Snails In Silico: A Review of Computational Studies on the Conopeptides

 , , , and
, , , and
Abstract
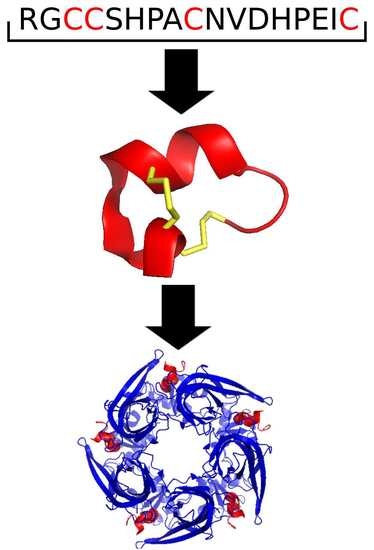
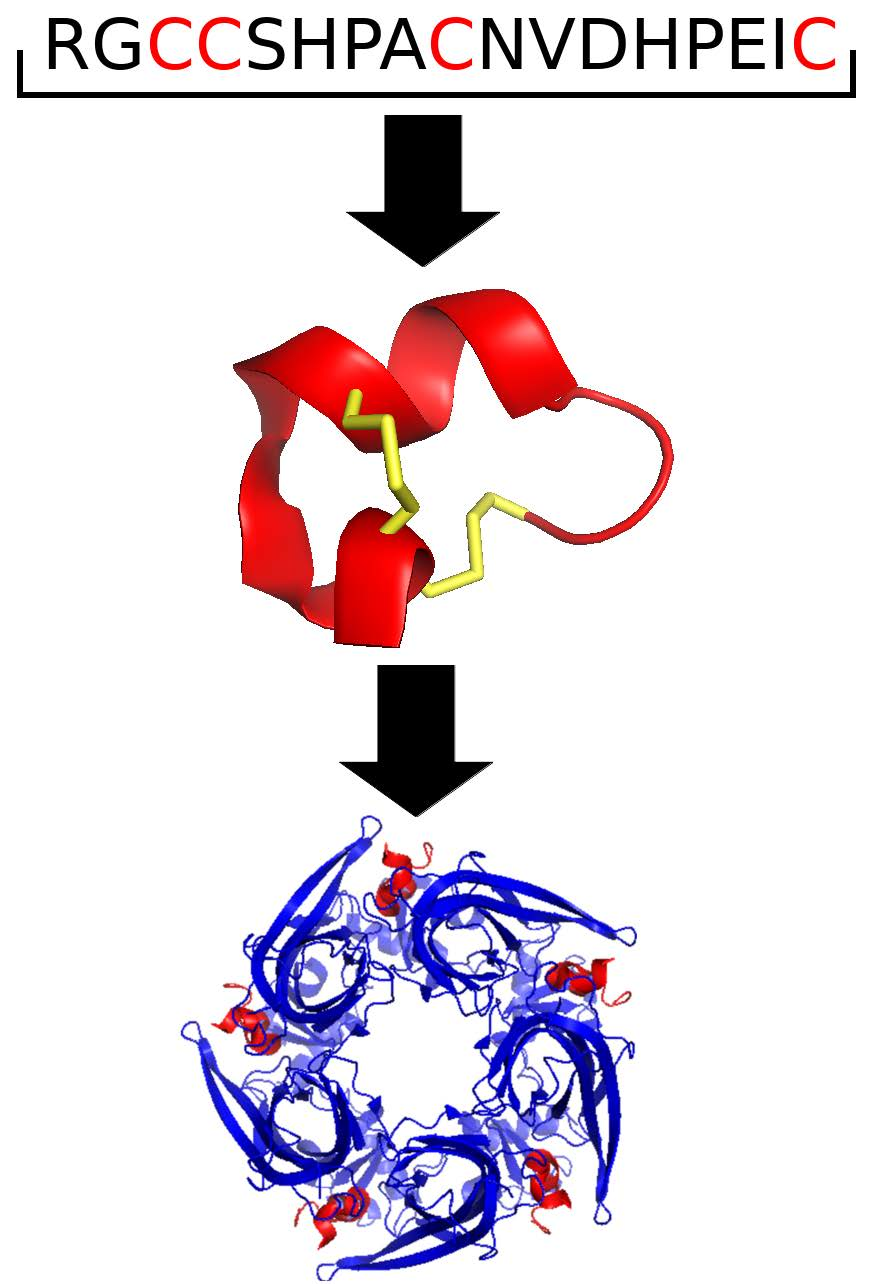
:
1. Introduction
2. Background
2.1. Categories of Conopeptide Classification
2.1.1. Gene Superfamily
