1. Introduction
Rhamnolipids belong to a class of biosurfactants composed of rhamnose linked to β-hydroxylated fatty acid chains [
1]. Rhamnolipids are classified as mono-rhamnolipids, which contain a single rhamnose molecule and di-rhamnolipids, which contain two rhamnose sugar rings [
2]. These biosurfactants are mainly produced by
Pseudomonas species [
3] such as
P. aeruginosa [
4],
P. chlororaphis,
P. plantarii,
P. putida, and
P. fluorescens [
5]. Over the past three decades, rhamnolipids have been broadly investigated and extensively reviewed due to their biodegradability and reduced toxicity compared to synthetic surfactants as well as their various applications [
6,
7,
8]. It has been widely recognized that rhamnolipids have surface active properties such as emulsification, dispersion, foaming, detergency, wetting and stabilization [
9]. Moreover, various researchers have demonstrated that rhamnolipids display low toxicity, antimicrobial activities and the ability to suppress the growth of breast cancer cells [
10,
11]. These unique and diverse properties make them suitable to be used in a wide range of industrial demands such as the bioremediation of pollutants, cosmetics, food, pharmaceuticals and therapeutics [
12].
The marine environment constitutes a significant reservoir of natural products which has offered the potential for new drug development over the last few decades [
13]. Specifically, marine microorganisms are considered efficient producers of lead compounds with biomedical potential [
