Efficacy and Mechanism of Action of Marine Alkaloid 3,10-Dibromofascaplysin in Drug-Resistant Prostate Cancer Cells

 ,
, 
 , ,
, ,
Abstract
1. Introduction
2. Results and Discussion
2.1. 3,10-Dibromofascaplysin (DBF) Induces Apoptotic Cell Death of Drug-Resistant Prostate Cancer Cells
2.2. DBF Induces Alterations of Protein Tyrosine Kinases Activity
2.3. Validation of Kinome Analysis Data
2.4. Role of JNK1/2 and Other MAPKs in Cytotoxic Effect of DBF
2.5. Effect of DBF in Combination with Platinum and Taxane Agents
2.6. Effect of DBF in Combination with Olaparib
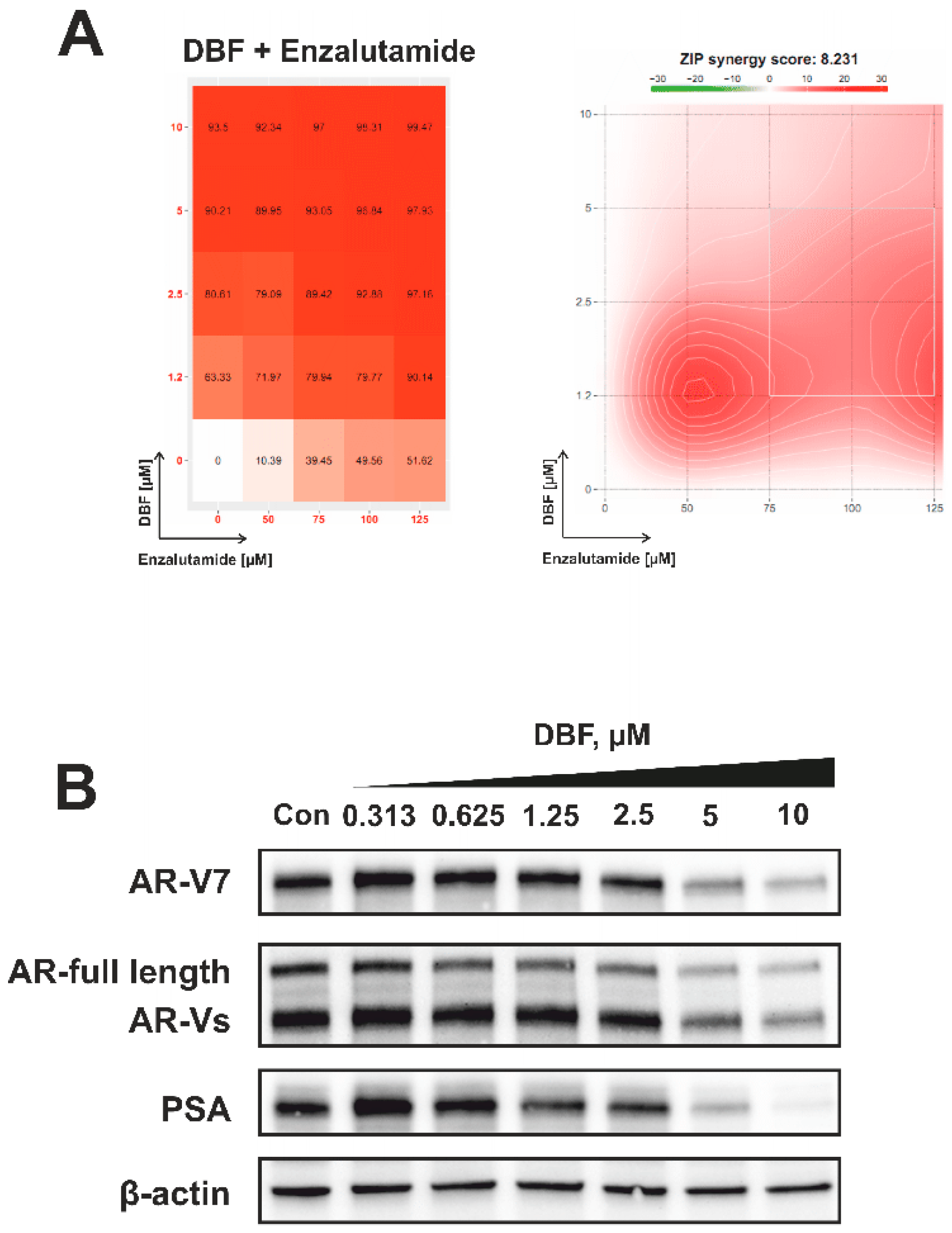
2.7. Effect of DBF on AR Signaling
3. Materials and Methods
3.1. 3,10-Dibromofascaplysin
3.2. Reagents and Antibodies
3.3. Cell Lines and Culture Conditions
3.4. MTT Assay
3.5. Kinase Activity Profiling
3.6. Western Blotting
3.7. Determination of Drug Combination Effects
3.8. Analysis of Intracellular ROS Level
3.9. Analysis of DNA Damage
3.10. Data and Statistical Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Beyer, J.; Albers, P.; Altena, R.; Aparicio, J.; Bokemeyer, C.; Busch, J.; Cathomas, R.; Cavallin-Stahl, E.; Clarke, N.W.; Claßen, J.; et al. Maintaining success, reducing treatment burden, focusing on survivorship: Highlights from the third European consensus conference on diagnosis and treatment of germ-cell cancer. Ann. Oncol. 2013, 24, 878–888. [Google Scholar] [CrossRef]
- Caffo, O.; De Giorgi, U.; Fratino, L.; Alesini, D.; Zagonel, V.; Facchini, G.; Gasparro, D.; Ortega, C.; Tucci, M.; Verderame, F.; et al. Clinical outcomes of castration-resistant prostate cancer treatments administered as third or fourth line following failure of docetaxel and other second-line treatment: Results of an Italian multicentre study. Eur. Urol. 2015, 68, 147–153. [Google Scholar] [PubMed]
- Armstrong, C.M.; Gao, A.C. Drug resistance in castration resistant prostate cancer: Resistance mechanisms and emerging treatment strategies. Am. J. Clin. Exp. Urol. 2015, 3, 64–76. [Google Scholar] [PubMed]
- Stonik, V.A. Marine natural products: A way to new drugs. Acta Nat. 2009, 2, 15–25. [Google Scholar]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar] [CrossRef]
- Dyshlovoy, S.A.; Honecker, P.D.D.F. Marine Compounds and Cancer: The First Two Decades of XXI Century. Mar. Drugs 2019, 18, 20. [Google Scholar] [CrossRef]
- Mayer, A. Marine Pharmaceutical: The Clinical Pipeline. 2020. Available online: https://www.midwestern.edu/departments/marinepharmacology/clinical-pipeline.xml (accessed on 2 November 2020).
- Dyshlovoy, S.A.; Honecker, P.D.D.F. Marine Compounds and Cancer: Where Do We Stand? Mar. Drugs 2015, 13, 5657–5665. [Google Scholar] [CrossRef]
