Fucoxanthin and Rosmarinic Acid Combination Has Anti-Inflammatory Effects through Regulation of NLRP3 Inflammasome in UVB-Exposed HaCaT Keratinocytes


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
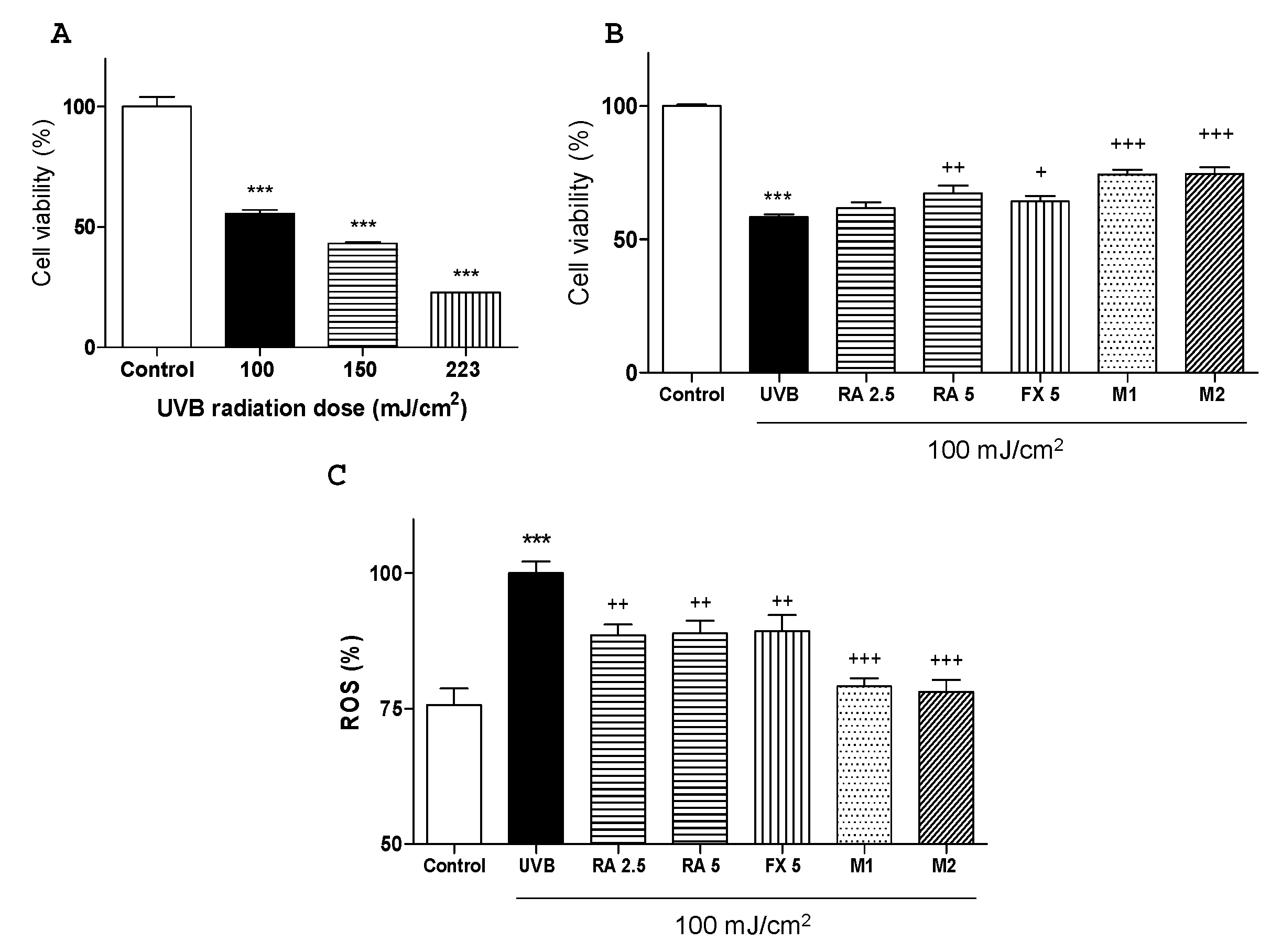
2.1. Effects of RA and FX on Cell Viability
2.2. Effect of RA and FX on UVB-induced ROS Production.
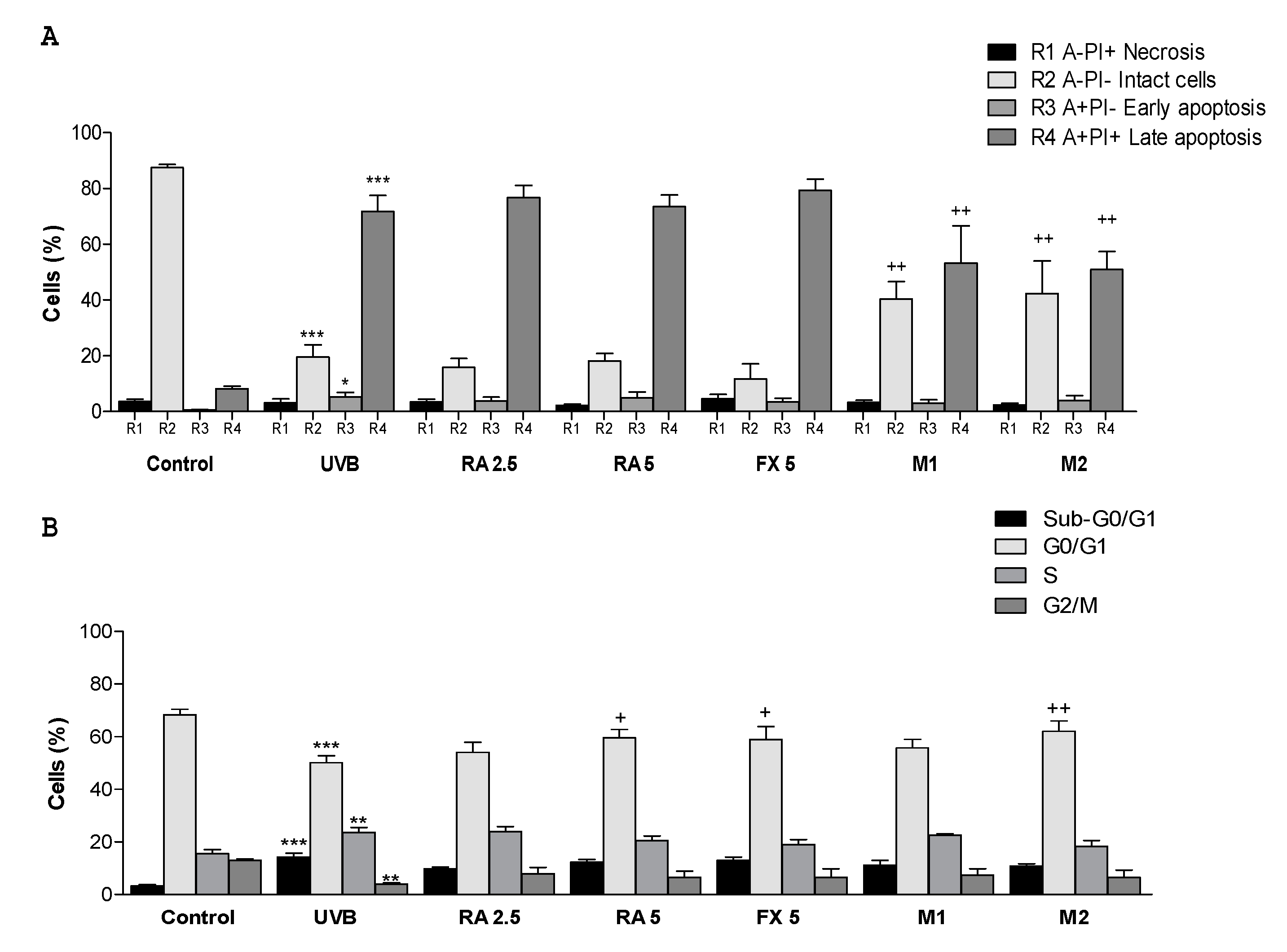
2.3. Effect of RA and FX on Apoptosis
2.4. Effect of RA and FX on Cell Cycle
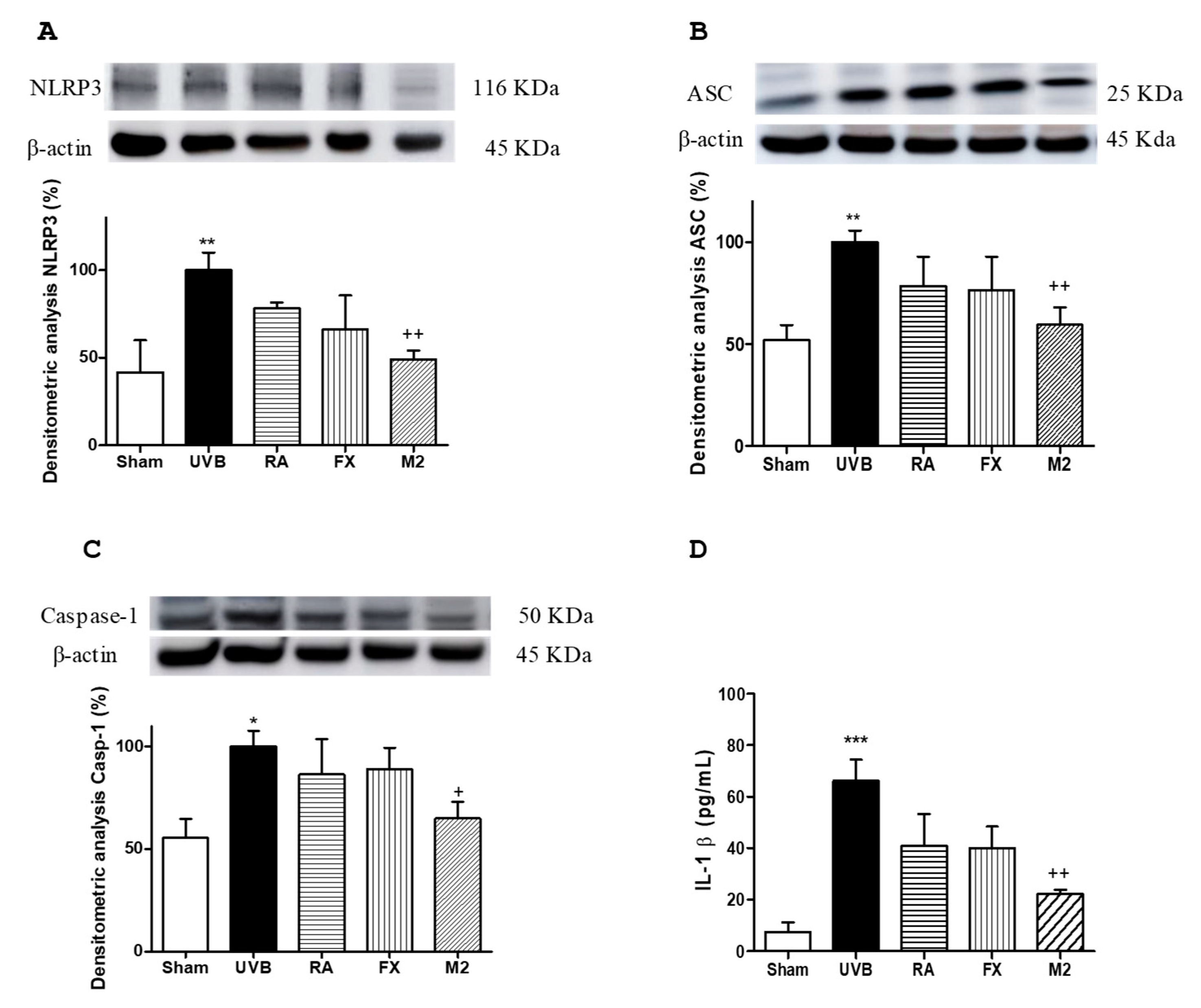
2.5. Effect of RA and FX on Inflammasome Regulation
2.6. Effect of RA and FX on Nrf-2 Antioxidant Signaling Pathway
3. Discussion
4. Materials and Methods
4.1. Compounds
4.2. Cell Line
4.3. UVB-Irradiation
4.4. Cell Proliferation Assay
4.5. Intracellular ROS Scavenging Activity
4.6. Apoptosis Determination
4.7. Cell Cycle Determination
4.8. Determination of IL-1β Production
4.9. Western Blot Analysis
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ASC | apoptosis-attached-speck-like protein containing a CARD |
| DCFH-DA | 2’,7’-dichlorodihydrofluorescein diacetate |
| FX | Fucoxanthin |
| HO-1 | heme oxygenase 1 |
| IL | Interleukin |
| NMSC | non-melanoma skin cancer |
| NLRP3 | nucleotide-binding domain, leucine-rich-repeat-containing family, pyrin domain- containing 3 |
| Nrf2 | nuclear factor E2-related factor 2 |
| ROS | reactive oxygen species |
| SRB | sulforhodamine B |
| TNF-α | tumour necrosis factor alpha |
| TPA | 12-O-tetradecanoylphorbol-13-acetate |
| UV | ultraviolet |
References
- Alam, S.; Pal, A.; Singh, D.; Ansari, K.M. Topical Application of Nexrutine Inhibits UVB-induced Cutaneous Inflammatory Responses in SKH-1 Hairless Mouse. Photodermatol. Photoimmunol. Photomed. 2018, 34, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Katiyar, S.K.; Pal, H.C.; Prasad, R. Dietary Proanthocyanidins Prevent Ultraviolet Radiation-Induced Non-Melanoma Skin Cancer Through Enhanced Repair of Damaged DNA-Dependent Activation of Immune Sensitivity. Semin. Cancer Biol. 2017, 46, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, M.; Fukunaga, A.; Washio, K.; Taguchi, K.; Oda, Y.; Ogura, K.; Nishigori, C. Anti-Inflammatory Role of Langerhans Cells and Apoptotic Keratinocytes in Ultraviolet-B-Induced Cutaneous Inflammation. J. Immunol. 2017, 199, 2937–2947. [Google Scholar] [CrossRef] [PubMed]
