2.1. Structural Elucidation of the Glycosides
The concentrated ethanolic extract of
P. fabricii was re-extracted with СHCl
3/MeOH, concentrated, and delipidized with EtOAc/H
2O. The water layer was chromatographed on a Polychrom-1 (powdered Teflon, Biolar, Latvia) in 50% EtOH and on Si gel columns using CHCl
3/EtOH/H
2O (100:75:10), (100:100:17) and (100:125:25) as mobile phases to give fractions I–VIII. The obtained fractions III–VIII were subjected to HPLC on reversed-phase or silica-based columns to give psolusosides: B (
1) (67 mg), E (
2) (10 mg), F (
3) (1.4 mg), G (
4) (46.5 mg), H (
5) (1.4 mg), H
1 (
6) (1.4 mg), and I (
7) (1.1 mg) (
Figure 1) as well as two known earlier compounds, psolusoside A (36.5 mg) found earlier in this species of sea cucumbers [
10,
11] and colochiroside D (2.5 mg) isolated first from
Colochirus robustus [
16]. The known compounds were identified by comparison of their
1H and
13C NMR spectra with those reported for psolusoside A (3β-
O-[6-
O-sodium sulfate-3-
O-methyl-β-
d-glucopyranosyl-(1→3)-6-
O-sodium-sulfate-β-
d-glucopyranosyl-(1→4)-β-
d-quinovopyranosyl-(1→2)-β-
d-xylopyranosyl]-16-ketoholosta-9(11),25-diene) and colochiroside D (3β-
O-[3-
O-methyl-β-
d-glucopyranosyl-(1→3)-6-
O-sodium-sulfate-β-
d-glucopyranosyl-(1→4)-β-
d-glucopyranosyl-(1→2)-β-
d-xylopyranosyl]-16-ketoholosta-9(11),25-diene).
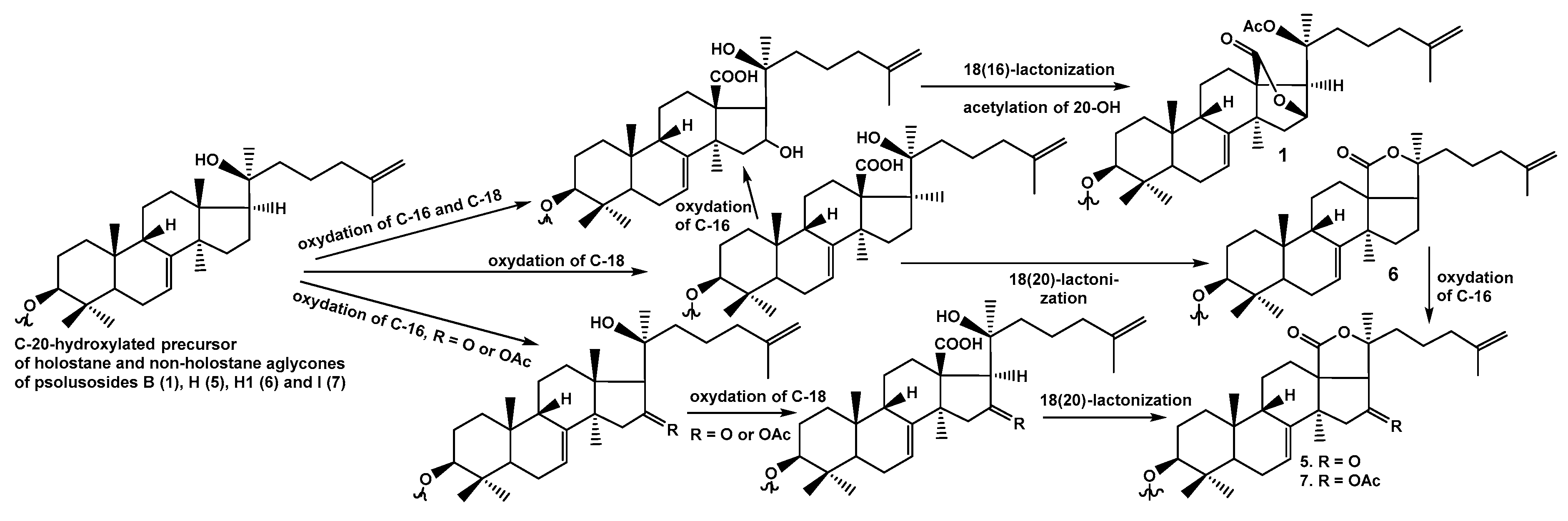
The structure of psolusoside B assigned earlier [
12,
13] was shown to be monosulfated branched tetraoside with non-holostane aglycone, namely 3β-
O-{β-
d-glucopyranosyl-(1→4)-β-
d-glucopyranosyl-(1→2)-[6-
O-sodium-sulfate-β-
d-glucopyranosyl-(1→4)]-β-
d-xylopyranosyl}-9βH,20(
S)-acetoxylanosta-7,25-diene-18(16)-lactone.
