. Long-Term Treatment with ASX Protects SH-SY5Y Cells Against Neurotoxic Stimuli
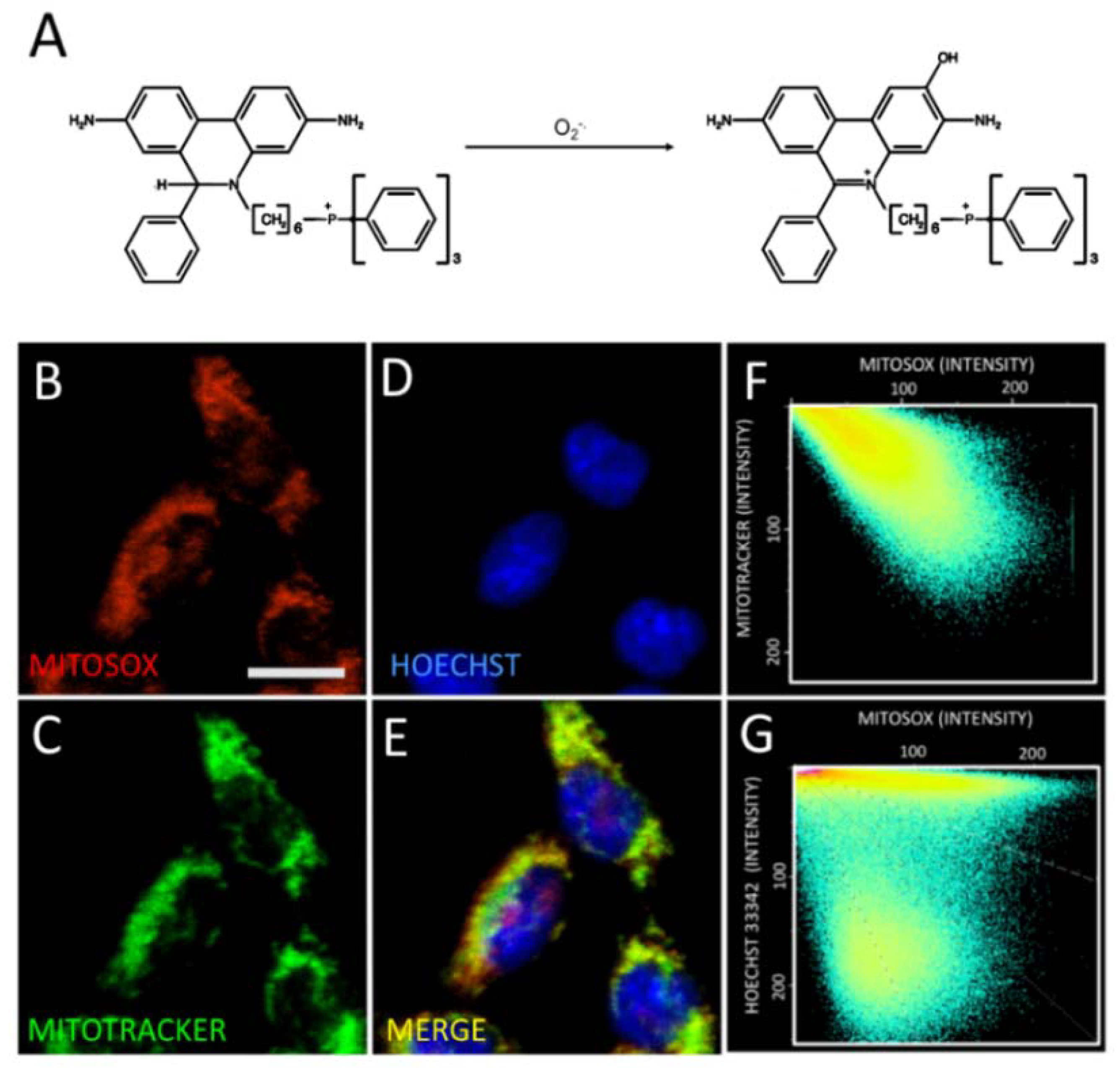
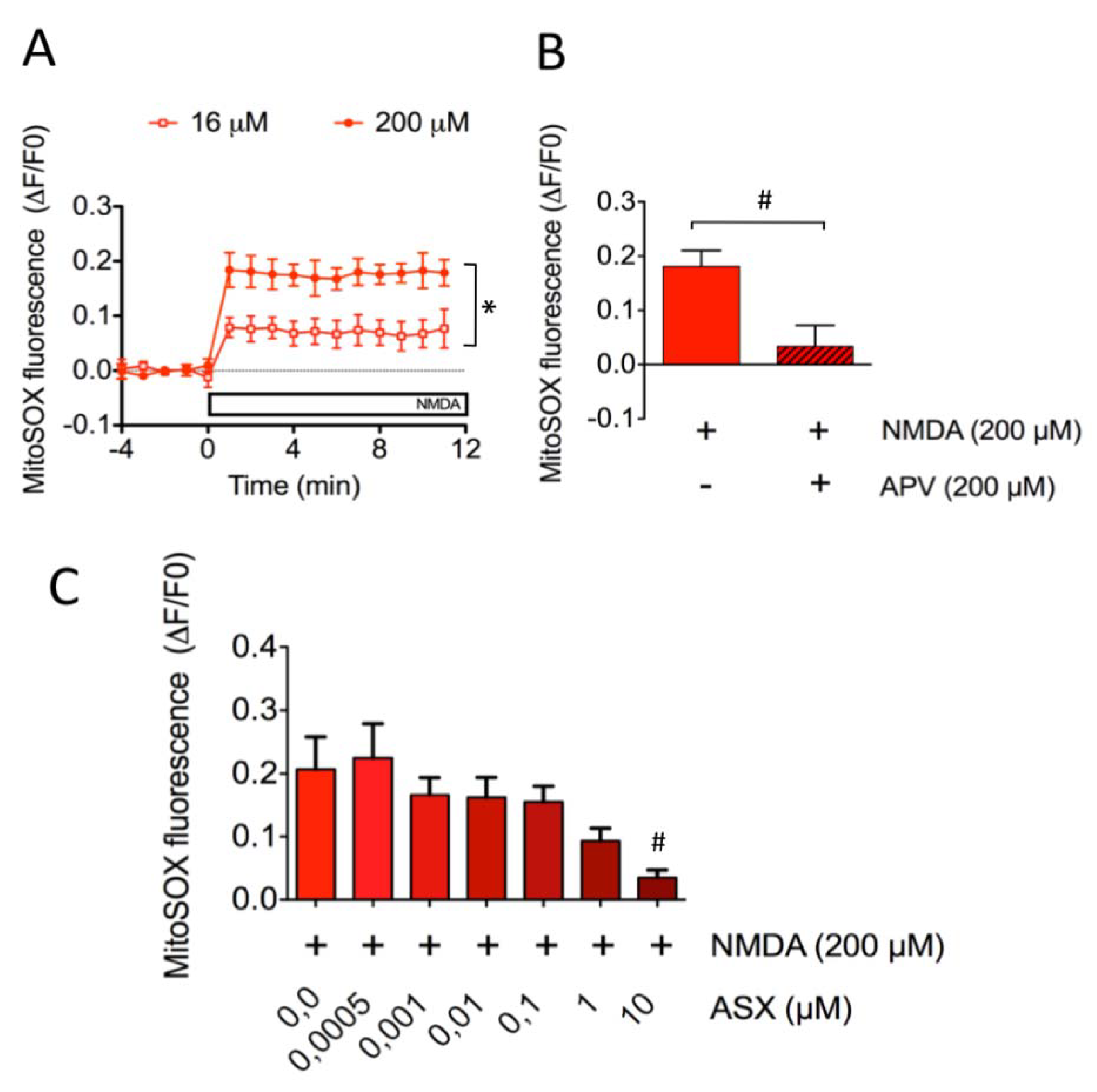
2.3. Astaxanthin Decreases Mitochondrial ROS Levels in SH-SY5Y Cells
2.4. Astaxanthin Attenuates the Generation of Excitotoxic Ca2+ Signals in Primary Hippocampal Neurons
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. SH-SY5Y Cell Cultures
4.3. Primary Hippocampal Cultures
4.4. Pharmacological Stimulation of NMDAR
4.5. Immunocytochemistry
4.6. Cell Metabolic Activity Assay
4.7. Intracellular Ca2+ Measurements
4.8. Mitochondrial ROS Measurements
4.9. Determination of Cytoplasmic Ca2+ Signals in Primary Hippocampal Neurons
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shah, M.M.; Liang, Y.; Cheng, J.J.; Daroch, M. Astaxanthin-Producing Green Microalga Haematococcus pluvialis: From Single Cell to High Value Commercial Products. Front. Plant Sci. 2016, 7, 531. [Google Scholar] [CrossRef] [PubMed]
- Guerin, M.; Huntley, M.E.; Olaizola, M. Haematococcus astaxanthin: Applications for human health and nutrition. Trends Biotechnol. 2003, 21, 210–216. [Google Scholar] [CrossRef]
- Ke, Y.; Bu, S.; Ma, H.; Gao, L.; Cai, Y.; Zhang, Y.; Zhou, W. Preventive and Therapeutic Effects of Astaxanthin on Depressive-Like Behaviors in High-Fat Diet and Streptozotocin-Treated Rats. Front. Pharmacol. 2019, 10, 1621. [Google Scholar] [CrossRef] [PubMed]
- Hongo, N.; Takamura, Y.; Nishimaru, H.; Matsumoto, J.; Tobe, K.; Saito, T.; Saido, T.C.; Nishijo, H. Astaxanthin Ameliorated Parvalbumin-Positive Neuron Deficits and Alzheimer’s Disease-Related Pathological Progression in the Hippocampus of App(NL-G-F/NL-G-F) Mice. Front. Pharmacol. 2020, 11, 307. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.P.; Liu, S.Y.; Sun, H.; Wu, X.M.; Li, J.J.; Zhu, L. Neuroprotective effect of astaxanthin on H2O2-induced neurotoxicity in vitro and on focal cerebral ischemia in vivo. Brain Res. 2010, 1360, 40–48. [Google Scholar] [CrossRef]
- Zhang, X.; Pan, L.; Wei, X.; Gao, H.; Liu, J. Impact of astaxanthin-enriched algal powder of Haematococcus pluvialis on memory improvement in BALB/c mice. Environ. Geochem. Health 2007, 29, 483–489. [Google Scholar] [CrossRef]