- Subedi, L.; Lee, T.H.; Wahedi, H.M.; Baek, S.H.; Kim, S.Y. Resveratrol-Enriched Rice Attenuates UVB-ROS-Induced Skin Aging Via Downregulation of Inflammatory Cascades. Oxid. Med. Cell. Longev. 2017, 2017, 8379539. [Google Scholar] [CrossRef] [PubMed]
- Kondo, S.; Sauder, D.N.; Kono, T.; Galley, K.A.; McKenzie, R.C. Differential Modulation of Interleukin-1 Alpha (IL-1α) and Interleukin-1 beta (IL-1β) in Human Epidermal Keratinocytes by UVB. Exp. Dermatol. 1994, 3, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Feldmeyer, L.; Keller, M.; Niklaus, G.; Hohl, D.; Werner, S.; Beer, H.D. The Inflammasome Mediates UVB-Induced Activation and Secretion of Interleukin-1β by Keratinocytes. Curr. Biol. 2007, 17, 1140–1145. [Google Scholar] [CrossRef]
- Hasegawa, T.; Nakashima, M.; Suzuki, Y. Nuclear DNA Damage-Triggered NLRP3 Inflammasome Activation Promotes UVB-Induced Inflammatory Responses in Human Keratinocytes. Biochem. Biophys. Res. Commun. 2016, 477, 329–335. [Google Scholar] [CrossRef]
- Ahmad, I.; Muneer, K.M.; Chang, M.E.; Nasr, H.M.; Clay, J.M.; Huang, C.C.; Yusuf, N. Ultraviolet Radiation-Induced Downregulation of SERCA2 Mediates Activation of NLRP3 Inflammasome in Basal Cell Carcinoma. Photochem. Photobiol. 2017, 93, 1025–1033. [Google Scholar] [CrossRef]
- Huang, C.F.; Chen, L.; Li, Y.C.; Wu, L.; Yu, G.T.; Zhang, W.F.; Sun, Z.J. NLRP3 Inflammasome Activation Promotes Inflammation-Induced Carcinogenesis in Head and Neck Squamous Cell Carcinoma. J. Exp. Clin. Cancer Res. 2017, 36, 116. [Google Scholar] [CrossRef]
- Zink, A.; Koch, E.; Seifert, F.; Rotter, M.; Spinner, C.D.; Biedermann, T. Non-Melanoma Skin Cancer in Mountain Guides: High Prevalence and Lack of Awareness Warrant Development of Evidence-Based Prevention Tools. Swiss Med. Wkly. 2016, 146, w14380. [Google Scholar]
