New Alkaloids and Polyketides from the Marine Sponge-Derived Fungus Penicillium sp. SCSIO41015

 ,
,  ,
,  ,
, 
Abstract
1. Introduction
2. Results and Discussion
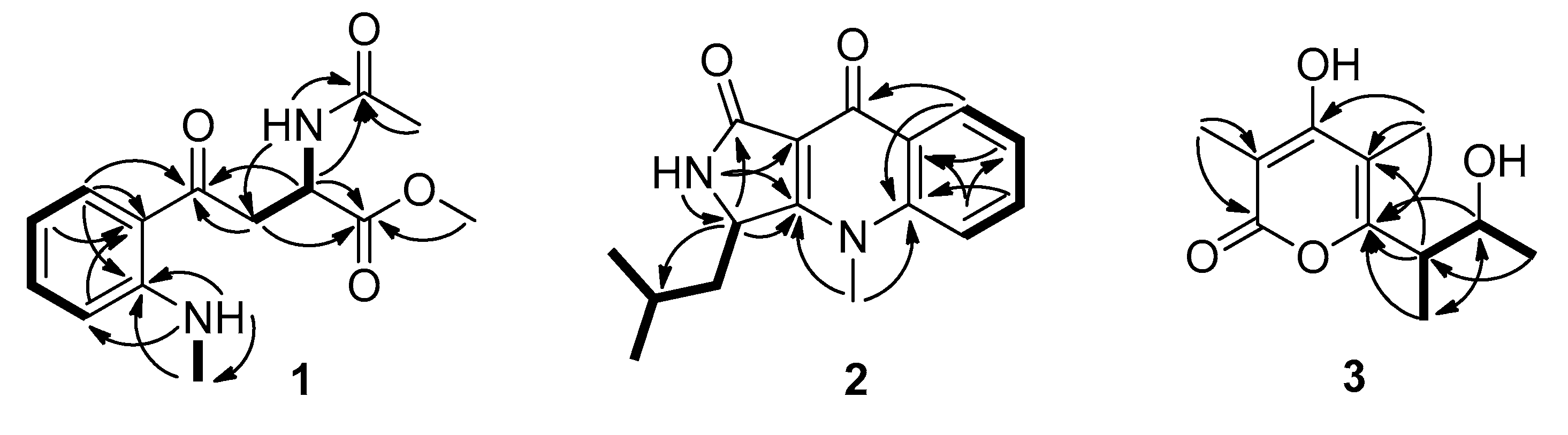
2.1. Isolation and Structural Elucidation
2.2. Biological Activity
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Fermentation and Extraction
3.4. Isolation and Purification
3.5. Spectral Data
3.6. X-ray Crystal Structure Analysis
3.7. Antibacterial Activity Assay
3.8. Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jin, L.M.; Quan, C.S.; Hou, X.Y.; Fan, S.D. Potential pharmacological resources: Natural bioactive compounds from marine-derived fungi. Mar. Drugs 2016, 14, 76. [Google Scholar] [CrossRef] [PubMed]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2019, 36, 122–173. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Su, M.Z.; Song, S.J.; Jung, J.H. Marine-derived Penicillium species as producers of cytotoxic metabolites. Mar. Drugs 2017, 15, 329. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.G.; Liu, Q.; Zhu, G.L.; Liu, H.S.; Zhu, W.M. Marine natural products sourced from marine-derived Penicillium fungi. J. Asian Nat. Prod. Res. 2016, 18, 92–115. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.Y.; Lin, X.P.; Yang, J.; Zhou, X.F.; Yang, B.; Wang, J.F.; Liu, Y.H. Spiro-phthalides and isocoumarins isolated from the marine-sponge-derived fungus Setosphaeria sp. SCSIO41009. J. Nat. Prod. 2018, 81, 1860–1868. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.Y.; Lin, X.P.; Wang, P.; Zhou, X.F.; Yang, B.; Wang, J.F.; Liu, Y.H. Perylenequione derivatives with anticancer activities isolated from the marine sponge-derived fungus, Alternaria sp. SCSIO41014. Mar. Drugs 2018, 16, 280. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.Y.; Lin, X.P.; Wang, J.; Liang, R.; Tian, Y.Q.; Salendra, L.; Luo, X.W.; Zhou, X.F.; Yang, B.; Tu, Z.C.; et al. Three new highly oxygenated sterols and one new dihydroisocoumarin from the marine sponge-derived fungus Cladosporium sp. SCSIO41007. Steroids 2018, 129, 41–46. [Google Scholar] [CrossRef] [PubMed]
- King, R.R.; Calhoun, L.A. Synthesis and NMR characteristics of N-acetyl-4-nitro, N-acetyl-5-nitro, N-acetyl-6-nitro and N-acetyl-7-nitrotryptophan methyl esters. Magn. Reson. Chem. 2009, 47, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Heiss, C.; Anderson, J.; Phillips, R.S. Differential effects of bromination on substrates and inhibitors of kynureninase from Pseudomonas fluorescens. Org. Biomol. Chem. 2003, 1, 288–295. [Google Scholar] [CrossRef]
- Clark, B.; Capon, R.J.; Lacey, E.; Tennant, S.; Gill, J.H. Quinolactacins revisited: From lactams to imide and beyond. Org. Biomol. Chem. 2006, 4, 1512–1519. [Google Scholar] [CrossRef]