- Wen, X.; Huang, A.; Hu, J.; Zhong, Z.; Liu, Y.; Li, Z.; Pan, X.; Liu, Z. Neuroprotective effect of astaxanthin against glutamate-induced cytotoxicity in HT22 cells: Involvement of the Akt/GSK-3beta pathway. Neuroscience 2015, 303, 558–568. [Google Scholar] [CrossRef]
- Chang, C.H.; Chen, K.C.; Liaw, K.C.; Peng, C.C.; Peng, R.Y. Astaxanthin Protects PC12 Cells against Homocysteine- and Glutamate-Induced Neurotoxicity. Molecules 2020, 25, 214. [Google Scholar] [CrossRef]
- Ikeda, Y.; Tsuji, S.; Satoh, A.; Ishikura, M.; Shirasawa, T.; Shimizu, T. Protective effects of astaxanthin on 6-hydroxydopamine-induced apoptosis in human neuroblastoma SH-SY5Y cells. J. Neurochem. 2008, 107, 1730–1740. [Google Scholar] [CrossRef]
- Yan, T.; Zhao, Y.; Zhang, X.; Lin, X. Astaxanthin Inhibits Acetaldehyde-Induced Cytotoxicity in SH-SY5Y Cells by Modulating Akt/CREB and p38MAPK/ERK Signaling Pathways. Mar. Drugs 2016, 14, 56. [Google Scholar] [CrossRef]
- Lin, X.; Zhao, Y.; Li, S. Astaxanthin attenuates glutamate-induced apoptosis via inhibition of calcium influx and endoplasmic reticulum stress. Eur. J. Pharmacol. 2017, 806, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.M.; Asoh, S.; Hiranuma, H.; Ohsawa, I.; Iio, K.; Satou, A.; Ishikura, M.; Ohta, S. Astaxanthin protects mitochondrial redox state and functional integrity against oxidative stress. J. Nutr. Biochem. 2010, 21, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, P. Molecular basis of NMDA receptor functional diversity. Eur. J. Neurosci. 2011, 33, 1351–1365. [Google Scholar] [CrossRef] [PubMed]
- Bashir, Z.I.; Alford, S.; Davies, S.N.; Randall, A.D.; Collingridge, G.L. Long-term potentiation of NMDA receptor-mediated synaptic transmission in the hippocampus. Nature 1991, 349, 156–158. [Google Scholar] [CrossRef]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Morris, R.G. NMDA receptors and memory encoding. Neuropharmacology 2013, 74, 32–40. [Google Scholar] [CrossRef]