- Dyshlovoy, S.A.; Honecker, F. Marine Compounds and Cancer: 2017 Updates. Mar. Drugs 2018, 16, 41. [Google Scholar] [CrossRef]
- Roll, D.M.; Ireland, C.M.; Lu, H.S.M.; Clardy, J. Fascaplysin, an unusual antimicrobial pigment from the marine sponge Fascaplysinopsis sp. J. Org. Chem. 1988, 53, 3276–3278. [Google Scholar] [CrossRef]
- Bharate, S.S.; Manda, S.; Mupparapu, N.; Battini, N.; Vishwakarma, R.A. Chemistry and Biology of Fascaplysin, a Potent Marine-Derived CDK-4 Inhibitor. MiniRev. Med. Chem. 2012, 12, 650–664. [Google Scholar] [CrossRef]
- Lin, J.; Yan, X.-J.; Chen, H. Fascaplysin, a selective CDK4 inhibitor, exhibit anti-angiogenic activity in vitro and in vivo. Cancer Chemother. Pharmacol. 2006, 59, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Kuzmich, A.S.; Fedorov, S.N.; Shastina, V.V.; Shubina, L.K.; Radchenko, O.S.; Balaneva, N.N.; Zhidkov, M.E.; Park, J.I.; Kwak, J.Y.; Stonik, V.A. The anticancer activity of 3-and 10-bromofascaplysins is mediated by caspase-8,-9,-3-dependent apoptosis. Bioorg. Med. Chem. 2010, 18, 3834–3840. [Google Scholar] [CrossRef] [PubMed]
- Popov, A.M.; A Stonik, V. Physiological activity of fascaplisine—An unusual pigment from tropical sea fishes. Antibiot. Khimioterapiia Antibiot. Chemoterapy 1991, 36, 12–14. [Google Scholar]
- Dembitsky, V.M.; Gloriozova, T.; Poroikov, V.V. Novel Antitumor Agents: Marine Sponge Alkaloids, their Synthetic Analogs and Derivatives. MiniRev. Med. Chem. 2005, 5, 319–336. [Google Scholar] [CrossRef]
- Hamilton, G. Cytotoxic Effects of Fascaplysin against Small Cell Lung Cancer Cell Lines. Mar. Drugs 2014, 12, 1377–1389. [Google Scholar] [CrossRef]
- Oh, T.-I.; Lee, Y.-M.; Nam, T.-J.; Ko, Y.-S.; Mah, S.; Kim, J.; Kim, Y.; Reddy, R.H.; Kim, Y.J.; Hong, S.; et al. Fascaplysin Exerts Anti-Cancer Effects through the Downregulation of Survivin and HIF-1α and Inhibition of VEGFR2 and TRKA. Int. J. Mol. Sci. 2017, 18, 2074. [Google Scholar] [CrossRef]
- Sharma, S.; Guru, S.K.; Manda, S.; Kumar, A.; Mintoo, M.J.; Prasad, V.D.; Sharma, P.R.; Mondhe, D.M.; Bharate, S.B.; Bhushan, S. A marine sponge alkaloid derivative 4-chloro fascaplysin inhibits tumor growth and VEGF mediated angiogenesis by disrupting PI3K/Akt/mTOR signaling cascade. Chem. Interact. 2017, 275, 47–60. [Google Scholar] [CrossRef]
- Meng, N.; Mu, X.; Lv, X.; Wang, L.; Li, N.; Gong, Y. Autophagy represses fascaplysin-induced apoptosis and angiogenesis inhibition via ROS and p8 in vascular endothelia cells. Biomed. Pharmacother. 2019, 114, 108866. [Google Scholar] [CrossRef]