- Sasaki, T.; Takahashi, S.; Uchida, K.; Funayama, S.; Kainosho, M.; Nakagawa, A. Biosynthesis of Quinolactacin A, a TNF production inhibitor. J. Antibiot. 2006, 59, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.Q.; Sun, J.; Gong, Q.H.; Wang, Y.; Fu, P.; Zhu, W.M. New alpha-pyridones with quorum-sensing inhibitory activity from diversity-enhanced extracts of a Streptomyces sp. derived from marine algae. J. Agric. Food Chem. 2018, 66, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.M.; Qi, S.H.; Yin, H.; Gao, C.H.; Zhang, S. Alkaloids from the stem bark of Micromelum falcatum. Chem. Pharm. Bull. 2009, 57, 600–602. [Google Scholar] [CrossRef] [PubMed]
- Goddard, M.L.; Mottier, N.; Jeanneret-Gris, J.; Christen, D.; Tabacchi, R.; Abou-Mansour, E. Differential production of phytotoxins from Phomopsis sp. from grapevine plants showing esca symptoms. J. Agric. Food Chem. 2014, 62, 8602–8607. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.Q.; Zhu, G.L.; Liu, H.S.; Jiang, G.L.; Wang, Y.; Zhu, W.M. Diversity and function of the Antarctic krill microorganisms from Euphausia superba. Sci. Rep. 2016, 6, 36496. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Li, A. Total syntheses and structural revision of alpha- and beta-diversonolic esters and total syntheses of diversonol and blennolide C. Angew. Chem. Int. Ed. Engl. 2008, 47, 6579–6582. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Zhang, X.Y.; Dong, J.J.; Xu, X.Y.; Nong, X.H.; Qi, S.H. Cyclopentane-condensed chromones from marine-derived fungus Penicillium oxalicum. Chem. Lett. 2014, 43, 837–839. [Google Scholar] [CrossRef]
- Xin, Z.H.; Tian, L.; Zhu, T.J.; Wang, W.L.; Du, L.; Fang, Y.C.; Gu, Q.Q.; Zhu, W.M. Isocoumarin derivatives from the sea squirt-derived fungus Penicillium stoloniferum QY2-10 and the halotolerant fungus Penicillium notatum B-52. Arch. Pharm. Res. 2007, 30, 816–819. [Google Scholar] [CrossRef]
- Xu, L.X.; Xue, J.H.; Xu, H.H.; Liu, X.Z.; Ma, W.Z.; Wei, X.Y. Three new isochromans from the mycelial culture of a Cylindrocarpon fungus. Heterocycles 2006, 68, 1955–1959. [Google Scholar]
- Ronsberg, D.; Debbab, A.; Mandi, A.; Vasylyeva, V.; Bohler, P.; Stork, B.; Engelke, L.; Hamacher, A.; Sawadogo, R.; Diederich, M.; et al. Pro-apoptotic and immunostimulatory tetrahydroxanthone dimers from the endophytic fungus Phomopsis longicolla. J. Org. Chem. 2013, 78, 12409–12425. [Google Scholar] [CrossRef]
- Meselhy, M.R. Constituents from moghat, the roots of Glossostemon bruguieri (Desf.). Molecules 2003, 8, 614–621. [Google Scholar] [CrossRef]
- Chen, L.; Huang, K.; Zhong, P.; Hu, X.; Fang, Z.X.; Wu, J.L.; Zhang, Q.Q. Tumonoic acids K and L, novel metabolites from the marine-derived fungus Penicillium citrinum. Heterocycles 2012, 85, 413–419. [Google Scholar] [CrossRef]
- Han, Z.; Mei, W.L.; Zhao, Y.X.; Deng, Y.Y.; Dai, H.F. A new cytotoxic isocoumarin from endophytic fungus Penicillium sp. 091402 of the mangrove plant Bruguiera sexangula. Chem. Nat. Compd. 2009, 45, 805–807. [Google Scholar] [CrossRef]
- Chen, L.L.; Wang, P.; Wang, H.; Gai, C.J.; Guo, Z.K.; Dai, H.F.; Mei, W.L. Studyon the secondary metabolites from the endophytic fungus Coriolopsis sp. J5 of Ceriops tagal and their biological activities. Chin. J. Mar. Drugs 2016, 35, 7–12. [Google Scholar]
- Wang, M.L.; Lu, C.H.; Xu, Q.Y.; Song, S.Y.; Hu, Z.Y.; Zheng, Z.H. Four new citrinin derivatives from a marine-derived Penicillium sp. fungal strain. Molecules 2013, 18, 5723–5735. [Google Scholar] [CrossRef]