14]. In addition, structurally diverse and impressive bioactive natural products have been identified from marine microbes [
15]. In our continuous search for secondary metabolites from marine-derived bacteria, the
Actinoalloteichus hymeniacidonis strain 179DD−027 was isolated from a deep-sea sediment sample collected off the coasts of Dokdo Island, East Sea, Republic of Korea. Dokdo Island is a large volcanic island with 89 small islets and rocks containing rich and well-preserved biodiversity [
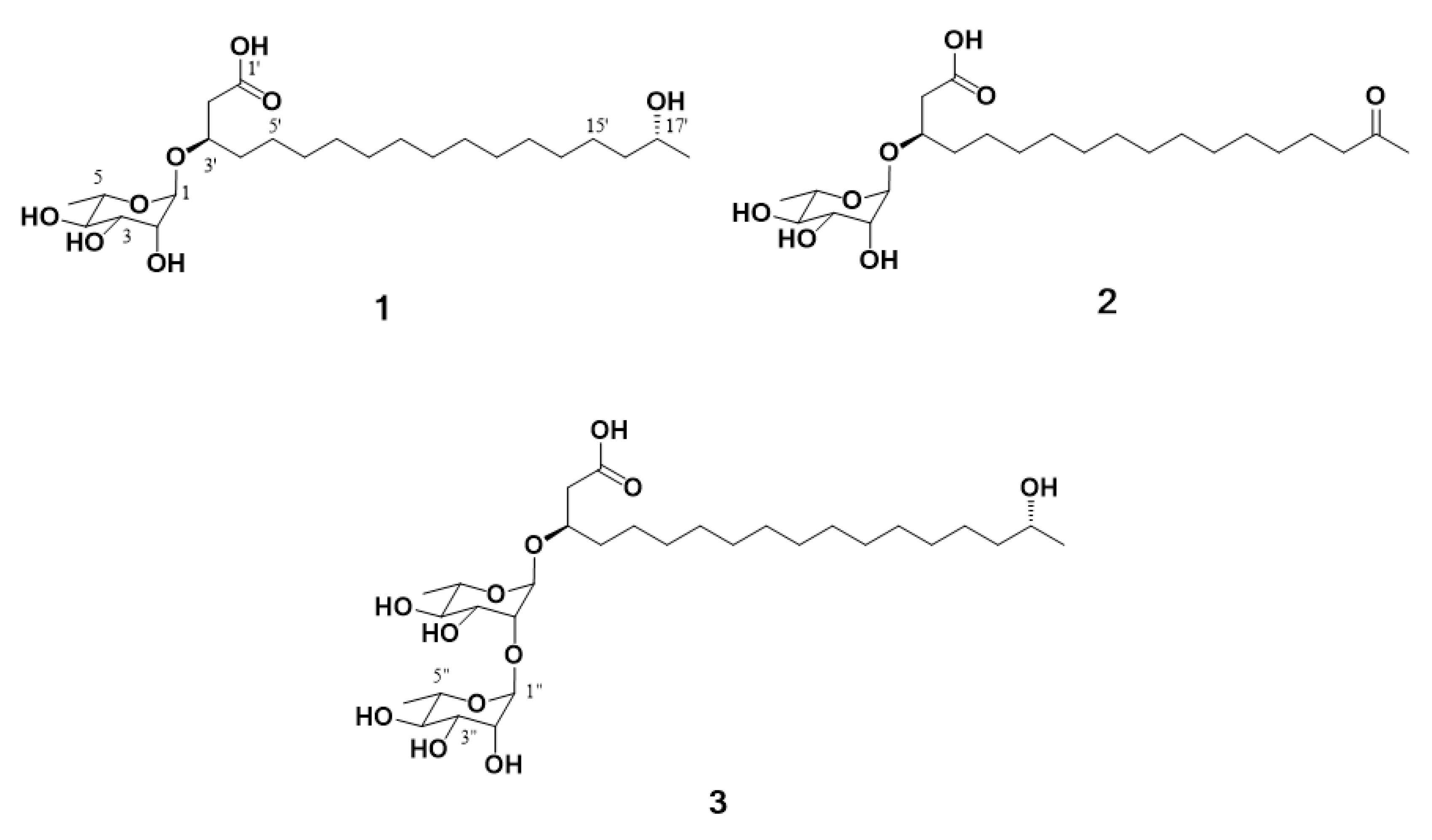
16]. Subsequent fermentation of the producing strain, solvent extraction and chemical investigation procedures led to the isolation of three new rhamnolipids, named dokdolipids A−C (
1−
3) (
Figure 1). Dokdolipids represent the first rhamnolipids containing a hydroxyl (
1 and
3) group and a ketone group (
2) in the side chains. In this paper, we describe the isolation, structure elucidation and bioactivities of dokdolipids A−C (
1−
3).
2. Results and Discussion
Compound
1 was obtained as a dark brown oil and gave a [M + Na]
+ ion peak at
m/z 485.3094 (calcd 485.3090) in the HRESIMS, consistent with a molecular formula C
24H
46O
8. The
1H NMR spectrum of
1 showed the signals of seven oxygenated methines (
δH 4.80, 4.08, 3.75, 3.70, 3.65, 3.60 and 3.35), 14 methylene protons (
δH 2.49, 1.55 and 12 overlapped protons at 1.29–1.45) and two methyls (
δH 1.23 and 1.13). The
1H and
13C NMR data, in conjunction with HSQC of
1, supported the presence of 24 carbons, which can be classified as one carbonyl (
δC 173.9), seven sp
3 methines (
δC 98.9, 75.2, 72.5, 71.2, 70.9, 68.7 and 67.1), 14 sp