- Adasme, T.; Haeger, P.; Paula-Lima, A.C.; Espinoza, I.; Casas-Alarcon, M.M.; Carrasco, M.A.; Hidalgo, C. Involvement of ryanodine receptors in neurotrophin-induced hippocampal synaptic plasticity and spatial memory formation. Proc. Natl. Acad. Sci. USA 2011, 108, 3029–3034. [Google Scholar] [CrossRef]
- Kamsler, A.; Segal, M. Hydrogen peroxide modulation of synaptic plasticity. J. Neurosci. 2003, 23, 269–276. [Google Scholar] [CrossRef]
- Zheng, S.; Eacker, S.M.; Hong, S.J.; Gronostajski, R.M.; Dawson, T.M.; Dawson, V.L. NMDA-induced neuronal survival is mediated through nuclear factor I-A in mice. J. Clin. Investig. 2010, 120, 2446–2456. [Google Scholar] [CrossRef] [PubMed]
- Knafo, S.; Esteban, J.A. Common pathways for growth and for plasticity. Curr. Opin. Neurobiol. 2012, 22, 405–411. [Google Scholar] [CrossRef]
- Munoz, P.; Humeres, A.; Elgueta, C.; Kirkwood, A.; Hidalgo, C.; Nunez, M.T. Iron mediates N-methyl-D-aspartate receptor-dependent stimulation of calcium-induced pathways and hippocampal synaptic plasticity. J. Biol. Chem. 2011, 286, 13382–13392. [Google Scholar] [CrossRef] [PubMed]
- Weilinger, N.L.; Maslieieva, V.; Bialecki, J.; Sridharan, S.S.; Tang, P.L.; Thompson, R.J. Ionotropic receptors and ion channels in ischemic neuronal death and dysfunction. Acta Pharmacol. Sin. 2013, 34, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.G.; Zukin, R.S. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat. Rev. Neurosci. 2007, 8, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Sanelli, T.; Ge, W.; Leystra-Lantz, C.; Strong, M.J. Calcium mediated excitotoxicity in neurofilament aggregate-bearing neurons in vitro is NMDA receptor dependant. J. Neurol. Sci. 2007, 256, 39–51. [Google Scholar] [CrossRef]
- von Engelhardt, J.; Coserea, I.; Pawlak, V.; Fuchs, E.C.; Kohr, G.; Seeburg, P.H.; Monyer, H. Excitotoxicity in vitro by NR2A- and NR2B-containing NMDA receptors. Neuropharmacology 2007, 53, 10–17. [Google Scholar] [CrossRef]
- Kritis, A.A.; Stamoula, E.G.; Paniskaki, K.A.; Vavilis, T.D. Researching glutamate-induced cytotoxicity in different cell lines: A comparative/collective analysis/study. Front. Cell Neurosci. 2015, 9, 91. [Google Scholar] [CrossRef]