- Zhidkov, M.E.; Smirnova, P.A.; Tryapkin, O.A.; Kantemirov, A.V.; Khudyakova, Y.V.; Malyarenko, O.S.; Ermakova, S.; Grigorchuk, V.P.; Kaune, M.; Von Amsberg, G.; et al. Total Syntheses and Preliminary Biological Evaluation of Brominated Fascaplysin and Reticulatine Alkaloids and Their Analogues. Mar. Drugs 2019, 17, 496. [Google Scholar] [CrossRef]
- Soni, R.; Müller, L.; Furet, P.; Schoepfer, J.; Stephan, C.; Zumstein-Mecker, S.; Fretz, H.; Chaudhuri, B. Inhibition of Cyclin-Dependent Kinase 4 (Cdk4) by Fascaplysin, a Marine Natural Product. Biochem. Biophys. Res. Commun. 2000, 275, 877–884. [Google Scholar] [CrossRef]
- Hörmann, A.; Chaudhuri, B.; Fretz, H. DNA binding properties of the marine sponge pigment fascaplysin. Bioorg. Med. Chem. 2001, 9, 917–921. [Google Scholar] [CrossRef]
- Oh, T.-I.; Lee, J.H.; Kim, S.; Nam, T.-J.; Kim, Y.-S.; Kim, B.M.; Yim, W.J.; Lim, J.-H. Fascaplysin Sensitizes Anti-Cancer Effects of Drugs Targeting AKT and AMPK. Molecules 2017, 23, 42. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Guru, S.K.; Pathania, A.S.; Manda, S.; Kumar, A.; Bharate, S.B.; Vishwakarma, R.A.; Malik, F.; Bhushan, S. Fascaplysin Induces Caspase Mediated Crosstalk Between Apoptosis and Autophagy Through the Inhibition of PI3K/AKT/mTOR Signaling Cascade in Human Leukemia HL-60 Cells. J. Cell. Biochem. 2015, 116, 985–997. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef]
- Kung, H.-J.; Changou, C.; Nguyen, H.G.; Yang, J.C.; Evans, C.P.; Bold, R.J.; Chuang, F. Autophagy and Prostate Cancer Therapeutics. In Advanced Structural Safety Studies; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2013; pp. 497–518. [Google Scholar]
- Lyakhova, I.A.; Bryukhovetsky, I.S.; Kudryavtsev, I.V.; Khotimchenko, Y.S.; Zhidkov, M.E.; Kantemirov, A.V. Antitumor Activity of Fascaplysin Derivatives on Glioblastoma Model In Vitro. Bull. Exp. Biol. Med. 2018, 164, 666–672. [Google Scholar] [CrossRef]
- Segraves, N.L.; Robinson, S.J.; Garcia, D.; Said, S.A.; Fu, X.; Schmitz, F.J.; Pietraszkiewicz, H.; Valeriote, A.F.A.; Crews, P. Comparison of Fascaplysin and Related Alkaloids: A Study of Structures, Cytotoxicities, and Sources. J. Nat. Prod. 2004, 67, 783–792. [Google Scholar] [CrossRef]
- Sampson, N.; Neuwirt, H.; Puhr, M.; Klocker, H.; Eder, I.E. In vitro model systems to study androgen receptor signaling in prostate cancer. Endocr. Relat. Cancer 2013, 20, R49–R64. [Google Scholar] [CrossRef]
- Nelson, P.S. Targeting the androgen receptor in prostate cancer—A resilient foe. N. Engl. J. Med. 2014, 371, 1067–1069. [Google Scholar] [CrossRef]