3 methylene (
δC 40.0–24.5) and two sp
3 methyl (
δC 22.0 and 16.2) carbon (
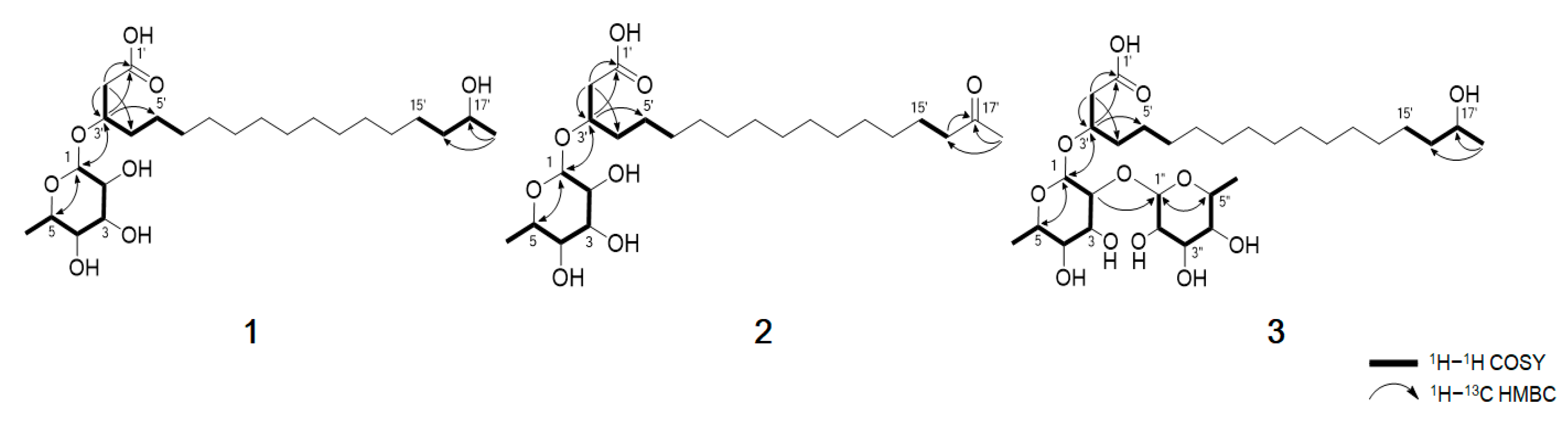
Table 1). The planar structure of compound
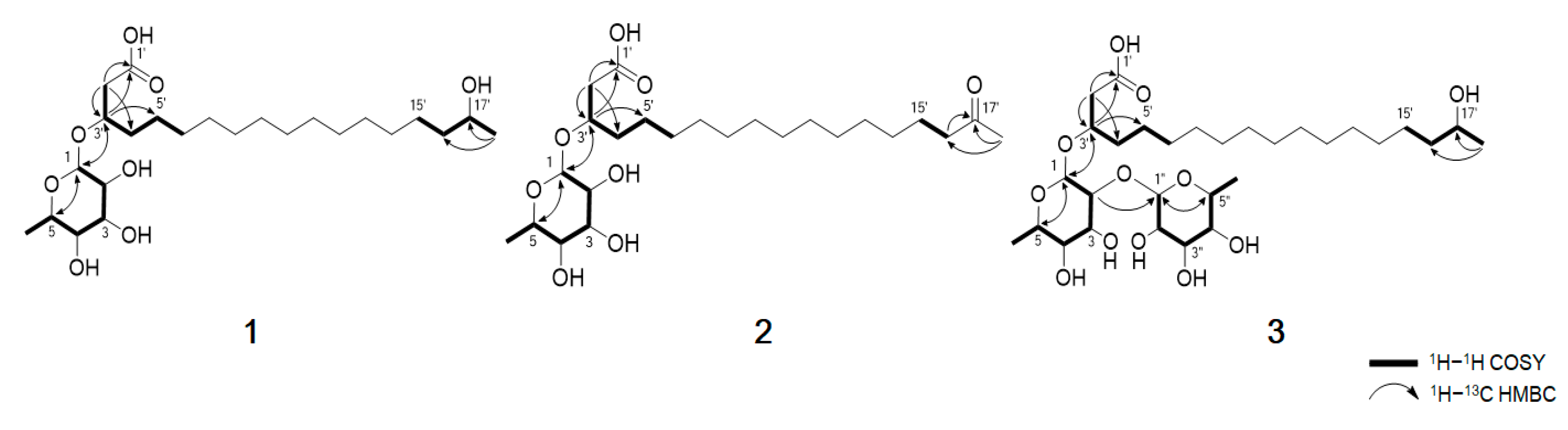
1 was elucidated by analyzing the 2D NMR data, including the
1H–
1H COSY and
1H–
13C HMBC spectra (
Figure 2). The COSY correlation from H
2-2′ to highly overlapped proton signals, a terminal methyl group and methylene carbons at
δC 24.5-29.4 suggested the presence of an aliphatic chain. The HMBC correlations from H
2-2′ (
δH 2.48, 2.53) to C-1′ (
δC 173.9), C-3′ (
δC 74.2) and C-4′ (
δC 33.1) and from H-3′ (
δH 4.08) to C-1′ (
δC 173.9), C-2′ (
δC 40.0) and C-5′ (
δC 24.5) established the position of the carbonyl carbon C-1′ at
δC 173.9 and the secondary alcohol H-3′ at
δH 4.08. In addition, the chemical shift value of H-17′ (
δH 3.70) and the HMBC correlation from the methyl doublet H
3-18′ (
δH 1.13, d,
J = 6.2 Hz) to C-17′ (
δC 67.1) and C-16′ (
δC 38.8) indicated that a hydroxyl group was attached to C-17′. Detailed analysis of the 2D NMR spectra revealed the presence of a linear hydroxylated and saturated fatty acid as a 3, 17-dihydroxyoctadecanoic acid. Another spin system was identified from the H-1/H-2/H-3/H-4/H-5/H
3-6 COSY correlations. A hexose moiety was confirmed by H-1/C-5 and H-5/C-1 HMBC correlations. The hexose ring was connected to C-3′ through an ether linkage which was confirmed by the H-3′/C-1 and H-1/C-3′ HMBC correlations. Thus, the planar structure of dokdolipid A (
1) was elucidated as a new rhamnolipid.