- Talero, E.; García-mauriño, S.; Ávila-román, J.; Rodríguez-Luna, A.; Alcaide, A.; Motilva, V. Bioactive Compounds Isolated from Microalgae in Chronic Inflammation and Cancer. Mar. Drugs 2015, 13, 6152–6209. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Kim, J.H.; Park, J.G.; Yi, Y.S.; Park, K.W.; Rho, H.S.; Lee, M.S.; Yoo, J.W.; Kang, S.H.; Hong, Y.D.; et al. Radical Scavenging Activity-Based and AP-1-Targeted Anti-Inflammatory Effects of Lutein in Macrophage-Like and Skin Keratinocytic Cells. Mediat. Inflamm. 2013, 2013, 787042. [Google Scholar] [CrossRef] [PubMed]
- Yoshihisa, Y.; Andoh, T.; Matsunaga, K.; Rehman, M.U.; Maoka, T.; Shimizu, T. Efficacy of Astaxanthin for the Treatment of Atopic Dermatitis in a Murine Model. PLoS ONE 2016, 11, e0152288. [Google Scholar] [CrossRef] [PubMed]
- Amicucci, A.; Barbieri, E.; Sparvoli, V.; Gioacchini, A.M.; Calcabrini, C.; Palma, F.; Stocchi, V.; Zambonelli, A. Microbial and Pigment Profile of the Reddish Patch Occurring within Tuber Magnatum Ascomata. Fungal Biol. 2018, 122, 1134–1141. [Google Scholar] [CrossRef] [PubMed]
- D’Orazio, N.; Gemello, E.; Gammone, M.A.; de Girolamo, M.; Ficoneri, C.; Riccioni, G. Fucoxantin: A Treasure from the Sea. Mar. Drugs 2012, 10, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Saric, S.; Sivamani, R. Polyphenols and Sunburn. Int. J. Mol. Sci. 2016, 17, 1521. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M. Rosmarinic Acid: New Aspects. Phytochem. Rev. 2013, 12, 207–227. [Google Scholar] [CrossRef]
- Wang, J.; Pan, X.; Han, Y.; Guo, D.; Guo, Q.; Li, R. Rosmarinic Acid from Eelgrass Shows Nematicidal and Antibacterial Activities Against Pine Wood Nematode and Its Carrying Bacteria. Mar. Drugs 2012, 10, 2729–2740. [Google Scholar] [CrossRef]
- Guan, C.; Parrot, D.; Wiese, J.; Sönnichsen, F.D.; Saha, M.; Tasdemir, D.; Weinberger, F. Identification of Rosmarinic Acid and Sulfated Flavonoids as Inhibitors of Microfouling on the Surface of Eelgrass Zostera Marina. Biofouling 2017, 33, 867–880. [Google Scholar] [CrossRef]
- Amoah, S.K.S.; Sandjo, L.P.; Kratz, J.M.; Biavatti, M.W. Rosmarinic Acid-Pharmaceutical and Clinical Aspects. Planta Med. 2016, 82, 388–406. [Google Scholar] [CrossRef]