- Amagata, T.; Amagata, A.; Tenney, K.; Valeriote, F.A.; Lobkovsky, E.; Clardy, J.; Crews, P. Unusual C25 steroids produced by a sponge-derived Penicillium citrinum. Org. Lett. 2003, 5, 4393–4396. [Google Scholar] [CrossRef]
- Wang, J.F.; Wei, X.Y.; Qin, X.C.; Tian, X.P.; Liao, L.; Li, K.M.; Zhou, X.F.; Yang, X.W.; Wang, F.Z.; Zhang, T.Y.; et al. Antiviral merosesquiterpenoids produced by the antarctic fungus Aspergillus ochraceopetaliformis SCSIO 05702. J. Nat. Prod. 2016, 79, 59–65. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| No. | 1 a | No. | 2 a | No. | 3 b | |||
|---|---|---|---|---|---|---|---|---|
| δC | δH | δC | δH | δC | δH | |||
| 1 | 116.3 C | 1 | 167.9 C | 2 | 168.6 C | |||
| 2 | 151.4 C | 3 | 52.5 CH | 4.86 brd (8.4) | 3 | 98.9 C | ||
| 3 | 111.4 CH | 6.74 d (8.4) | 3a | 165.6 C | 4 | 168.0 C | ||
| 4 | 135.4 CH | 7.43 td (7.7, 0.7) | 4a | 141.2 C | 5 | 110.0 C | ||
| 5 | 114.0 CH | 6.60 td (7.7, 0.7) | 5 | 117.0 CH | 7.82 m | 6 | 161.6 C | |
| 6 | 131.9 CH | 7.81 dd (7.7, 1.4) | 6 | 132.6 CH | 7.82 m | 7 | 8.9 CH3 | 1.93 s |
| 7 | 198.1 C | 7 | 124.3 CH | 7.49 ddd (7.7, 4.9, 2.8) | 8 | 10.0 CH3 | 2.01 s | |
| 8a 8b | 40.2 CH2 | 3.44 dd (17.5, 7.0) 3.40 dd (17.5, 5.6) | 8 | 125.9 CH | 8.26 d (7.7) | 9 | 43.8 CH | 2.95 qui (7.0) |
| 9 | 47.9 CH | 4.76 td (7.0, 5.6) | 8a | 128.1 C | 10 | 70.5 CH | 3.94 qui (6.5) | |
| 10 | 172.2 C | 9 | 171.8 C | 11 | 21.2 CH3 | 1.27 d (6.5) | ||
| 11 | 29.0 CH3 | 2.85 d (4.9) | 9a | 109.3 C | 12 | 14.8 CH3 | 1.17 d (7.0) | |
| 12 | 169.2 C | 10 | 42.3 CH2 | 1.79–1.88 m 1.37 ddd (12.6, 9.1, 2.8) | ||||
| 13 | 22.3 CH3 | 1.82 s | 11 | 24.7 CH | 1.79–1.88 m | |||
| 14 | 52.0 CH3 | 3.61 s | 12 | 23.5 CH3 | 0.83 d (6.3) | |||
| NH-9 | 8.23 d (7.7) | 13 | 21.5 CH3 | 1.01 d (6.3) | ||||
| NH-11 | 8.59 dd (9.1, 4.2) | 14 | 35.8 CH3 | 3.79 s | ||||
| NH | 8.27 m | |||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pang, X.; Cai, G.; Lin, X.; Salendra, L.; Zhou, X.; Yang, B.; Wang, J.; Wang, J.; Xu, S.; Liu, Y. New Alkaloids and Polyketides from the Marine Sponge-Derived Fungus Penicillium sp. SCSIO41015. Mar. Drugs 2019, 17, 398. https://doi.org/10.3390/md17070398
Pang X, Cai G, Lin X, Salendra L, Zhou X, Yang B, Wang J, Wang J, Xu S, Liu Y. New Alkaloids and Polyketides from the Marine Sponge-Derived Fungus Penicillium sp. SCSIO41015. Marine Drugs. 2019; 17(7):398. https://doi.org/10.3390/md17070398
Chicago/Turabian StylePang, Xiaoyan, Guodi Cai, Xiuping Lin, Limbadri Salendra, Xuefeng Zhou, Bin Yang, Junjian Wang, Junfeng Wang, Shihai Xu, and Yonghong Liu. 2019. "New Alkaloids and Polyketides from the Marine Sponge-Derived Fungus Penicillium sp. SCSIO41015" Marine Drugs 17, no. 7: 398. https://doi.org/10.3390/md17070398
APA StylePang, X., Cai, G., Lin, X., Salendra, L., Zhou, X., Yang, B., Wang, J., Wang, J., Xu, S., & Liu, Y. (2019). New Alkaloids and Polyketides from the Marine Sponge-Derived Fungus Penicillium sp. SCSIO41015. Marine Drugs, 17(7), 398. https://doi.org/10.3390/md17070398