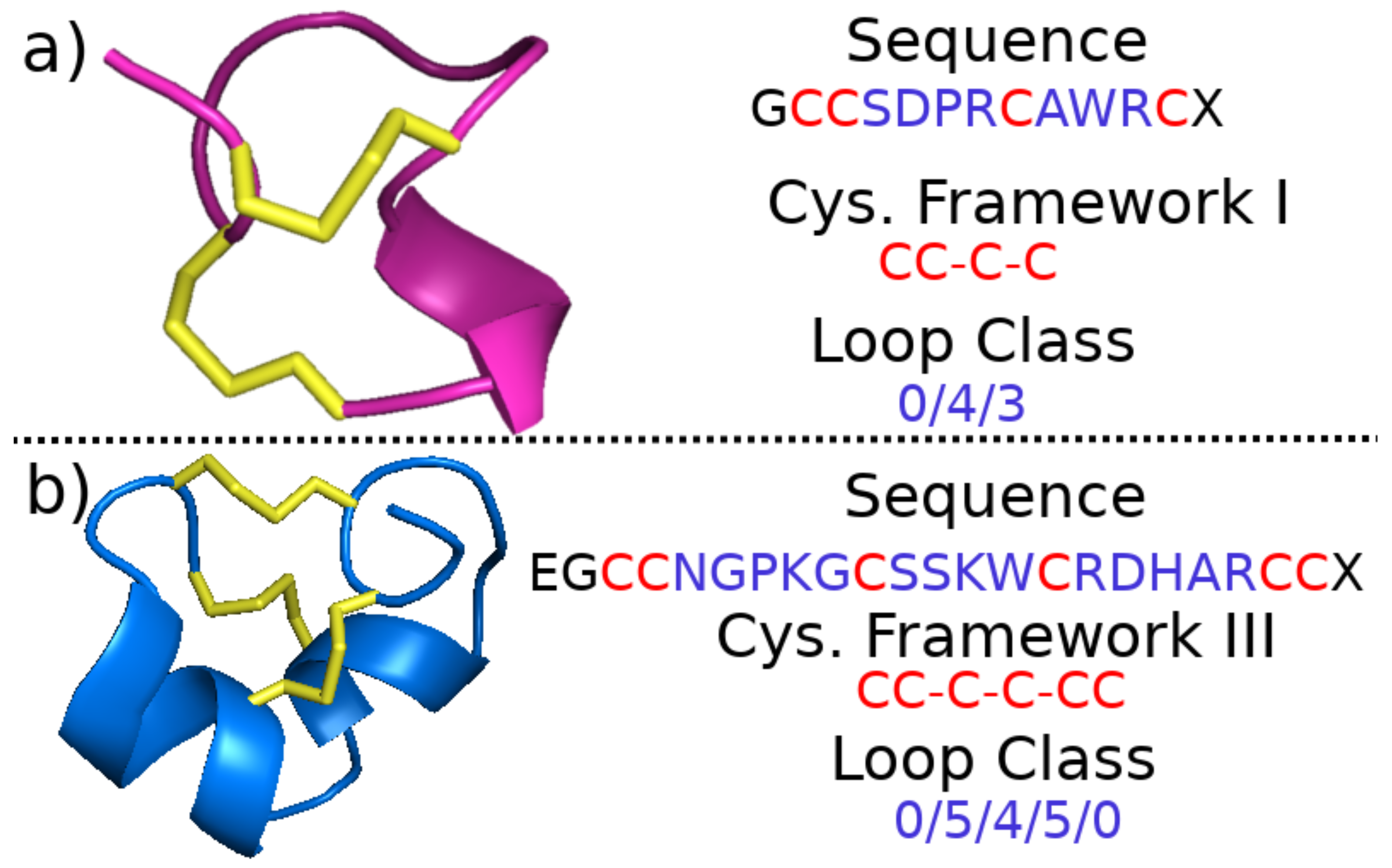
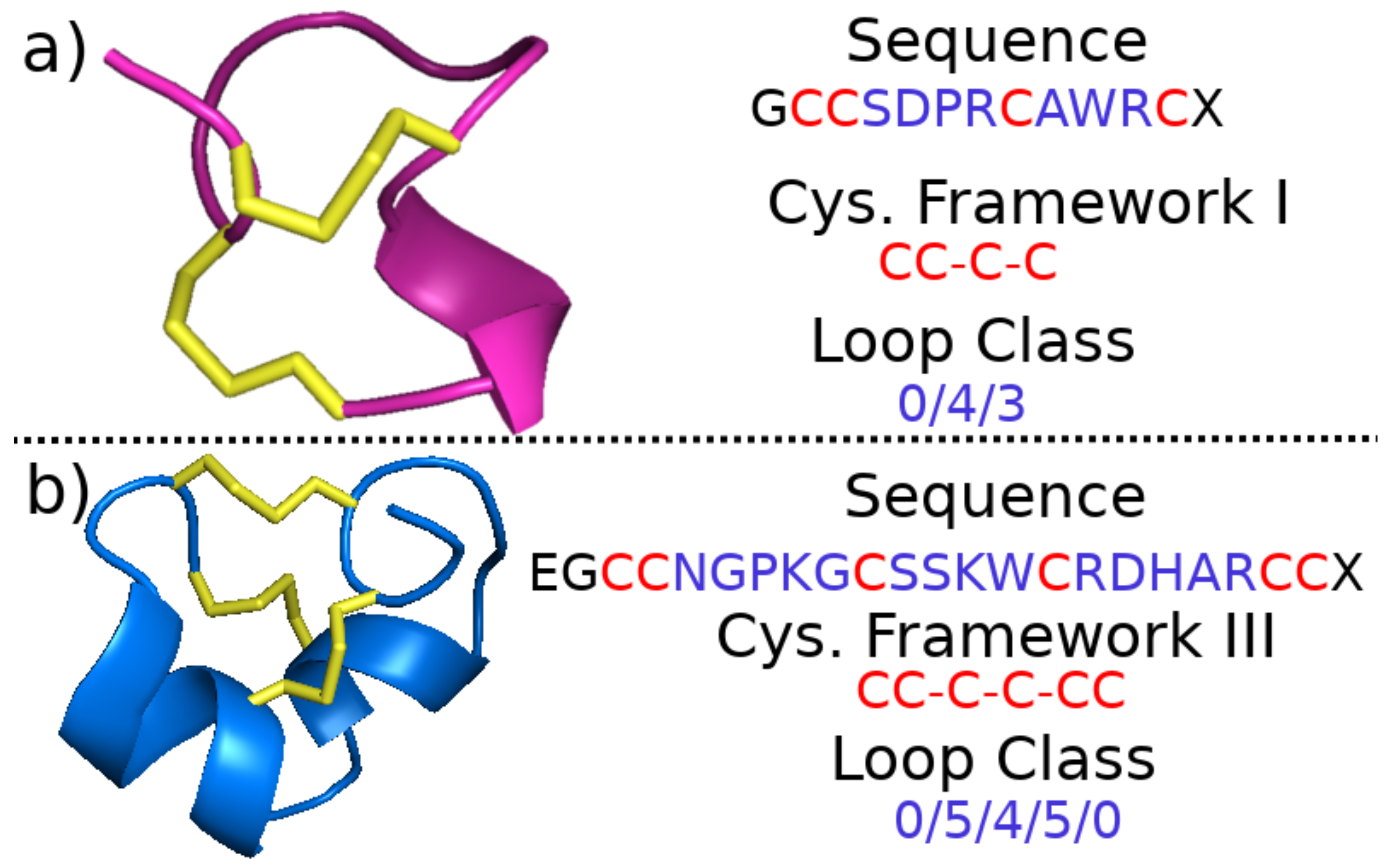
2.1.2. Cysteine Framework and Loop Class
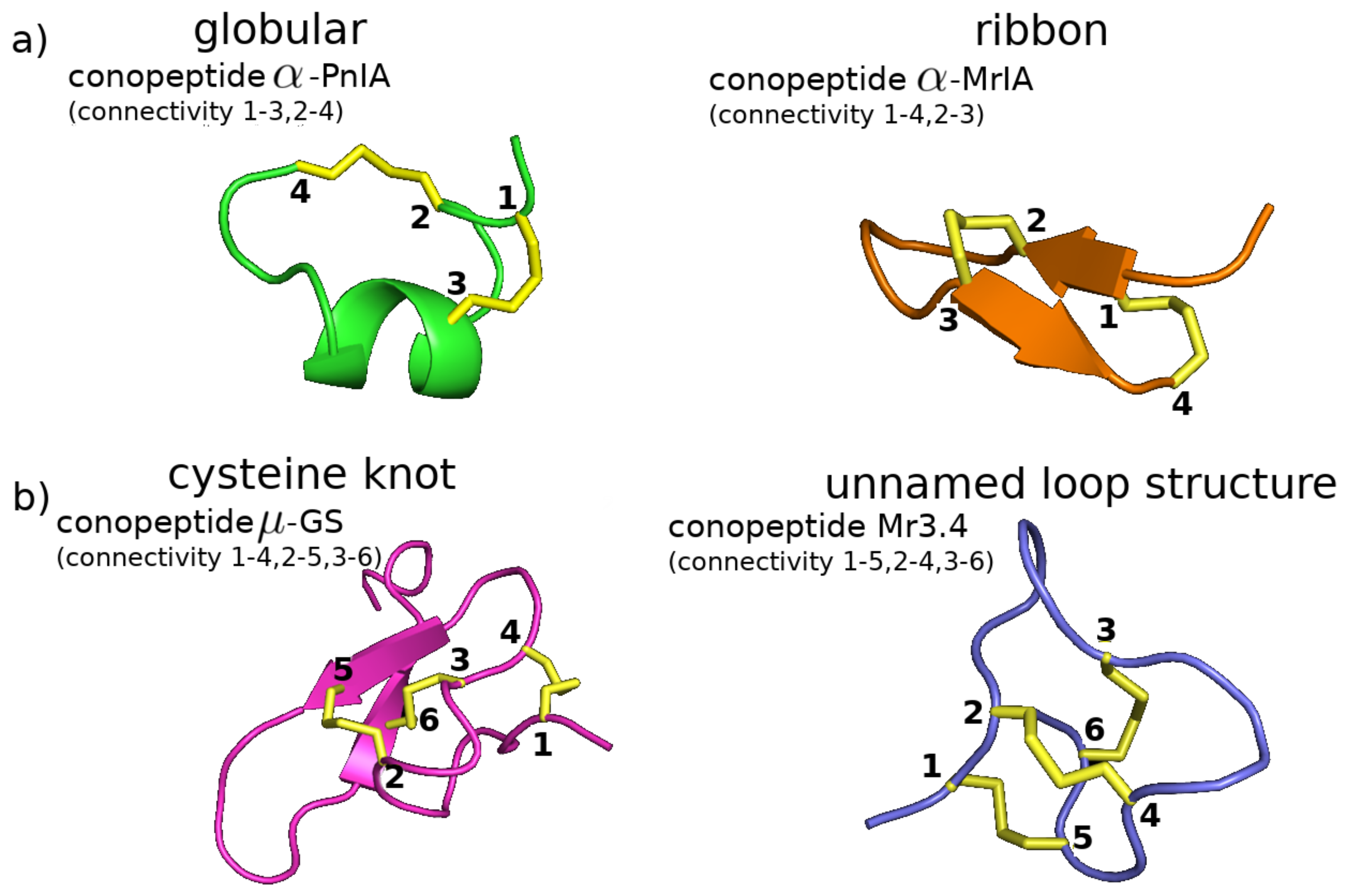