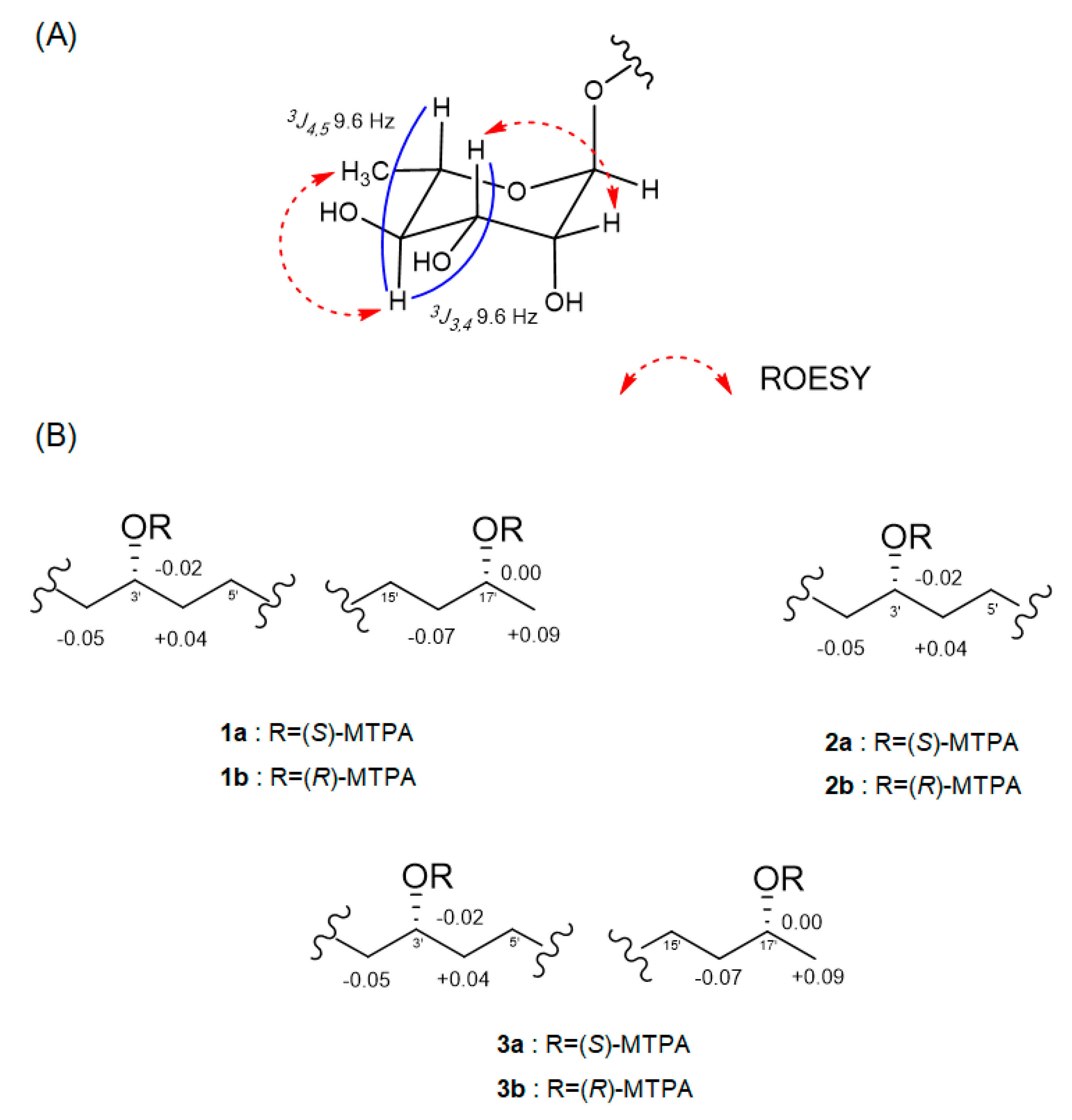
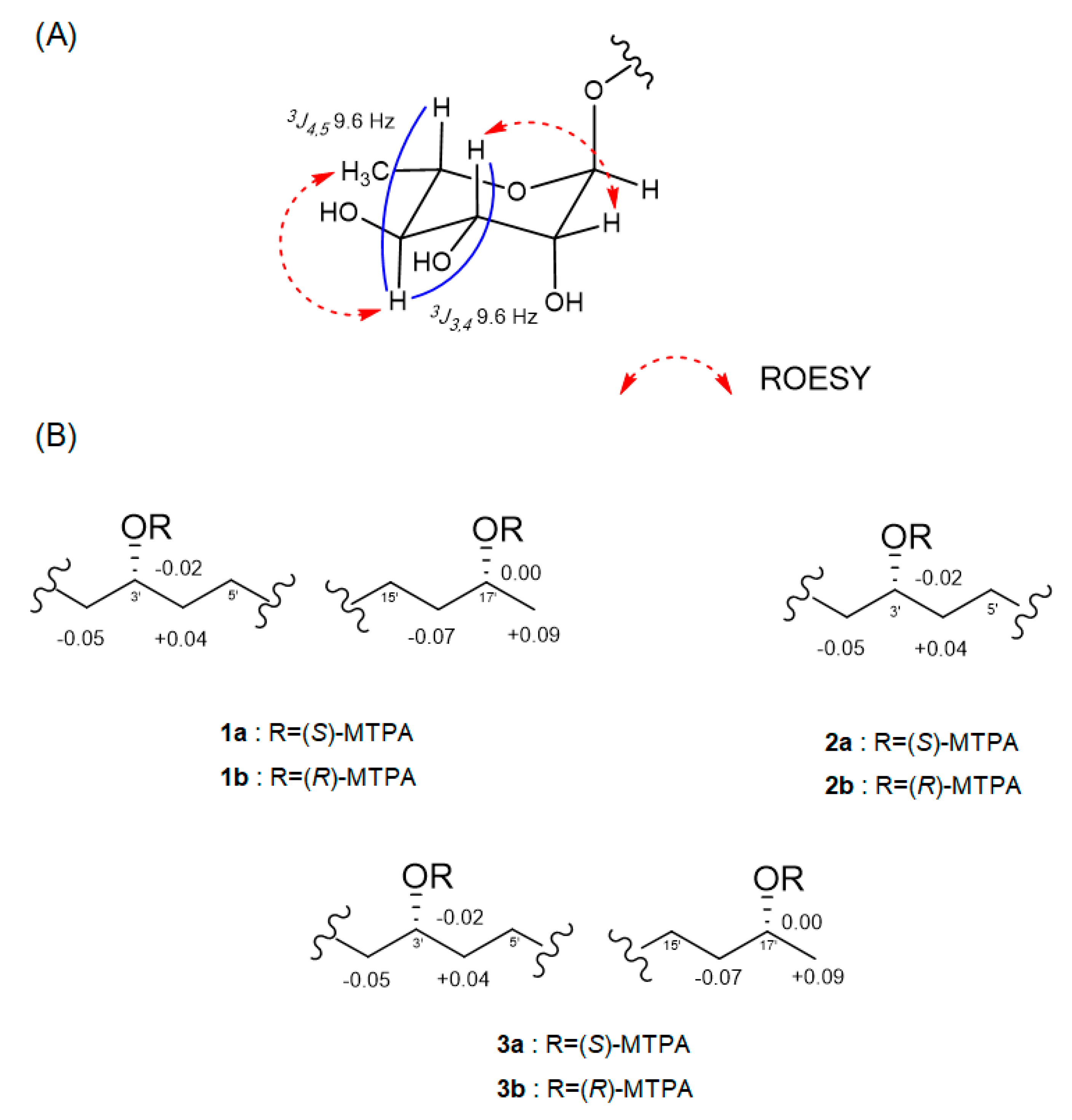
The relative configuration of the sugar moiety was established by analyzing the vicinal coupling constant
3JHH values and ROESY correlations, as seen in
Figure 3A. The axial positions of H-3, H-4 and H-5 were assigned by the large coupling constants (
3J3,4 = 9.6 Hz and
3J4,5 = 9.6 Hz). The broad singlet of the anomeric proton (H-1) and the ROESY correlations from H-2 to H-3 and from H-4 to H
3-6 also suggested that the sugar moiety was rhamnose. To determine the absolute configuration, dokdolipid A (
1) was subjected to a chemical degradation. The acid hydrolysis in MeOH of
1 afforded the methylated aglycone of
1 and rhamnopyranose. The absolute configuration of the methylated aglycone was confirmed using the modified Mosher’s method [
17]. The observed chemical shift differences Δ
δS−R suggested the 3′
R,17′
R configurations in
1 (
Figure 3B). In addition, the rhamnose obtained from hydrolysis and the authentic
l- and
d-rhamnose were separately derivatized with
l-cysteine methyl ester hydrochloride and σ-Tolyl isothiocyanate to establish the absolute configuration of rhamnose [
18]. On the basis of the HPLC analysis of the derivatives, the
l-rhamnose moiety in
1 was confirmed by chemical derivatization and comparison with standards. Thus, the structure of
1 was identified to be (3′
R,17′
R)-3′-
O-(α
-l-rhamnopyranosyl)-17′- dihydroxyoctadecanoic acid and named as dokdolipid A (
1).