- Mattson, M.P. Glutamate and neurotrophic factors in neuronal plasticity and disease. Ann. N. Y. Acad. Sci. 2008, 1144, 97–112. [Google Scholar] [CrossRef]
- More, J.Y.; Bruna, B.A.; Lobos, P.E.; Galaz, J.L.; Figueroa, P.L.; Namias, S.; Sanchez, G.L.; Barrientos, G.C.; Valdes, J.L.; Paula-Lima, A.C.; et al. Calcium Release Mediated by Redox-Sensitive RyR2 Channels Has a Central Role in Hippocampal Structural Plasticity and Spatial Memory. Antioxid. Redox Signal. 2018, 29, 1125–1146. [Google Scholar] [CrossRef]
- Goussakov, I.; Miller, M.B.; Stutzmann, G.E. NMDA-mediated Ca2+ influx drives aberrant ryanodine receptor activation in dendrites of young Alzheimer’s disease mice. J. Neurosci. 2010, 30, 12128–12137. [Google Scholar] [CrossRef] [PubMed]
- Arias-Cavieres, A.; Adasme, T.; Sanchez, G.; Munoz, P.; Hidalgo, C. Aging Impairs Hippocampal- Dependent Recognition Memory and LTP and Prevents the Associated RyR Up-regulation. Front. Aging Neurosci. 2017, 9, 111. [Google Scholar] [CrossRef]
- SanMartin, C.D.; Veloso, P.; Adasme, T.; Lobos, P.; Bruna, B.; Galaz, J.; Garcia, A.; Hartel, S.; Hidalgo, C.; Paula-Lima, A.C. RyR2-Mediated Ca(2+) Release and Mitochondrial ROS Generation Partake in the Synaptic Dysfunction Caused by Amyloid beta Peptide Oligomers. Front. Mol. Neurosci. 2017, 10, 115. [Google Scholar] [CrossRef] [PubMed]
- Biedler, J.L.; Roffler-Tarlov, S.; Schachner, M.; Freedman, L.S. Multiple neurotransmitter synthesis by human neuroblastoma cell lines and clones. Cancer Res. 1978, 38, 3751–3757. [Google Scholar]
- Sun, Z.W.; Zhang, L.; Zhu, S.J.; Chen, W.C.; Mei, B. Excitotoxicity effects of glutamate on human neuroblastoma SH-SY5Y cells via oxidative damage. Neurosci. Bull. 2010, 26, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.D.; Niznik, H.B.; Mishra, R.K. NMDA and dopamine D2L receptor interaction in human neuroblastoma SH-SY5Y cells involves tyrosine kinase and phosphatase. Neuroreport 1996, 7, 2937–2940. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; An, L.; Gao, L.Y.; Bai, J.P.; Wang, J.; Meng, W.H.; Ren, T.S.; Zhao, Q.C. Compound MQA, a Caffeoylquinic Acid Derivative, Protects Against NMDA-Induced Neurotoxicity and Potential Mechanisms In Vitro. CNS Neurosci. Ther. 2015, 21, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.V.; Tan, A.S. Characterization of the cellular reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT): Subcellular localization, substrate dependence, and involvement of mitochondrial electron transport in MTT reduction. Arch. Biochem. Biophys. 1993, 303, 474–482. [Google Scholar] [CrossRef]
- Naguib, Y.M. Antioxidant activities of astaxanthin and related carotenoids. J. Agric. Food Chem. 2000, 48, 1150–1154. [Google Scholar] [CrossRef]
- Berridge, M.J. Calcium regulation of neural rhythms, memory and Alzheimer’s disease. J. Physiol. 2014, 592, 281–293. [Google Scholar] [CrossRef]
- Brocard, J.B.; Tassetto, M.; Reynolds, I.J. Quantitative evaluation of mitochondrial calcium content in rat cortical neurones following a glutamate stimulus. J. Physiol. 2001, 531, 793–805. [Google Scholar] [CrossRef]
- Vergun, O.; Keelan, J.; Khodorov, B.I.; Duchen, M.R. Glutamate-induced mitochondrial depolarisation and perturbation of calcium homeostasis in cultured rat hippocampal neurones. J. Physiol. 1999, 519, 451–466. [Google Scholar] [CrossRef]
- McCormack, J.G.; Denton, R.M. The role of intramitochondrial Ca2+ in the regulation of oxidative phosphorylation in mammalian tissues. Biochem. Soc. Trans. 1993, 21, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Lobos, P.; Bruna, B.; Cordova, A.; Barattini, P.; Galaz, J.L.; Adasme, T.; Hidalgo, C.; Munoz, P.; Paula-Lima, A. Astaxanthin Protects Primary Hippocampal Neurons against Noxious Effects of Abeta-Oligomers. Neural Plast. 2016, 3456783, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bindokas, V.P.; Jordan, J.; Lee, C.C.; Miller, R.J. Superoxide production in rat hippocampal neurons: Selective imaging with hydroethidine. J. Neurosci. 1996, 16, 1324–1336. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Prabhakar, M.; Kumar, P.; Deshmukh, R.; Sharma, P.L. Excitotoxicity: Bridge to various triggers in neurodegenerative disorders. Eur. J. Pharmacol. 2013, 698, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Shibata, T.; Hisaka, S.; Osawa, T. Astaxanthin inhibits reactive oxygen species-mediated cellular toxicity in dopaminergic SH-SY5Y cells via mitochondria-targeted protective mechanism. Brain Res. 2009, 1254, 18–27. [Google Scholar] [CrossRef]