- Puhr, M.; Hoefer, J.; Schäfer, G.; Erb, H.H.; Oh, S.J.; Klocker, H.; Heidegger, I.; Neuwirt, H.; Culig, Z. Epithelial-to-Mesenchymal Transition Leads to Docetaxel Resistance in Prostate Cancer and Is Mediated by Reduced Expression of miR-200c and miR-205. Am. J. Pathol. 2012, 181, 2188–2201. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef] [PubMed]
- Struve, N.; Binder, Z.A.; Stead, L.F.; Brend, T.; Bagley, S.J.; Faulkner, C.; Ott, L.; Müller-Goebel, J.; Weik, A.-S.; Hoffer, K.; et al. EGFRvIII upregulates DNA mismatch repair resulting in increased temozolomide sensitivity of MGMT promoter methylated glioblastoma. Oncogene 2020, 39, 3041–3055. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Berriguete, G.; Fraile, B.; Martínez-Onsurbe, P.; Olmedilla, G.; Paniagua, R.; Royuela, M. MAP Kinases and Prostate Cancer. J. Signal Transduct. 2012, 2012, 169170. [Google Scholar] [CrossRef]
- Xu, R.; Hu, J. The role of JNK in prostate cancer progression and therapeutic strategies. Biomed. Pharmacother. 2020, 121, 109679. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-M.; Bost, F.; Charbono, W.; Dean, N.; McKay, R.; Rhim, J.S.; Depatie, C.; Mercola, D. C-Jun NH(2)-terminal kinase mediates proliferation and tumor growth of human prostate carcinoma. Clin. Cancer Res. 2003, 9, 391. [Google Scholar] [PubMed]
- Hu, J.; Wang, G.; Sun, T. Dissecting the roles of the androgen receptor in prostate cancer from molecular perspectives. Tumor Biol. 2017, 39, 1010428317692259. [Google Scholar] [CrossRef]
- Liu, P.-Y.; Lin, S.-Z.; Sheu, J.J.-C.; Lin, C.-T.; Lin, P.-C.; Chou, Y.-W.; Liu, C.-A.; Chiou, T.-W.; Harn, H.-J. Regulation of androgen receptor expression byZ-isochaihulactone mediated by the JNK signaling pathway and might be related to cytotoxicity in prostate cancer. Prostate 2012, 73, 531–541. [Google Scholar] [CrossRef]
- Tang, F.; Kokontis, J.; Lin, Y.; Liao, S.; Lin, A.; Xiang, J. Androgen via p21 Inhibits Tumor Necrosis Factor α-induced JNK Activation and Apoptosis. J. Biol. Chem. 2009, 284, 32353–32358. [Google Scholar] [CrossRef]
- Liu, J.; Lin, A. Role of JNK activation in apoptosis: A double-edged sword. Cell Res. 2005, 15, 36–42. [Google Scholar] [CrossRef]
- Samatar, A.A.; Poulikakos, P.I. Targeting RAS–ERK signalling in cancer: Promises and challenges. Nat. Rev. Drug Discov. 2014, 13, 928–942. [Google Scholar] [CrossRef]