However, the reinvestigation has shown that this glycoside has two sulfate groups instead of the one reported earlier. In fact, the more accurate molecular formula of psolusoside B (
1) was determined to be C
55H
84O
30S
2Na
2 from the [M
2Na + Na]
+ ion peak at
m/z 1357.4169 (calc. 1357.4176) and [M
2Na + 2Na]
2+ at
m/z 690.2039 (calc. 690.2034) in the (+)HR-ESI-MS and indicated the presence of two sulfate groups in
1. The comparison of 1D and 2D NMR spectra of the aglycone part of psolusoside B (
1) (
Table 1,
Figures S1–S8) with those of the aglycone part of colochiroside E, isolated from the sea cucumber
Colochirus robustus [
17], has confirmed their identity with the aglycone of psolusoside B elucidated earlier [
12]. Thus, psolusoside B (
1), isolated by us, actually contains non-holostane aglycone with 18(16)-lactone and
O-acetic group at C-20, which was described earlier as onekotanogenin.
In the
1H and
13C NMR spectra (
Table 2,
Figures S1 and S2) of the carbohydrate part of
1, four characteristic doublets at δ (H) 4.56–5.11 (
J = 7.3–7.9 Hz) and, corresponding to them, four signals of anomeric carbons at δ(C) 100.9–104.8 (
Figure S4) were indicative of a tetrasaccharide chain and
β-configurations of glycosidic bonds. The
1H,
1H-COSY and 1D TOCSY spectra of
1 showed the signals of the isolated spin systems assigned to one xylose and three glucose residues (
Figures S3, S7 and S8). The positions of interglycosidic linkages were established by the ROESY and HMBC spectra of
1 (
Table 2,
Figures S5 and S6) where the cross-peaks between H(1) of the xylose and H(3) (C(3)) of an aglycone, H(1) of the second residue (glucose) and H(2) (C(2)) of the xylose, H(1) of the third residue (glucose) and H(4) (C(4)) of the second residue (glucose), and H(1) of the fourth residue (glucose) and H-4 (C(4)) of the first residue (xylose), were observed. These data indicated the same architecture (tetrasaccharide branched chain) and monosaccharide composition of sugar chain of
1 as it has been reported earlier [
13]. Thorough analysis of the NMR spectra of
1 showed the glucose residue (the third sugar unit, in which signals were deduced by
1H,
1H-COSY, and confirmed by 1D TOCSY) attached to C(4) of the second sugar unit (glucose) was sulfated by C(6) due to α- and β-shifting effects observed in the
13C NMR spectrum. Really, the signal of C(6) was observed at δ(C) 67.5 and the signal of C(5) at δ(C) 75.5. Hence, these signals were shifted in comparison with corresponding signals (δC
(6) at 62.1, δC
(5) at 77.8) in non-sulfated glucose residue in the same position of carbohydrate chains of psolusosides, belonging to the group D [
15]. The analogous shifting effects were observed for the signals C(2) and C(1) of the glucose occupying the fourth position of the carbohydrate chain of
1 and attached to C(4) of the xylose unit. The signal of C(2) of this residue was observed at δ 80.6 due to the attachment of a sulfate group to this position while the signal of C(1) was shifted upfield to δ(C) 100.9 due to β-effect of a sulfate group. Moreover, the signals in the
13C NMR spectrum of
1 assigning to this glucose residue were coincident with the corresponding signals of glucose residue sulfated by C-2 and attached to C-4 of the first xylose unit in the spectrum of colochiroside E [
17] corroborating the unusual position of one of sulfate groups in psolusoside B (
1). So, the both spectroscopic methods—HR-ESI-MS and NMR—confirmed the presence of two sulfate groups in the carbohydrate chain of psolusoside B (
1).