- Zhou, M.W.; Jiang, R.H.; Kim, K.D.; Lee, J.H.; Kim, C.D.; Yin, W.T.; Lee, J.H. Rosmarinic Acid Inhibits Poly(I:C)-Induced Inflammatory Reaction of Epidermal Keratinocytes. Life Sci. 2016, 155, 189–194. [Google Scholar] [CrossRef]
- Wei, Y.; Chen, J.; Hu, Y.; Lu, W.; Zhang, X.; Wang, R.; Chu, K. Rosmarinic Acid Mitigates Lipopolysaccharide-Induced Neuroinflammatory Responses through the Inhibition of TLR4 and CD14 Expression and NF-κB and NLRP3 Inflammasome Activation. Inflammation 2018, 41, 732–740. [Google Scholar] [CrossRef]
- Yao, Y.; Mao, J.; Xu, S.; Zhao, L.; Long, L.; Chen, L.; Li, D.; Lu, S. Rosmarinic Acid Inhibits Nicotine-Induced C-Reactive Protein Generation by Inhibiting NLRP3 Inflammasome Activation in Smooth Muscle Cells. J. Cell. Physiol. 2019, 234, 1758–1767. [Google Scholar] [CrossRef]
- Kovacs, D.; Raffa, S.; Flori, E.; Aspite, N.; Briganti, S.; Cardinali, G.; Torrisi, M.R.; Picardo, M. Keratinocyte Growth Factor Down-Regulates Intracellular ROS Production Induced by UVB. J. Dermatol. Sci. 2009, 54, 106–113. [Google Scholar] [CrossRef]
- Feehan, R.P.; Shantz, L.M. Molecular Signaling Cascades Involved in Nonmelanoma Skin Carcinogenesis. Biochem. J. 2016, 473, 2973–2994. [Google Scholar] [CrossRef]
- Ascenso, A.; Pedrosa, T.; Pinho, S.; Pinho, F.; de Oliveira, J.M.; Cabral-Marques, H.; Oliveira, H.; Simões, S.; Santos, C. The Effect of Lycopene Preexposure on UV-B-Irradiated Human Keratinocytes. Oxid. Med. Cell. Longev. 2016, 2016, 8214631. [Google Scholar] [CrossRef]
- Lee, J.J.; Kim, K.B.; Heo, J.; Cho, D.H.; Kim, H.S.; Han, S.H.; Ahn, K.J.; An, I.S.; An, S.; Bae, S. Protective Effect of Arthrospira platensis Extracts Against Ultraviolet B-Induced Cellular Senescence Through Inhibition of DNA Damage and Matrix Metalloproteinase-1 Expression in Human Dermal Fibroblasts. J. Photochem. Photobiol. B Biol. 2017, 173, 196–203. [Google Scholar] [CrossRef]
- Furue, M.; Uchi, H.; Mitoma, C.; Hashimoto-Hachiya, A.; Chiba, T.; Ito, T.; Nakahara, T.; Tsuji, G. Antioxidants for Healthy Skin: The Emerging Role of Aryl Hydrocarbon Receptors and Nuclear Factor-Erythroid 2-Related Factor-2. Nutrients 2017, 9, 223. [Google Scholar] [CrossRef]