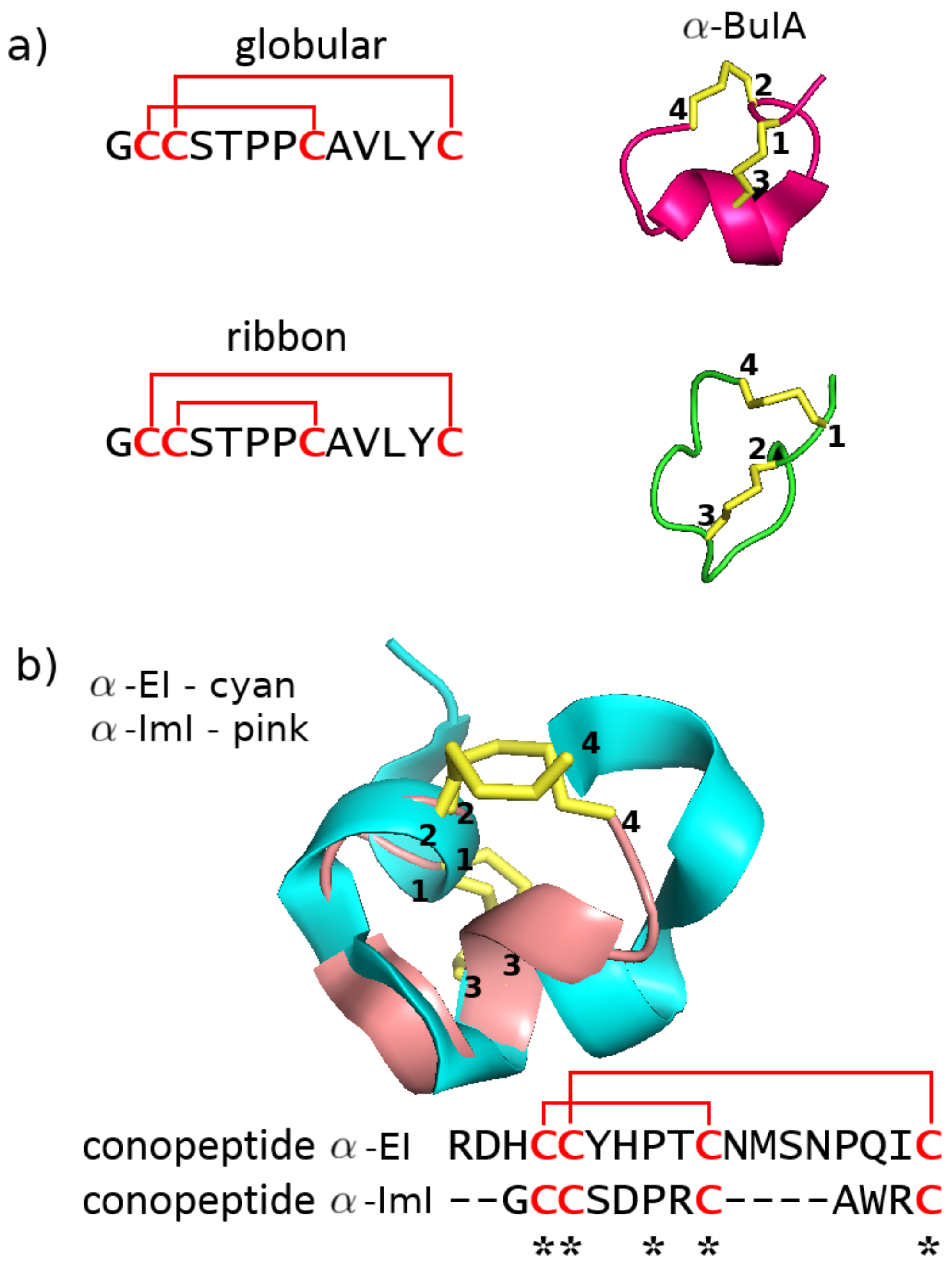
2.1.3. Fold and Subfold Class
2.1.4. Disulfide Connectivity
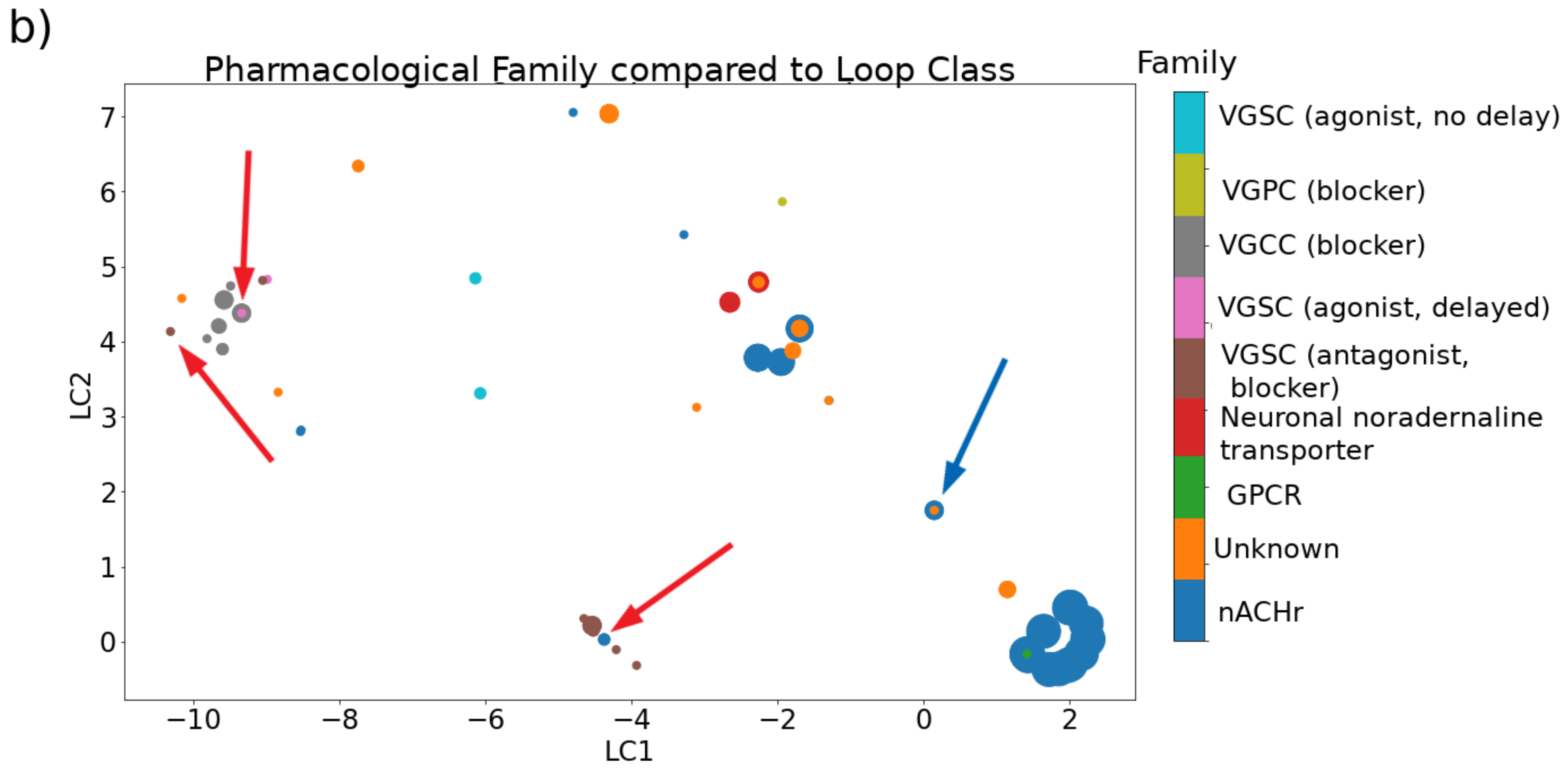