The structure of 1 was also confirmed by the (+)ESI-MS/MS of the [M2Na + Na]+ ion at m/z 1357.4, in which the peaks of fragment ions were observed at m/z 1297.4 [M2Na + Na − CH3COOH]+, 1237.4 [M2Na + Na − NaHSO4]+, 1177.4 [M2Na + Na − CH3COOH − NaHSO4]+, 1117.4 [M2Na + Na − 2NaHSO4]+, 1075.5 [M2Na + Na − C6H10O9SNa (GlcSO3Na)]+, 913.4 [M2Na + Na − GlcSO3Na − Glc]+, 863.1 [M2Na + Na − C32H47O4 (Agl) + H]+, 743.1 [M2Na + Na − C32H47O4 (Agl) − NaHSO4]+, 581.1 [M2Na + Na − C32H47O4 (Agl) − C6H10O9SNa (GlcSO3Na)]+, 449.0 [M2Na + Na − C32H47O4 (Agl) − C6H10O9SNa (GlcSO3Na) − Xyl]+, 287.0 [M2Na + Na − C32H47O4 (Agl) − C6H10O9SNa (GlcSO3Na) − Xyl − Glc]+.
Based on these results, the structure of psolusoside B (1) was determined as 3β-O-{6-O-sodium sulfate-β-d-glucopyranosyl-(1→4)-β-d-glucopyranosyl-(1→2)-[2-O-sodium sulfate-β-d-glucopyranosyl-(1→4)]-β-d-xylopyranosyl}-9βH,20(S)-acetoxylanosta-7,25-diene-18(16)-lactone.
Colochiroside E [
17], having trisaccharide sugar chain with terminal (glucose) residue sulfated by C(2), differed from psolusoside B (
1) only by the lack of a terminal glucose residue attached to C(4) of the glucose (the second unit in the chain). This fact indicates the biogenetic interconnection of these compounds: colochiroside E seems to be a biosynthetic precursor of psolusoside B (
1) that, additionally, corroborates the new structure of
1. The incorrect structure elucidation of psolusoside B in 1989 [
13] could be explained by an ambiguity of interpretation of the
13C NMR signals. The use of FAB-MS for the molecular formula calculation obviously resulted in the desulfation of the glycoside during the spectrum registration.
The
1H and
13C NMR spectra of aglycone parts of psolusosides E (
2), F (
3), and G (
4) were coincident to each other showing the identity of the aglycones in these glycosides. In the aglycone part of the
13C NMR spectra of
2–
4, the signals characteristic of 18(20)-lactone (δ(C) 175.8 C(18) and 82.9 (C(20)), 9(11)- (δ(C) 151.2 C(9) and 110.9 C(11)), and 25(26)-double bonds (δ(C) 145.4 C(25) and 110.3 C(26)), as well as the signal of C-16 keto-group (δ(C) 212.9) were observed (
Table 3,
Figures S9, S17 and S25). Based on the analysis of the NMR spectra, the aglycone of compounds
2–
4 was identified as earlier known 16-ketoholosta-9(11),25-dien-3β-ol, found first in holotoxins A
1 and B
1 from the sea cucumbers belonging to the family Stichopodidae [
18], and frequently occurred in the glycosides of different sea cucumber taxa.
The molecular formula of psolusoside E (
2) was determined to be C
54H
83O
25SNa from the [M
Na − Na]
− ion peak at
m/z 1163.4945 (calc. 1163.4950) in the (−)HR-ESI-MS. In the
1H and
13C NMR spectra of the carbohydrate part of psolusoside E (
2), four characteristic doublets at δ(H) 4.77–5.22 (
J = 7.3–7.7 Hz) and, corresponding to them, signals of anomeric carbons at δ(C) 104.6–105.4 were indicative of a tetrasaccharide chain and β-configurations of glycosidic bonds (
Table 4,
Figures S9, S10 and S12). The
1H,
1H-COSY, and 1D TOCSY spectra of
2 showed the signals of four isolated spin systems assigned to the xylose, quinovose, glucose, and 3-
O-methylglucose residues (
Figures S11, S15 and S16). The positions of interglycosidic linkages were elucidated by the ROESY and HMBC spectra of
2 (
Table 4,
Figures S13 and S14) by same manner as for
1, indicating the presence of linear tetrasaccharide chain in psolusoside E (
2). The signals of C(6) and C(5) of the glucose residue (the third sugar), observed at δ(C) 67.6 and 75.0, correspondingly, were characteristic of the sulfated by C(6) glucopyranose residue. Thus, psolusoside E (
2) is a monosulfated tetraoside, with the glucose residue, sulfated by C(6), as the third monosaccharide unit. Such carbohydrate chain was not found earlier in the glycosides from sea cucumbers.