- Sajo, M.E.J.; Kim, C.S.; Kim, S.K.; Shim, K.Y.; Kang, T.Y.; Lee, K.J. Antioxidant and Anti-Inflammatory Effects of Shungite Against Ultraviolet B Irradiation-Induced Skin Damage in Hairless Mice. Oxid. Med. Cell. Longev. 2017, 2017, 7340143. [Google Scholar] [CrossRef]
- Chen, L.; Hu, J.Y.; Wang, S.Q. The Role of Antioxidants in Photoprotection: A Critical Review. J. Am. Acad. Dermatol. 2012, 67, 1013–1024. [Google Scholar] [CrossRef]
- Psotova, J.; Svobodova, A.; Kolarova, H.; Walterova, D. Photoprotective Properties of Prunella vulgaris and Rosmarinic Acid on Human Keratinocytes. J. Photochem. Photobiol. B Biol. 2006, 84, 167–174. [Google Scholar] [CrossRef]
- Dreher, F.; Gabard, B.; Schwindt, D.; Maibach, H.I. Topical Melatonin in Combination with Vitamins E and C Protects Skin from Ultraviolet-Induced Erythema: A Human Study In Vivo. Br. J. Dermatol. 1998, 139, 332–339. [Google Scholar] [CrossRef]
- Leerach, N.; Yakaew, S.; Phimnuan, P.; Soimee, W.; Nakyai, W.; Luangbudnark, W.; Viyoch, J. Effect of Thai Banana (Musa AA group) in Reducing Accumulation of Oxidation End Products in UVB-Irradiated Mouse Skin. J. Photochem. Photobiol. B Biol. 2017, 168, 50–58. [Google Scholar] [CrossRef]
- Heo, S.J.; Jeon, Y.J. Protective Effect of Fucoxanthin Isolated from Sargassum Siliquastrum on UV-B Induced Cell Damage. J. Photochem. Photobiol. B Biol. 2009, 95, 101–107. [Google Scholar] [CrossRef]
- Zheng, J.; Piao, M.J.; Keum, Y.S.; Kim, H.S.; Hyun, J.W. Fucoxanthin Protects Cultured Human Keratinocytes Against Oxidative Stress by Blocking free Radicals and Inhibiting Apoptosis. Biomol. Ther. (Seoul) 2013, 21, 270–276. [Google Scholar] [CrossRef]
- Vostálová, J.; Zdařilová, A.; Svobodová, A. Prunella Vulgaris Extract and Rosmarinic Acid Prevent UVB-Induced DNA Damage and Oxidative Stress in HaCaT Keratinocytes. Arch. Dermatol. Res. 2010, 302, 171–181. [Google Scholar] [CrossRef]
- Pavey, S.; Russell, T.; Gabrielli, B. G2 Phase Cell Cycle Arrest in Human Skin Following UV Irradiation. Oncogene 2001, 20, 6103–6110. [Google Scholar] [CrossRef]