Compound
2 was isolated as a dark brown oil, and its molecular formula was determined as C
24H
44O
8 by the [M + Na]
+ ion peak at
m/z 483.2936 (calcd 483.2934) in the HRESIMS. The
1H and
13C NMR spectra of
2 were similar to those of
1, suggesting that
2 shared the same carbon skeleton as
1. The obvious differences were the disappearance of a doublet methyl and appearance of a singlet methyl. In addition, a
13C NMR signal of a carbonyl carbon at
δC 210.8 was observed. The HMBC correlations from H
3-18′ (
δH 2.12) to C-16′ (
δC 42.9) and C-17′ (
δC 210.8) and from H
2-16′ (
δH 2.47) to C-17′ (
δC 210.8) suggested that the oxygenated methine C-17′ in
1 was replaced by the carbonyl carbon in
2. The absolute configuration of C-3′ in
2 was confirmed by the same method as that of
1, as seen in
Figure 3B. The results indicated the
R-configuration of C-3′ in
2. The comparison of the specific rotation values of
1 and
2 ([α]
D25 −33.3 (c 0.3, MeOH) and [α]
D25 −10.0 (c 0.3, MeOH), respectively) and identical chemical shifts also supported that
2 had the same absolute configuration as
1. Thus, the structure of
2 was determined as a new derivative of
1 and named dokdolipid B (
2).
Compound
3 was purified as a dark brown oil and gave a [M + Na]
+ ion peak at
m/z 631.3669 (calcd 631.3669) in the HRESIMS, consistent with a molecular formula C
30H
56O
12. Comparison of the NMR spectroscopic data of
3 with
1 revealed that
3 has a very similar structure to that of
1. However, the singlet at
δH 1.23 corresponded to two methyl groups while the proton signal at
δH 4.79 belonged to two anomeric protons. In addition, the presence of eight methine protons at
δH 3.35–3.72 suggested that
3 possessed two hexose units. Further analysis of its 2D NMR data and coupling constants
3JHH confirmed that the structure of
3 was analogous to that of
1 with two rhamnoses. The absolute configurations of C-3′ and C-17′ in
3 were also determined using the same method as
1 and the comparison of the specific rotation values of
1 and
3, as well as similar chemical shifts, as seen in
Figure 3B. By considering all the experimental data and the biosynthetic pathway of
1 and
3, the absolute configuration of
3 was determined to be the same as
1. Thus, the structure of
3 was elucidated and named dokdolipid C (
3).
Compounds
1–
3 were tested for their cytotoxicity against cancer cell lines including HCT-15, NUGC-3, NCI-H23, ACHN, PC-3 and MDA-MB-231 using sulforhodamine B (SRB) assay, with adriamycin as a positive control. As shown in
Table 2,
1−
3 showed moderate activity against these cells, with GI
50 values ranging from 13.7 to 41.5 μM. Among the tested compounds,
2 displayed the strongest cytotoxicity in all the cell lines except for MDA-MB-231 (Breast cancer), whereas
3 showed better activity against the MDA-MB-231 cell line than other compounds.
3. Materials and Methods
3.1. General Experimental Procedures
The 1D (1H and 13C) and 2D (COSY, ROESY, HSQC, and HMBC) NMR spectra were acquired on a Bruker 600 MHz spectrometer. UV spectra were obtained on a Shimadzu UV-1650PC spectrophotometer. IR spectra were recorded on a JASCO FT/IR-4100 spectrophotometer. Optical rotations were measured on a Rudolph Research Analytical (Autopol III) polarimeter. HRESIMS spectra were recorded on a hybrid ion-trap time-of-flight mass spectrometer (Shimadzu LC/MS-IT-TOF). HPLC was performed on a PrimeLine Binary pump with RI-101 (Shodex). Analytical HPLC was conducted on an ODS column (YMC-Pack-ODS-A, 250 × 4.6 mm i.d, 5 µm).
3.2. Isolation and Cultivation of the Strain 179DD-027 (Actinoalloteichus hymeniacidonis)
The strain 179DD-027 was isolated from a sediment sample, collected off the coasts of Dokdo island, Republic of Korea. The strain was identified as Actinoalloteichus hymeniacidonis on the basis of the 16s rRNA gene sequence analysis (GenBank accession number MH681580). The strain 179DD-027 was grown on a Bennett’s (BN) agar plate for 7 days at 28 °C and then incubated in BN medium (composed of 10 g of glucose, 1 g of yeast extract, 2 g of tryptone, 1 g of beef extract, 5 g of glycerol and 32 g of NaCl in 1 L of H2O) in a 50 mL flask. After a four-day cultivation at 28 °C with shaking at 130 rpm, 10 mL of the seed culture in a 50 mL flask was used to inoculate 1 L of the culture medium in a 2 L flask for four days. For mass culture, 1 L of the culture in a 2 L flask was used to inoculate 40 L cultivation in BN medium in a 100 L fermenter. A total of 40 L of bacterial culture was incubated at 28 °C for 7 days.