The (−)ESI-MS/MS of 2 demonstrated the fragmentation of [MNa − Na]− ion at m/z 1163.5. The peaks of fragment ions were observed at m/z: 987.4 [MNa − Na − MeGlc + H]−, 695.2 [MNa − Na − C30H43O4 (Agl) − H]−, 563.1 [MNa − Na − C30H43O4 (Agl) − Xyl]−, 417.1 [MNa − Na − C30H43O4 (Agl) − Xyl − Qui]−, 241.0 [MNa − Na − C30H43O4 (Agl) − Xyl − Qui − MeGlc]− (corresponds to desodiated sulfated glucose residue) corroborating the structure of psolusoside E (2).
All these data indicate that psolusoside E (2) is 3β-O-[3-O-methyl-β-d-glucopyranosyl-(1→3)-6-O-sodium-sulfate-β-d-glucopyranosyl-(1→4)-β-d-quinovopyranosyl-(1→2)-β-d-xylopyranosyl]-16-ketoholosta-9(11),25-diene.
The molecular formula of psolusoside F (
3) was determined to be C
54H
83O
25SNa from the [M
Na − Na]
− ion peak at
m/z 1163.4952 (calc. 1163.4950) in the (−)HR-ESI-MS and was coincident with the formula of psolusoside E (
2). In the
1H and
13C NMR spectra of the carbohydrate part of psolusoside F (
3), four characteristic doublets at δ(H) 4.71–5.12 (
J = 7.3–8.2 Hz) and corresponding signals of anomeric carbons at δ(C) 104.0–105.0 were indicative of a tetrasaccharide chain and
β-configurations of glycosidic bonds (
Figures S17, S18 and S20). The positions of interglycosidic linkages were elucidated by the ROESY and HMBC spectra of
3 (
Table 5,
Figures S21 and S22) as described above, indicating the presence of linear tetrasaccharide carbohydrate chain. The monosaccharide composition of
3, deduced from the
1H,
1H-COSY, and 1D TOCSY spectra (
Figures S19, S23 and S24), was the same as in
2. The comparison of the
13C NMR spectra of these compounds showed the coincidence of their signals corresponding to xylose and quinovose residues. The signals of C(6) and C(5) of the glucose residue (the third unit in the chain) in the
13C NMR spectrum of
3 were observed at δ(C) 61.7 (shielded as compared with corresponding signal in the spectrum of
2) and 77.1 (de-shielded as compared with C(5) of the glucose in the spectrum of
2), correspondingly, indicating the absence of a sulfate group in this residue. The signal of C(6) of 3-
O-methyl-glucose residue was observed at δ(C) 67.0 and the signal C(5) of the same residue—at δ(C) 75.6 in the
13C NMR spectrum of
3 indicating the attachment of a sulfate group to C(6) of terminal 3-
O-methyl-glucose unit in the carbohydrate chain of psolusoside F (
3). So, psolusosides E (
2) and F (
3) differed from each other only in the position of a sulfate group. The carbohydrate chain of
3 is a new one.
The (−)ESI-MS/MS of 3 demonstrated the fragmentation of [MNa − Na]− ion at m/z 1163.5. The peaks of fragment ions were observed at m/z: 695.2 [MNa − Na − C30H43O4 (Agl) − H]−, 563.1 [MNa − Na − C30H43O4 (Agl) − Xyl]−, 417.1 [MNa − Na − C30H43O4 (Agl) − Xyl − Qui]−, 255.0 [MNa − Na – C30H43O4 (Agl) – Xyl – Qui – Glc]−, confirming the sequence of monosaccharides in the sugar chain.