- Jang, Y.; Lee, A.Y.; Jeong, S.H.; Park, K.H.; Paik, M.K.; Cho, N.J.; Kim, J.E.; Cho, M.H. Chlorpyrifos Induces NLRP3 Inflammasome and Pyroptosis/Apoptosis Via Mitochondrial Oxidative Stress in Human Keratinocyte HaCaT Cells. Toxicology 2015, 338, 37–46. [Google Scholar] [CrossRef]
- Drexler, S.K.; Bonsignore, L.; Masin, M.; Tardivel, A.; Jackstadt, R.; Hermeking, H. Tissue-Specific Opposing Functions of the Inflammasome Adaptor ASC in the Regulation of Epithelial Skin Carcinogenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 18384–18389. [Google Scholar] [CrossRef]
- Lin, C.; Zhang, J. Inflammasomes in Inflammation-Induced Cancer. Front. Immunol. 2017, 8, 271. [Google Scholar] [CrossRef]
- Choi, J.H.; Kim, N.H.; Kim, S.J.; Lee, H.J.; Kim, S. Fucoxanthin Inhibits the Inflammation Response in Paw Edema Model Through Suppressing MAPKs, Akt, and NFκB. J. Biochem. Mol. Toxicol. 2016, 30, 111–119. [Google Scholar] [CrossRef]
- Tan, C.; Hou, Y. First Evidence for the Anti-Inflammatory Activity of Fucoxanthin in High-Fat-Diet-Induced Obesity in Mice and the Antioxidant Functions in PC12 Cells. Inflammation 2014, 37, 443–450. [Google Scholar] [CrossRef]
- Gong, D.; Chu, W.; Jiang, L.; Geng, C.; Li, J.; Ishikawa, N.; Kajima, K.; Zhong, L. Effect of Fucoxanthin Alone and in Combination with D-Glucosamine Hydrochloride on Carrageenan/Kaolin-Induced Experimental Arthritis in Rats. Phyther. Res. 2014, 28, 1054–1063. [Google Scholar] [CrossRef]
- Rodríguez-Luna, A.; Ávila-Román, J.; González-Rodríguez, M.L.; Cózar, M.J.; Rabasco, A.M.; Motilva, V.; Talero, E. Fucoxanthin-Containing Cream Prevents Epidermal Hyperplasia and UVB-Induced Skin Erythema in Mice. Mar. Drugs 2018, 16, 378. [Google Scholar] [CrossRef]
- Lee, J.; Jung, E.; Kim, Y.; Lee, J.; Park, J.; Hong, S.; Hyun, C.G.; Park, D.; Kim, Y.S. Rosmarinic Acid as a Downstream Inhibitor of IKK-Beta in TNF-Alpha-Induced Upregulation of CCL11 and CCR3. Br. J. Pharmacol. 2006, 148, 366–375. [Google Scholar]