3.3. Isolation of Compounds
The culture broth (40 L) was separated into cells and supernatant by centrifugation. The supernatant was extracted with EtOAc (40 L × 2) at room temperature and then concentrated under reduced pressure to yield the crude extract (3 g). The crude extract was fractionated by flash column chromatography on ODS using a stepwise elution (each fraction 300 mL × 3) with combinations of MeOH/H2O (1:4, 2:3, 3:2, 4:1 and 100% MeOH). The second fraction eluted with MeOH/H2O (4:1) was purified by an analytical, reversed-phase HPLC (YMC-Pack-ODS-A, 250 × 4.6 mm i.d, 5 µm, flow rate 2.0 mL/min, RI detector) using isocratic elution with 40% MeCN in H2O to yield 1 (35.2 mg, tR = 14 min), 2 (4.5 mg, tR = 20 min), and 3 (5.8 mg, tR = 9 min).
3.4. Spectral Data
Dokdolipid A(
1): dark brown oil; [α]
D25 −33.3 (c 0.3, MeOH); IR ν
max 3345, 2918, 2851, 1710, 1646, 1127, 1049 cm
−1; UV(MeOH) λ
max (log ε) 318 (3.09), 218 (3.42) nm; HRESIMS
m/z 485.3094 [M + Na]
+ (calcd for 485.3090, C
24H
46O
8Na);
1H NMR (CD
3OD, 600 MHz) and
13C NMR (CD
3OD, 125 MHz) see
Table 1.
Dokdolipid B (
2): dark brown oil; [α]
D25 −10.0 (c 0.3, MeOH); IR ν
max 3377, 2910, 2851, 1710, 1371, 1068, 1017 cm
−1; UV(MeOH) λ
max (log ε) 406 (3.29), 312 (3.50), 238 (3.60) nm; HRESIMS
m/z 483.2936 [M + Na]
+ (calcd for 483.2934, C
24H
44O
8Na);
1H NMR (CD
3OD, 600 MHz) and
13C NMR (CD
3OD, 125 MHz) see
Table 1.
Dokdolipid C (
3): dark brown oil; [α]
D25 −40.0 (c 0.3, MeOH); IR ν
max 3693, 3328, 2971, 2858, 1632, 1349, 1058, 1010 cm
−1; UV(MeOH) λ
max (log ε) 310 (3.15), 216 (3.51) nm; HRESIMS
m/z 631.3669 [M + Na]
+ (calcd for 631.3669, C
30H
56O
12Na);
1H NMR (CD
3OD, 600 MHz) and
13C NMR (CD
3OD, 125 MHz) see
Table 1.
3.5. Acid Hydrolysis and Determination of Absolute Configuration of Rhamnose
Each of the dokdolipids A−C (1−3) was dissolved in 3 N HCl (0.5 mL) in methanol and heated to 100 °C for 30 min. The solution was cooled and extracted with EtOAc twice. The EtOAc layer and the aqueous layer gave a methylated aglycone and a sugar residue after removal of the solvent respectively. The sugar residue was dissolved in pyridine (0.5 mL) containing l-cysteine methyl ester hydrochloride (0.5 mg) and heated to 60 °C for 1 h. σ-Tolylisothiocyanate (10 μL) was added to the mixture and heating was continued for an additional 1 h. The reaction mixture was directly analyzed using HPLC (10 to 100% MeCN gradient with 0.1% formic acid over 40 min). The sugar residue was detected at 17.9 min. The retention times of the authentic rhamnose samples were 15.5 (d-rhamnose) and 17.9 (l-rhamnose) min under the same HPLC conditions. Therefore, the absolute configuration of the rhamnose unit was established as l-configuration. All dokdolipids were also assigned using the chemical derivatization and HPLC analysis as described above.
3.6. Preparation of MTPA and Esters of 1−3 using the Modified Mosher’s Method
(R)-MTPA-Cl (10 μL) or (S)-MTPA-Cl (10 μL) and anhydrous pyridine (200 μL) were added to a methylated aglycone (0.6 mg for each). The mixture was stirred overnight at room temperature. The reaction mixture was evaporated to dryness and extracted with EtOAc twice. The EtOAc extracts were purified using an analytical reversed-phase HPLC (YMC-Pack-ODS-A, 250 × 4.6 mm i.d, 5 µm, flow rate 2.0 mL/min, RI detector) using gradient elution from 70% to 100% MeOH in 40 min to yield 1a (0.4 mg, tR = 33 min) and 1b (0.5 mg, tR = 33 min). Using the same procedure, 2a (0.3 mg, tR = 27 min), 2b (0.3 mg, tR = 28 min), 3a (0.3 mg, tR = 32 min) and 3b (0.4 mg, tR = 33 min) were prepared from dokdolipids B and C (2 and 3, 1.0 mg for each), respectively.