All these data indicate that psolusoside F (3) is 3β-O-[6-O-sodium-sulfate-3-O-methyl-β-d-glucopyranosyl-(1→3)-β-d-glucopyranosyl-(1→4)-β-d-quinovopyranosyl-(1→2)-β-d-xylopyranosyl]-16-ketoholosta-9(11),25-diene.
The molecular formula of psolusoside G (
4) was determined to be C
54H
82O
29S
2Na
2 from the [M
2Na − Na]
− ion peak at
m/z 1281.4313 (calc. 1281.4286) in the (−)HR-ESI-MS indicating the presence of two sulfate groups in this glycoside. In the
1H and
13C NMR spectra of the carbohydrate part of psolusoside G (
4), four characteristic doublets at δ(H) 4.72–5.16 (
J = 7.2–8.4 Hz) and, corresponding to them, signals of anomeric carbons at δ(C) 103.8–105.0 were indicative of a tetrasaccharide chain and β-configurations of glycosidic bonds (
Table 6,
Figures S25, S26 and S28).
Analysis of the
1H,
1H-COSY and 1D TOCSY spectra of psolusoside G (
4) showed the availability of one xylose, two glucose, and one 3-O-methyl-glucose residues (
Figures S27, S31 and S32). So, the quinovose unit was absent in the chain of
4 that was corroborated by the lack of characteristic doublet of methyl group of quinovose residue at δ(H) ≈1.70 in the
1H NMR spectrum and the corresponding signal at δ(C) ≈18.0 in the
13C NMR spectrum of
4. It was supposed that the second position of carbohydrate moiety was occupied by the glucose residue and confirmed by the appearance of the additional signal at δ(C) 61.2 corresponding to C(6) of a glucopyranose residue. Two signals at δ(C) 67.4 and 67.1 corresponding to sulfated hydroxy-methylene carbons of glucopyranose residues were observed in the
13C NMR spectrum of
4 indicating the presence of two sulfate groups. The positions of interglycosidic linkages and the consequence of monosaccharides in the chain of
4 were established by analysis of the ROESY and HMBC spectra (
Table 6,
Figures S29 and S30), indicating the presence of linear carbohydrate moiety with the glucose as second unit and sulfated glucose and 3-
O-methyl-glucose residues as third and terminal monosaccharides, correspondingly. The comparison of the
13C NMR spectrum of sugar part of psolusoside G (
4) with that of earlier known okhotoside B
3, isolated from
Cucumaria okhotensis [
19] showed the coincidence of their signals, suggesting the identity of the linear disulfated carbohydrate moieties.
The (−)ESI-MS/MS of 4 demonstrated the fragmentation of [M2Na − Na]− ion at m/z 1281.4. The peaks of fragment ions were observed at m/z: 1161.5 [M2Na − Na − HSO4Na]−, 1003.4 [M2Na − Na − HSO4Na − C7H12O8SNa (MeGlcSO3Na)]−, 813.2 [M2Na − Na − C30H43O4 (Agl) − H]−, 681.1 [M2Na − Na − C30H43O4 (Agl) − Xyl]−, 519.0 [M2Na − Na − C30H43O4 (Agl) − Xyl − Glc]−, 255.0 [M2Na − Na − C30H43O4 (Agl) − Xyl − Glc − C6H9O8SNa (GlcSO3Na)]−, corroborating the aglycone structure and consequence of monosaccharides in psolusoside G (4).
All these data indicate that psolusoside G (4) is 3β-O-[6-O-sodium-sulfate-3-O-methyl-β-d-glucopyranosyl-(1→3)-6-O-sodium-sulfate-β-d-glucopyranosyl-(1→4)-β-d-glucopyranosyl-(1→2)-β-d-xylopyranosyl]-16-ketoholosta-9(11),25-diene.
The sulfation of third or/and fourth monosaccharide residues in the carbohydrate chain when C(4) position of the first xylose residue is not sulfated as in psolusosides E (
2), F (
3) and G (
4) is probably characteristic structural feature of the glycosides of
Psolus fabricii. It was also observed in a disulfated tetraoside psolusoside A, with sulfate groups at C-6 of the third (glucose) and fourth (3-
O-methyl-glucose) residues. Monosulfated colochiroside D, isolated first from the sea cucumber
Colochirus robustus [
16] and later from
Psolus fabricii as well as the disulfated okhotoside B
3 from
Cucumaria okhotensis [
19], are the glycosides found in the sea cucumbers belonging to other genera, sharing the same structural peculiarity. However, the majority of known sulfated glycosides contain a sulfate group at C-4 of the first xylose residue.