- Oh, H.A.; Park, C.S.; Ahn, H.J.; Park, Y.S.; Kim, H.M. Effect of Perilla Frutescens Var. Acuta Kudo and Rosmarinic Acid on Allergic Inflammatory Reactions. Exp. Biol. Med. (Maywood) 2011, 236, 99–106. [Google Scholar] [CrossRef]
- Jeong, H.J.; Choi, Y.; Kim, M.H.; Kang, I.C.; Lee, J.H.; Park, C.; Park, R.; Kim, H.M. Rosmarinic Acid, Active Component of Dansam-Eum Attenuates Ototoxicity of Cochlear Hair Cells Through Blockage of Caspase-1 Activity. PLoS ONE 2011, 6, e18815. [Google Scholar] [CrossRef]
- Zheng, J.; Piao, M.J.; Kim, K.C.; Yao, C.W.; Cha, J.W.; Hyun, J.W. Fucoxanthin Enhances the Level of Reduced Glutathione Via the Nrf2-Mediated Pathway in Human Keratinocytes. Mar. Drugs 2014, 12, 4214–4230. [Google Scholar] [CrossRef]
- Liu, Y.; Zheng, J.; Zhang, Y.; Wang, Z.; Yang, Y.; Bai, M.; Dai, Y. Fucoxanthin Activates Apoptosis Via Inhibition of PI3K/Akt/mTOR Pathway and Suppresses Invasion and Migration by Restriction of p38-MMP-2/9 Pathway in Human Glioblastoma Cells. Neurochem. Res. 2016, 41, 2728–2751. [Google Scholar] [CrossRef]
- Urikura, I.; Sugawara, T.; Hirata, T. Protective Effect of Fucoxanthin Against UVB-Induced Skin Photoaging in Hairless Mice. Biosci. Biotechnol. Biochem. 2011, 75, 757–760. [Google Scholar] [CrossRef]
- Shimoda, H.; Tanaka, J.; Shan, S.J.; Maoka, T. Anti-Pigmentary Activity of Fucoxanthin and Its Influence on Skin mRNA Expression of Melanogenic Molecules. J. Pharm. Pharmacol. 2010, 62, 1137–1145. [Google Scholar] [CrossRef]
- Matsui, M.; Tanaka, K.; Higashiguchi, N.; Okawa, H.; Yamada, Y.; Tanaka, K.; Taira, S.; Aoyama, T.; Takanishi, M.; Natsume, C.; et al. Protective and Therapeutic Effects of Fucoxanthin Against Sunburn Caused by UV Irradiation. J. Pharmacol. Sci. 2016, 132, 55–64. [Google Scholar] [CrossRef]
- Sun, Z.; Park, S.Y.; Hwang, E.; Park, B.; Seo, S.A.; Cho, J.G.; Zhang, M.; Yi, T.H. Dietary Foeniculum Vulgare Mill Extract Attenuated UVB Irradiation-Induced Skin Photoaging by Activating of Nrf2 and Inhibiting MAPK Pathways. Phytomedicine 2016, 23, 1273–1284. [Google Scholar] [CrossRef]