- Yadav, B.; Wennerberg, K.; Aittokallio, T.; Tang, J. Searching for Drug Synergy in Complex Dose–Response Landscapes Using an Interaction Potency Model. Comput. Struct. Biotechnol. J. 2015, 13, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Tomida, T. Visualization of the spatial and temporal dynamics of MAPK signaling using fluorescence imaging techniques. J. Physiol. Sci. 2014, 65, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Ventura, J.J.; Hübner, A.; Zhang, C.; Flavell, R.A.; Shokat, K.M.; Davis, R.J. Chemical genetic analysis of the time course of signal transduction by JNK. Mol. Cell 2006, 21, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Qiu, M.-S.; Green, S.H. PC12 cell neuronal differentiation is associated with prolonged p21ras activity and consequent prolonged ERK activity. Neuron 1992, 9, 705–717. [Google Scholar] [CrossRef]
- Traverse, S.; Gomez, N.; Paterson, H.; Marshall, C.; Cohen, P. Sustained activation of the mitogen-activated protein (MAP) kinase cascade may be required for differentiation of PC12 cells. Comparison of the effects of nerve growth factor and epidermal growth factor. Biochem. J. 1992, 288, 351–355. [Google Scholar] [CrossRef]
- Sachdev, E.; Tabatabai, R.; Roy, V.; Rimel, B.J.; Mita, M. PARP Inhibition in Cancer: An Update on Clinical Development. Target. Oncol. 2019, 14, 657–679. [Google Scholar] [CrossRef]
- Bochum, S.; Berger, S.; Martens, U.M. Olaparib. Recent Results Cancer Res. 2018, 211, 217–233. [Google Scholar]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Li, L.; Karanika, S.; Yang, G.; Wang, J.; Park, S.; Broom, B.M.; Manyam, G.C.; Wu, W.; Luo, Y.; Basourakos, S.; et al. Androgen receptor inhibitor–induced “BRCAness” and PARP inhibition are synthetically lethal for castration-resistant prostate cancer. Sci. Signal. 2017, 10, eaam7479. [Google Scholar] [CrossRef]
- Feiersinger, G.E.; Trattnig, K.; Leitner, P.D.; Guggenberger, F.; Oberhuber, A.; Peer, S.; Hermann, M.; Skvortsova, I.; Vrbková, J.; Bouchal, J.; et al. Olaparib is effective in combination with, and as maintenance therapy after, first-line endocrine therapy in prostate cancer cells. Mol. Oncol. 2018, 12, 561–576. [Google Scholar] [CrossRef]
- Lee, J.; Kim, Y.; Lim, S.; Jo, K. Single-molecule visualization of ROS-induced DNA damage in large DNA molecules. Analyst 2016, 141, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Lin, Y.; Kim, Y.-S.; Asharani, P.V.; Liu, Z.-G.; Shen, H.-M. c-Jun N-terminal kinase mediates hydrogen peroxide-induced cell death via sustained poly(ADP-ribose) polymerase-1 activation. Cell Death Differ. 2007, 14, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Shafi, A.A.; Yen, A.E.; Weigel, N.L. Androgen receptors in hormone-dependent and castration-resistant prostate cancer. Pharmacol. Ther. 2013, 140, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.A.; Madanchi, R.; Hauschild, J.; Otte, K.; Alsdorf, W.H.; Schumacher, U.; Kalinin, V.I.; Silchenko, A.S.; Avilov, S.A.; Honecker, F.; et al. The marine triterpene glycoside