The
1H and
13C NMR spectra of carbohydrate parts of psolusosides H (
5) and H
1 (
6) were coincident to each other indicating the identity of carbohydrate chains of these glycosides. The presence of three characteristic doublets at δ(H) 4.73 (
J = 7.5 Hz), 5.19 (
J = 6.8 Hz), and 4.88 (
J = 7.9 Hz) in the
1H NMR spectra of the carbohydrate chains of
5,
6 correlated with the HSQC spectra with the signals of anomeric carbons at δ(C) 104.9, 104.4, and 104.5, correspondingly, were indicative of a trisaccharide chain and β-configurations of glycosidic bonds (
Table 7,
Figures S33, S34, S36, S40 and S41, S43). The
1H,
1H-COSY, and 1D TOCSY spectra of
5 and
6 showed the signals of three isolated spin systems assigned to two glucose and one xylose residues (
Figures S35, S39 and S42). The positions of interglycosidic linkages established by the ROESY and HMBC spectra of
5 and
6 (
Table 7,
Figures S37, S38, S44 and S45) demonstrated cross-peaks between H(1) of the xylose and H(3) (C(3)) of an aglycone, H(1) of the glucose and H(2), (C(2)) of the xylose and H(1) of the terminal unit (glucose), and H(4) (C(4)) of the second unit (glucose). The terminal glucose unit was sulfated by C(6), which was deduced from character signal at δ(C) 67.2 in comparison with the analogous signal of C(6) of the glucose in the second position of the carbohydrate chain, which was observed at δ(C) 61.6. Therefore, the carbohydrate chain of psolusosides H (
5) and H
1 (
6) differed from that of psolusoside G (
4) in the loss of terminal 3-O-methyl-glucose residue. Actually, the signals in their
13C NMR spectra assigning to xylose and glucose (the second unit) residues were coincident. The signal of C(3) of terminal glucose in the spectra of
5,
6 was shifted up-field to δ(C) 76.9 due to the absence of the glycosylation effect that was observed in the spectrum of
4 (δ(C) 86.4 C(3) of terminal glucose). The carbohydrate chain of psolusosides H (
5) and H
1 (
6) has never been found earlier in the glycosides from sea cucumbers.
The molecular formula of psolusoside H (
5) was determined to be C
47H
71O
21SNa from the [M
Na−Na]
− ion peak at
m/z 1003.4213 (calc. 1003.4214) in the (−)HR-ESI-MS. The
1H and
13C NMR spectra of aglycone part of psolusoside H (
5) demonstrated the signals characteristic of the holostane-type aglycone (the signals of 18(20)-lactone at δ(C) 179.0 (C(18)) and 83.6 (C(20))) with 16-keto-group (the signals of C(16) at δ(C) 213.8, C(15) at δ(C) 51.8, and C(17) at δ(C) 63.3 with corresponding proton signals at δ(H) 2.65 (d,
J = 16.0 Hz, H(15)), and 2.32 (d,
J = 16.0 Hz, H(15)), as well as 2.87 (s, H(17)) (
Table 8,
Figures S33 and S34). The characteristic signals at δ(C) 121.7 (C(7)), 143.9 (C(8)), and at δ(H) 5.63 (m, H(7)) in the
13C and
1H NMR spectra of
5 were assigned to 7(8)-double bond in the polycyclic system. The availability of terminal double bond in the side chain of
5 was deduced from the signals at δ(C) 145.4 (C(25)) and 110.3 (C(26)) observed in the
13C NMR and two broad singlets at δ(H) 4.70 and 4.69 (H
2-26) in the
1H NMR spectra of psolusoside H (
5). So, the aglycone of psolusoside H (
5) is a positional isomer (by the double bond position in polycyclic nucleus) of the aglycone comprising psolusosides E (
2), F (
3), and G (
4). This aglycone was found earlier in the glycosides of sea cucumbers belonging to different orders:
Cucumaria japonica [
20,
21],
Pseudocolochirus violaceus [
22] (Cucumariidae, Dendrochirotida), and
Australostichopus mollis [
23] (Stichopodidae, Synallactida) [
24].