2.1.5. Pharmacological Family
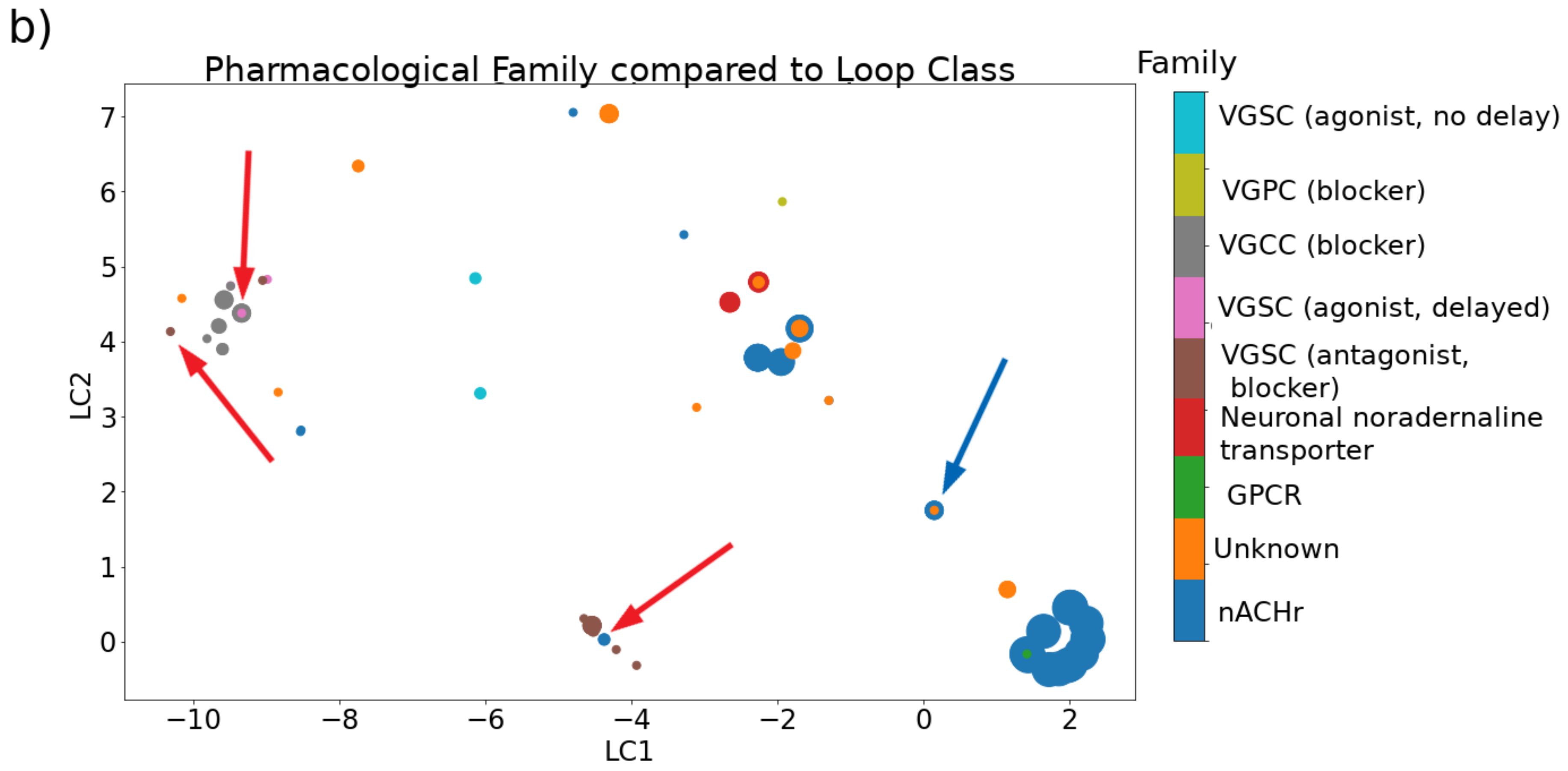
2.2. Relationships between Categories
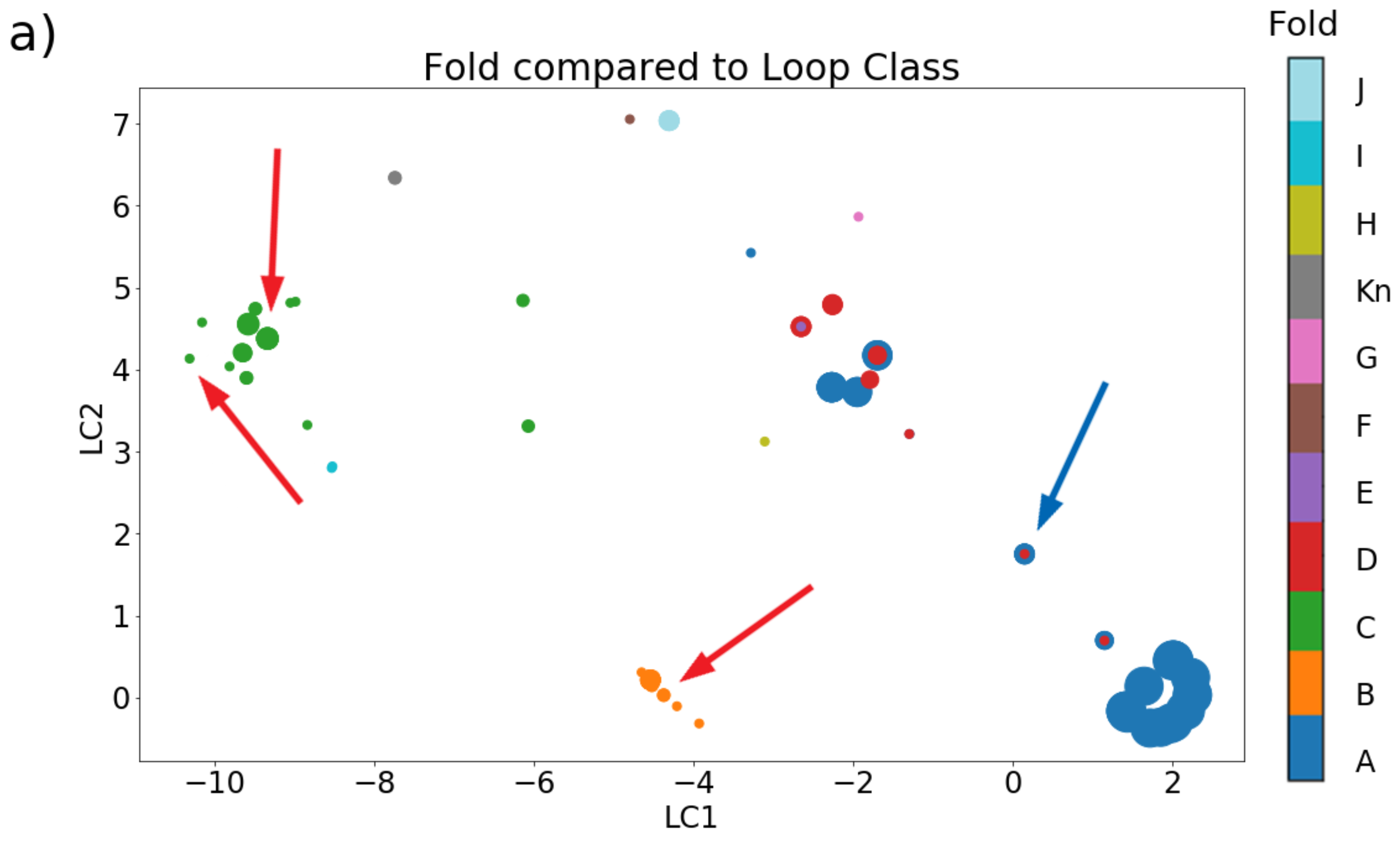