frondoside A induces p53-independent apoptosis and inhibits autophagy in urothelial carcinoma cells. BMC Cancer 2017, 17, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Arni, S.; Le, T.H.N.; De Wijn, R.; García-Villegas, R.; Dankers, M.; Weder, W.; Hillinger, S. Ex vivo multiplex profiling of protein tyrosine kinase activities in early stages of human lung adenocarcinoma. Oncotarget 2017, 8, 68599–68613. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dyshlovoy, S.A.; Kaune, M.; Kriegs, M.; Hauschild, J.; Busenbender, T.; Shubina, L.K.; Makarieva, T.N.; Hoffer, K.; Bokemeyer, C.; Graefen, M.; et al. Marine alkaloid monanchoxymycalin C: A new specific activator of JNK1/2 kinase with anticancer properties. Sci. Rep. 2020, 10, 13178. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.A.; Pelageev, D.N.; Hauschild, J.; Sabutskii, Y.E.; Khmelevskaya, E.A.; Krisp, C.; Kaune, M.; Venz, S.; Borisova, K.L.; Busenbender, T.; et al. Inspired by Sea Urchins: Warburg Effect Mediated Selectivity of Novel Synthetic Non-Glycoside 1,4-Naphthoquinone-6S-Glucose Conjugates in Prostate Cancer. Mar. Drugs 2020, 18, 251. [Google Scholar] [CrossRef] [PubMed]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 2.0: Visual analytics of multi-drug combination synergies. Nucleic Acids Res. 2020, 48, W488–W493. [Google Scholar] [CrossRef]
- Dyshlovoy, S.A.; Tabakmakher, K.M.; Hauschild, J.; Shchekaleva, R.K.; Otte, K.; Guzii, A.G.; Makarieva, T.N.; Kudryashova, E.K.; Fedorov, S.N.; Shubina, L.K.; et al. Guanidine Alkaloids from the Marine Sponge Monanchora pulchra Show Cytotoxic Properties and Prevent EGF-Induced Neoplastic Transformation In Vitro. Mar. Drugs 2016, 14, 133. [Google Scholar] [CrossRef]
- Dyshlovoy, A.S.; Pelageev, N.D.; Hauschild, J.; Borisova, L.K.; Kaune, M.; Krisp, C.; Venz, S.; Sabutskii, E.Y.; Khmelevskaya, A.E.; Busenbender, T.; et al. Successful Targeting of the Warburg Effect in Prostate Cancer by Glucose-Conjugated 1,4-Naphthoquinones. Cancers 2019, 11, 1690. [Google Scholar] [CrossRef]
- Dyshlovoy, S.A.; Rast, S.; Hauschild, J.; Otte, K.; Alsdorf, W.H.; Madanchi, R.; Kalinin, V.I.; Silchenko, A.S.; Avilov, S.A.; Dierlamm, J.; et al. Frondoside A induces AIF-associated caspase-independent apoptosis in Burkitt lymphoma cells. Leuk. Lymphoma 2017, 58, 2905–2915. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | IC50, 72 h | |
|---|---|---|
| DBF [µM] | Docetaxel [nM] | |
| 22Rv1 | 0.29 ± 0.04 | 0.38 ± 0.08 |
| PC3 | 0.79 ± 0.17 | 8.55 ± 3.09 |
| PC3-DR | 1.51 ± 0.35 | 355.8 ± 148.7 |
| DU145 | 4.19 ± 0.81 | 1.72 ± 0.19 |
| DU145-DR | 1.25 ± 0.27 | 89.4 ± 13.2 |