The (−)ESI-MS/MS of 5 demonstrated the fragmentation of [MNa − Na]− ion at m/z 1003.4. The peaks of fragment ions were observed at m/z: 535.1 [MNa − Na − C30H43O4 (Agl) − H]−, 403.1 [MNa − Na − C30H43O4 (Agl) − Xyl]−, 241.0 [MNa − Na − C30H43O4 (Agl) − Xyl − Glc]− corroborating the structure of psolusoside H (5).
All these data indicate that psolusoside H (5) is 3β-O-[6-O-sodium-sulfate-β-d-glucopyranosyl-(1→4)-β-d-glucopyranosyl-(1→2)-β-d-xylopyranosyl]-16-ketoholosta-7,25-diene.
The molecular formula of psolusoside H
1 (
6) was determined to be C
47H
73O
20SNa from the [M
Na − Na]
− ion peak at
m/z 989.4432 (calc. 989.4421) in the (−)HR-ESI-MS and [M
Na + Na]
+ at
m/z 1035.4205 (calc. 1035.4206) in the (+)HR-ESI-MS. In the
1H and
13C NMR spectra of the aglycone part of psolusoside H
1 (
6), the signals characteristic of the holostane-type aglycone (the signals of 18(20)-lactone at δ(C) 181.0 (C(18)) and 84.7 (C(20))) with 7(8)-double bond in the polycyclic system (the signals at δ(C) 119.8 (C(7)), 146.5 (C(8)) in the
13C NMR, and at δ(H) 5.62 (m, H(7)) in the
1H NMR) and terminal double bond in the side chain (the signals at δ(C) 145.5 (C(25)) and 110.6 (C(26)) in the
13C NMR and two broad singlets at δ(H) 4.78 and 4.74 (H
2-26) in the
1H NMR) were observed (
Table 9,
Figures S40 and S41). The analysis of
1H,
1H-COSY spectrum of
6 showed the protons H
2(15)/H
2(16)/H(17) form the isolated spin system (
Figure S42). The signals of C(15), C(16), and C(17) in the
13C NMR spectrum of
6 were observed at δ(C) 34.1, 24.5, and 52.9, correspondingly, and were shielded when compared with the signals C(15)–C(17) in the spectrum of psolusoside H (
5) due to the absence of 16-keto-group in psolusoside H
1 (
6). So, the aglycone of psolusoside H
1 (
6) differed from that of psolusoside H (
5) only in the lack of 16-keto-group. Such aglycone was earlier found in the glycosides from sea cucumbers of the order Dendrochirotida:
Colochirus robustus [
16] and
Cucumaria japonica [
20].
The (−)ESI-MS/MS of 6 demonstrated the fragmentation of [MNa − Na]− ion at m/z 989.4. The peaks of fragment ions analogous to those for 5 were observed at m/z: 535.1 [MNa − Na − C30H45O3 (Agl) − H]−, 403.1 [MNa − Na − C30H45O3 (Agl) − Xyl]−, 241.0 [MNa − Na − C30H45O3 (Agl) − Xyl − Glc]− corroborating the structure of psolusoside H1 (6).
All these data indicate that psolusoside H1 (6) is 3β-O-[6-O-sodium-sulfate-β-d-glucopyranosyl-(1→4)-β-d-glucopyranosyl-(1→2)-β-d-xylopyranosyl]-holosta-7,25-diene.
The molecular formula of psolusoside I (
7) was determined to be C
54H
82O
29S
2Na
2 from the [M
2Na − Na]
− ion peak at
m/z 1281.4267 (calc. 1281.4286) in the (−)HR-ESI-MS and [M
2Na + Na]
+ at
m/z 1327.4065 (calc. 1327.4071) in the (+)HR-ESI-MS indicating the presence of two sulfate groups. In the
1H and
13C NMR spectra (
Table 10,
Figures S46 and S47) of the carbohydrate part of
7 four characteristic doublets at δ(H) 4.63–4.82 (
J = 7.3–8.1 Hz) and corresponding to them four signals of anomeric carbons at δ(C) 103.9–105.6 were indicative of a tetrasaccharide chain and
β-configurations of glycosidic bonds (
Figure S50). The
1H,
1H-COSY and 1D TOCSY spectra of
7 showed the signals of four isolated spin systems assigned to two xylose and two glucose residues (
Figures S49, S53 and S54). The positions of interglycosidic linkages established by the ROESY and HMBC spectra of
7 (
Table 10,
Figures S51 and S52) indicated the branched architecture of tetrasaccharide chain when the fourth glucose residue is attached to C(4) of the first (xylose) residue.