2.2.1. Cysteine Framework, Loop Class, Fold, and Pharmacological Family
2.2.2. Disulfide Connectivity Determines Fold
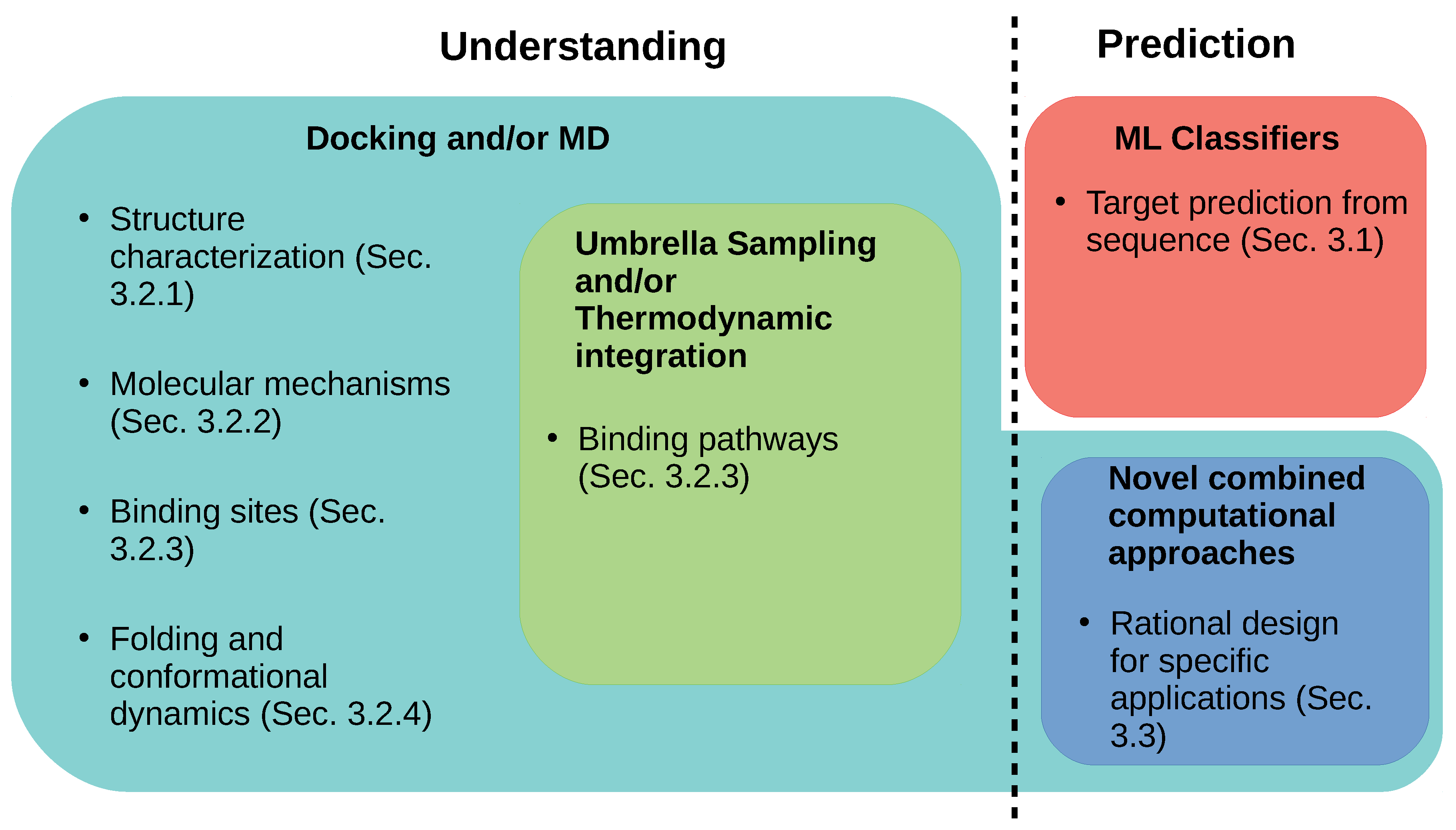