- Sun, Z.; Park, S.Y.; Hwang, E.; Zhang, M.; Seo, S.A.; Lin, P.; Yi, T.H. Thymus Vulgaris Alleviates UVB Irradiation Induced Skin Damage Via Inhibition of MAPK/AP-1 and Activation of Nrf2-ARE Antioxidant System. J. Cell. Mol. Med. 2016, 21, 336–348. [Google Scholar] [CrossRef]
- Lu, C.; Zou, Y.; Liu, Y.; Niu, Y. Rosmarinic Acid Counteracts Activation of Hepatic Stellate Cells Via Inhibiting the ROS-Dependent MMP-2 Activity: Involvement of Nrf2 Antioxidant System. Toxicol. Appl. Pharmacol. 2017, 318, 69–78. [Google Scholar] [CrossRef]
- Twentyman, P.R.; Luscombe, M. A Study of Some Variables in a Tetrazolium Dye (MTT) Based Assay for Cell Growth and Chemosensitivity. Br. J. Cancer 1987, 56, 279–285. [Google Scholar] [CrossRef]
- Wang, H.; Joseph, J.A. Quantifying Cellular Oxidative Stress by Dichlorofluorescein Assay Using Microplate Reader. Free Radic. Biol. Med. 1999, 27, 612–616. [Google Scholar] [CrossRef]
- Bradford, M. A Rapid and Sensitive Method for The Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Luna, A.; Ávila-Román, J.; Oliveira, H.; Motilva, V.; Talero, E. Fucoxanthin and Rosmarinic Acid Combination Has Anti-Inflammatory Effects through Regulation of NLRP3 Inflammasome in UVB-Exposed HaCaT Keratinocytes. Mar. Drugs 2019, 17, 451. https://doi.org/10.3390/md17080451
Rodríguez-Luna A, Ávila-Román J, Oliveira H, Motilva V, Talero E. Fucoxanthin and Rosmarinic Acid Combination Has Anti-Inflammatory Effects through Regulation of NLRP3 Inflammasome in UVB-Exposed HaCaT Keratinocytes. Marine Drugs. 2019; 17(8):451. https://doi.org/10.3390/md17080451
Chicago/Turabian StyleRodríguez-Luna, Azahara, Javier Ávila-Román, Helena Oliveira, Virginia Motilva, and Elena Talero. 2019. "Fucoxanthin and Rosmarinic Acid Combination Has Anti-Inflammatory Effects through Regulation of NLRP3 Inflammasome in UVB-Exposed HaCaT Keratinocytes" Marine Drugs 17, no. 8: 451. https://doi.org/10.3390/md17080451
APA StyleRodríguez-Luna, A., Ávila-Román, J., Oliveira, H., Motilva, V., & Talero, E. (2019). Fucoxanthin and Rosmarinic Acid Combination Has Anti-Inflammatory Effects through Regulation of NLRP3 Inflammasome in UVB-Exposed HaCaT Keratinocytes. Marine Drugs, 17(8), 451. https://doi.org/10.3390/md17080451