The second sugar unit in the chain of psolusoside I (
7) is a xylose connected to the first xylose residue by β-(1→2)-glycosidic bond. This feature is very rare occurred in the holothurians glycoside’s carbohydrate moieties [
25]. The third monosaccharide in the chain is a glucose attached to C(4) of the second (xylose) unit, the fourth residue (glucose) is attached to C-4 of the first xylose unit. Both glucose residues are sulfated by C(6) that was deduced from two signals—at δ(C) 67.6 and 67.9 in the
13C NMR spectrum of
7—demonstrating α-shifting effect of a sulfate group. The tetrasaccharide branched disulfated carbohydrate moiety of psolusoside I (
7) with the xylose as the second unit has never been found among the sea cucumber glycosides.
The aglycone of psolusoside I (
7) shared some structural features with the aglycones of psolusosides H (
5) and H
1 (
6). In the
1H and
13C NMR spectra of the aglycone part of
7, the signals of holostane-type aglycone (C(18) at δ(C) 180.2 and C(20) at δ(C) 85.5) with 7(8)-double bond in the polycyclic system (the signals at δ(C) 120.2 (C(7)), 145.6 (C(8)), and at δ(H) 5.60 (m, H(7)) and terminal double bond in the side chain (the signals at δ(C) 145.4 (C(25)) and 110.8 (C(26)) in the
13C NMR and two broad singlets at δ(H) 4.72 and 4.73 (H
2-26) in the
1H NMR spectra) were observed (
Table 11,
Figures S46 and S47). An isolated spin system formed by the protons H
2(15)/H(16)/H(17) was deduced from the
1H,
1H-COSY spectrum (
Figure S49). The signal of H(16) was observed at δ(H) 5.82 (brq,
J = 8.6 Hz) and the corresponding signal of C(16) at δ(C) 75.2 indicated the presence of β-
O-acetic group. Actually, the additional signals corresponding to this group were observed in the
13C NMR spectrum of
7 at δ(C) 170.7 (carboxyl carbon) and 21.2 (methyl carbon). The holostane aglycone with 7(8)- and 25(26)-double bonds and 16β-acetoxy group frequently occurred in the glycosides of sea cucumbers [
2,
16,
19].
The (−)ESI-MS/MS of 7 demonstrated the fragmentation of [M2Na − Na]− ion at m/z 1281.4. The peaks of fragment ions were observed at m/z: 769.1 [M2Na − Na − C32H47O5 (Agl) − H]−, 505.2 [M2Na − Na − C32H47O5 (Agl) − C6H10O8SNa (GlcSO3Na)]−, 373.0 2 [M2Na − Na − C32H47O5 (Agl) − GlcSO3Na − Xyl]−, 241.0 2 [M2Na − Na − C32H47O5 (Agl) − GlcSO3Na − 2Xyl]−. The (+)ESI-MS/MS of 7 demonstrated the fragmentation of [M2Na + Na]+ ion at m/z 1327.4. The peaks of fragment ions were observed at m/z: 1207.5 [M2Na + Na − NaHSO4]+, 1147.4 [M2Na + Na − NaHSO4 − CH3COOH]+, 1063.4 [M2Na + Na − C6H10O8SNa (GlcSO3Na)]+, 1003.4 [M2Na + Na – GlcSO3Na – CH3COO]+, 931.4 [M2Na + Na – GlcSO3Na − Xyl + H]+, 833.1 [M2Na + Na − C32H47O5 (Agl) +H]+. All these data exhaustively confirmed the structure psolusoside I (7) deduced by analyses of NMR data.
All these data indicate that psolusoside I (7) is 3β-O-{6-O-sodium sulfate-β-d-glucopyranosyl-(1→4)-β-d-xylopyranosyl-(1→2)-[6-O-sodium sulfate-β-d-glucopyranosyl-(1→4)]-β-d-xylopyranosyl}-16β-acetoxyholosta-7,25-diene.



 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}