| Antibodies | Clonality | Source | Cat.-No. | Dilution | Manufacturer |
|---|---|---|---|---|---|
| anti-AR | pAb | rabbit | sc-816 | 1:200 | Santa Cruz |
| anti-AR-V7 | mAb | rabbit | 198394 | 1:1000 | abcam |
| anti-cleaved Caspase-3 | mAb | rabbit | #9664 | 1:1000 | Cell Signaling |
| anti-ERK1/2 | mAb | mouse | #9107 | 1:2000 | Cell Signaling |
| anti-JNK1/2 | mAb | rabbit | #9258 | 1:1000 | Cell Signaling |
| anti-mouse IgG-HRP | sheep | NXA931 | 1:10,000 | GE Healthcare | |
| anti-p38 | mAb | rabbit | #9212 | 1:1000 | Cell Signaling |
| anti-PARP | pAb | rabbit | #9542 | 1:1000 | Cell Signaling |
| anti-phospho-ERK1/2 | mAb | rabbit | #4377 | 1:1000 | Cell Signaling |
| anti-phospho-JNK1/2 | mAb | rabbit | #4668 | 1:1000 | Cell Signaling |
| anti-phospho-p38 | mAb | rabbit | #4511 | 1:1000 | Cell Signaling |
| anti-PSA/KLK3 | mAb | rabbit | #5365 | 1:1000 | Cell Signaling |
| anti-rabbit IgG-HRP | goat | #7074 | 1:5000 | Cell Signaling | |
| anti-α-Tubulin | mAb | mouse | T5168 | 1:5000 | Sigma-Aldrich |
| anti-β-Actin-HRP | pAb | goat | sc-1616 | 1:10,000 | Santa Cruz |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dyshlovoy, S.A.; Kaune, M.; Hauschild, J.; Kriegs, M.; Hoffer, K.; Busenbender, T.; Smirnova, P.A.; Zhidkov, M.E.; Poverennaya, E.V.; Oh-Hohenhorst, S.J.; et al. Efficacy and Mechanism of Action of Marine Alkaloid 3,10-Dibromofascaplysin in Drug-Resistant Prostate Cancer Cells. Mar. Drugs 2020, 18, 609. https://doi.org/10.3390/md18120609
Dyshlovoy SA, Kaune M, Hauschild J, Kriegs M, Hoffer K, Busenbender T, Smirnova PA, Zhidkov ME, Poverennaya EV, Oh-Hohenhorst SJ, et al. Efficacy and Mechanism of Action of Marine Alkaloid 3,10-Dibromofascaplysin in Drug-Resistant Prostate Cancer Cells. Marine Drugs. 2020; 18(12):609. https://doi.org/10.3390/md18120609
Chicago/Turabian StyleDyshlovoy, Sergey A., Moritz Kaune, Jessica Hauschild, Malte Kriegs, Konstantin Hoffer, Tobias Busenbender, Polina A. Smirnova, Maxim E. Zhidkov, Ekaterina V. Poverennaya, Su Jung Oh-Hohenhorst, and et al. 2020. "Efficacy and Mechanism of Action of Marine Alkaloid 3,10-Dibromofascaplysin in Drug-Resistant Prostate Cancer Cells" Marine Drugs 18, no. 12: 609. https://doi.org/10.3390/md18120609
APA StyleDyshlovoy, S. A., Kaune, M., Hauschild, J., Kriegs, M., Hoffer, K., Busenbender, T., Smirnova, P. A., Zhidkov, M. E., Poverennaya, E. V., Oh-Hohenhorst, S. J., Spirin, P. V., Prassolov, V. S., Tilki, D., Bokemeyer, C., Graefen, M., & von Amsberg, G. (2020). Efficacy and Mechanism of Action of Marine Alkaloid 3,10-Dibromofascaplysin in Drug-Resistant Prostate Cancer Cells. Marine Drugs, 18(12), 609. https://doi.org/10.3390/md18120609