- Lee, D.H.; Kim, C.S.; Lee, Y.J. Astaxanthin protects against MPTP/MPP+-induced mitochondrial dysfunction and ROS production in vivo and in vitro. Food Chem. Toxicol. 2011, 49, 271–280. [Google Scholar] [CrossRef]
- Simola, N.; Morelli, M.; Carta, A.R. The 6-hydroxydopamine model of Parkinson’s disease. Neurotox. Res. 2007, 11, 151–167. [Google Scholar] [CrossRef]
- Wu, H.; Niu, H.; Shao, A.; Wu, C.; Dixon, B.J.; Zhang, J.; Yang, S.; Wang, Y. Astaxanthin as a Potential Neuroprotective Agent for Neurological Diseases. Mar. Drugs 2015, 13, 5750–5766. [Google Scholar] [CrossRef]
- Kim, Y.H.; Koh, H.K.; Kim, D.S. Down-regulation of IL-6 production by astaxanthin via ERK-, MSK-, and NF-kappaB-mediated signals in activated microglia. Int. Immunopharmacol. 2010, 10, 1560–1572. [Google Scholar] [CrossRef]
- Yan, Y.; Wei, C.L.; Zhang, W.R.; Cheng, H.P.; Liu, J. Cross-talk between calcium and reactive oxygen species signaling. Acta Pharmacol. Sin. 2006, 27, 821–826. [Google Scholar] [CrossRef]
- Hidalgo, C.; Donoso, P. Crosstalk between calcium and redox signaling: From molecular mechanisms to health implications. Antioxid. Redox Signal. 2008, 10, 1275–1312. [Google Scholar] [CrossRef] [PubMed]
- Paula-Lima, A.C.; Adasme, T.; Hidalgo, C. Contribution of Ca2+ release channels to hippocampal synaptic plasticity and spatial memory: Potential redox modulation. Antioxid. Redox Signal. 2014, 21, 892–914. [Google Scholar] [CrossRef] [PubMed]
- Arias-Cavieres, A.; Barrientos, G.C.; Sanchez, G.; Elgueta, C.; Munoz, P.; Hidalgo, C. Ryanodine Receptor-Mediated Calcium Release Has a Key Role in Hippocampal LTD Induction. Front. Cell Neurosci. 2018, 12, 403. [Google Scholar] [CrossRef] [PubMed]
- Girouard, H.; Park, L.; Anrather, J.; Zhou, P.; Iadecola, C. Cerebrovascular nitrosative stress mediates neurovascular and endothelial dysfunction induced by angiotensin II. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.S.; Zhang, X.; Wu, Q.; Li, W.; Wang, C.X.; Xie, G.B.; Zhou, X.M.; Shi, J.X.; Zhou, M.L. Astaxanthin offers neuroprotection and reduces neuroinflammation in experimental subarachnoid hemorrhage. J. Surg. Res. 2014, 192, 206–213. [Google Scholar] [CrossRef]