Compound
1a:
1H NMR (CD
3OD, 600 MHz)
δH 7.52−7.42 (10H, m, aromatic), 5.49 (1H, m, H-3′), 5.13 (1H, m, H-17′), 3.57 (3H, s, OMe), 3.55 (3H, s, OMe), 3.50 (3H, s, OMe), 2.64 (2H, dd, H-2′), 1.67 (2H, m, H-4′), 1.54 (2H, m, H-16′), 1.35−1.17 (22H, m), 1.32 (3H, d, H-18′); ESIMS
m/z 785.2 [M + Na]
+ (
Supporting information).
Compound
1b:
1H-NMR (CD
3OD, 600 MHz)
δH 7.52−7.42 (10H, m, aromatic), 5.46 (1H, m, H-3′), 5.13 (1H, m, H-17′), 3.66 (3H, s, OMe), 3.53 (3H, s, OMe), 3.52 (3H, s, OMe), 2.69 (2H, dd, H-2′), 1.63 (2H, m, H-4′), 1.61 (2H, m, H-16′), 1.35−1.17 (22H, m), 1.23 (3H, d, H-18′); ESIMS
m/z 785.4 [M + Na]
+ (
Supporting information).
Compound
2a:
1H NMR (CD
3OD, 600 MHz)
δH 7.52−7.42 (10H, m, aromatic), 5.50 (1H, m, H-3′), 3.57 (3H, s, OMe), 3.50 (3H, s, OMe), 2.64 (2H, dd, H-2′), 1.70 (2H, m, H-4′), 2.47 (2H, t, H-16′), 1.35−1.17 (22H, m), 2.12 (3H, s, H-18′); ESIMS
m/z 567.4 [M + Na]
+ (
Supporting information).
Compound
2b:
1H NMR (CD
3OD, 600 MHz)
δH 7.50−7.42 (10H, m, aromatic), 5.47 (1H, m, H-3′), 3.66 (3H, s, OMe), 3.53 (3H, s, OMe), 2.70 (2H, dd, H-2′), 1.62 (2H, m, H-4′), 2.47 (2H, t, H-16′), 1.35−1.17 (22H, m), 2.12 (3H, s, H-18′); ESIMS
m/z 567.3 [M + Na]
+ (
Supporting information).
Compound
3a:
1H NMR (CD
3OD, 600 MHz)
δH 7.52−7.42 (10H, m, aromatic), 5.49 (1H, m, H-3′), 5.13 (1H, m, H-17′), 3.57 (3H, s, OMe), 3.55 (3H, s, OMe), 3.50 (3H, s, OMe), 2.64 (2H, dd, H-2′), 1.70 (2H, m, H-4′), 1.55 (2H, m, H-16′), 1.35−1.17 (22H, m), 1.33 (3H, d, H-18′); ESIMS
m/z 785.5 [M + Na]
+ (
Supporting information).
Compound
3b:
1H NMR (CD
3OD, 600 MHz)
δH 7.52−7.42 (10H, m, aromatic), 5.46 (1H, m, H-3′), 5.13 (1H, m, H-17′), 3.66 (3H, s, OMe), 3.53 (3H, s, OMe), 3.52 (3H, s, OMe), 2.69 (2H, dd, H-2′), 1.66 (2H, m, H-4′), 1.60 (2H, m, H-16′), 1.35−1.17 (22H, m), 1.24 (3H, d, H-18′); ESIMS
m/z 785.2 [M + Na]
+ (
Supporting information).
3.7. Cytotoxicity Test by SRB Assay
Human cancer cell lines HCT-15 (colon), NUGC-3 (stomach), NCI-H23 (lung), ACHN (renal), PC-3 (prostate) and MDA-MB-231 (breast), were purchased from the American Type Culture Collection (Manassas, VA). The cell lines were cultured RPMI 1640 supplemented with 10% fetal bovine serum (FBS). Cell cultures were maintained at 37 °C under a humidified atmosphere of 5% CO
2. The growth inhibition assay against human cancer cell lines was carried out according to a sulforhodamine B (SRB) assay [
19]. In brief, 8000 cells/well were seeded in a 96-well plate. Next day, the cells were treated with compounds
1–
3 including vehicle control (0.1% DMSO) and positive control (adriamycin). After being incubated for 48 hours, cultures were fixed with 50% trichloroactetic acid (50 μg/mL) and stained with 0.4% sulforhodamine B in 1% acetic acid. Unbound dye was removed by washing with 1% acetic acid, and protein-bound dye was extracted with 10 mM Tris base (pH 10.5) for determination of optical density. The absorbance at 540 nm was determined using a VersaMax microplate reader (Molecular Devices, LLC, Sunnyvale, CA, USA). GI
50 values were calculated using GraphPad Prism 4.0 software (GraphPad Software, Inc., San Diego, CA, USA).



{kind=link}
{kind=link}
{kind=link}
{kind=link}