- Katagiri, M.; Satoh, A.; Tsuji, S.; Shirasawa, T. Effects of astaxanthin-rich Haematococcus pluvialis extract on cognitive function: A randomised, double-blind, placebo-controlled study. J. Clin. Biochem. Nutr. 2012, 51, 102–107. [Google Scholar] [CrossRef]
- Wu, D.; Xu, H.; Chen, J.; Zhang, L. Effects of Astaxanthin Supplementation on Oxidative Stress. Int. J. Vitam. Nutr. Res. 2020, 90, 179–194. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García, F.; Lobos, P.; Ponce, A.; Cataldo, K.; Meza, D.; Farías, P.; Estay, C.; Oyarzun-Ampuero, F.; Herrera-Molina, R.; Paula-Lima, A.; et al. Astaxanthin Counteracts Excitotoxicity and Reduces the Ensuing Increases in Calcium Levels and Mitochondrial Reactive Oxygen Species Generation. Mar. Drugs 2020, 18, 335. https://doi.org/10.3390/md18060335
García F, Lobos P, Ponce A, Cataldo K, Meza D, Farías P, Estay C, Oyarzun-Ampuero F, Herrera-Molina R, Paula-Lima A, et al. Astaxanthin Counteracts Excitotoxicity and Reduces the Ensuing Increases in Calcium Levels and Mitochondrial Reactive Oxygen Species Generation. Marine Drugs. 2020; 18(6):335. https://doi.org/10.3390/md18060335
Chicago/Turabian StyleGarcía, Francisca, Pedro Lobos, Alejandra Ponce, Karla Cataldo, Daniela Meza, Patricio Farías, Carolina Estay, Felipe Oyarzun-Ampuero, Rodrigo Herrera-Molina, Andrea Paula-Lima, and et al. 2020. "Astaxanthin Counteracts Excitotoxicity and Reduces the Ensuing Increases in Calcium Levels and Mitochondrial Reactive Oxygen Species Generation" Marine Drugs 18, no. 6: 335. https://doi.org/10.3390/md18060335
APA StyleGarcía, F., Lobos, P., Ponce, A., Cataldo, K., Meza, D., Farías, P., Estay, C., Oyarzun-Ampuero, F., Herrera-Molina, R., Paula-Lima, A., Ardiles, Á. O., Hidalgo, C., Adasme, T., & Muñoz, P. (2020). Astaxanthin Counteracts Excitotoxicity and Reduces the Ensuing Increases in Calcium Levels and Mitochondrial Reactive Oxygen Species Generation. Marine Drugs, 18(6), 335. https://doi.org/10.3390/md18060335