3. Computational Strategies to Understand and Predict Conopeptide Structure and Function
3.1. Predicting Function from Sequence through Machine Learning
3.2. Docking Studies and Molecular Dynamics Simulations for Understanding of Conopeptide Structure and Binding
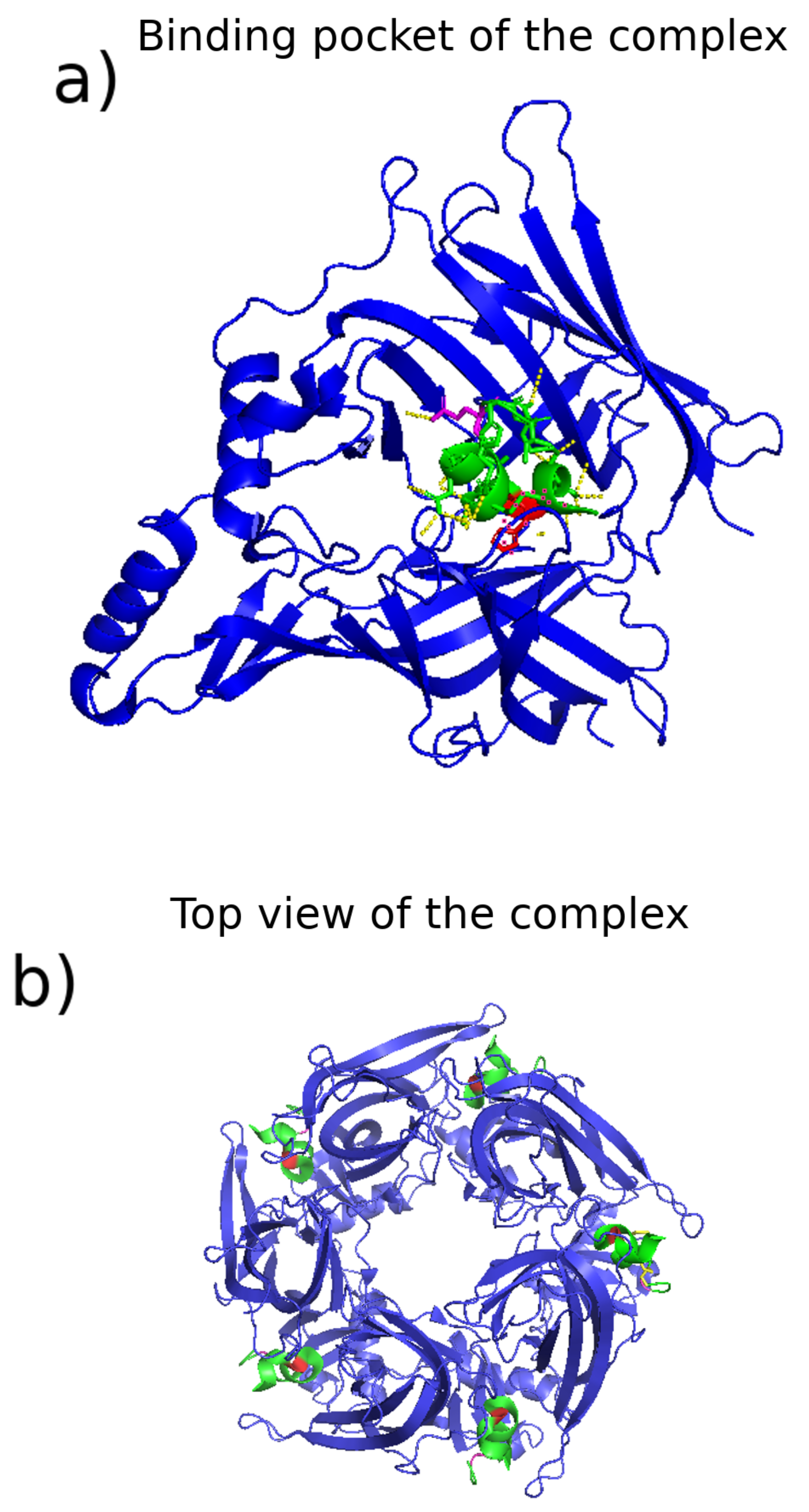
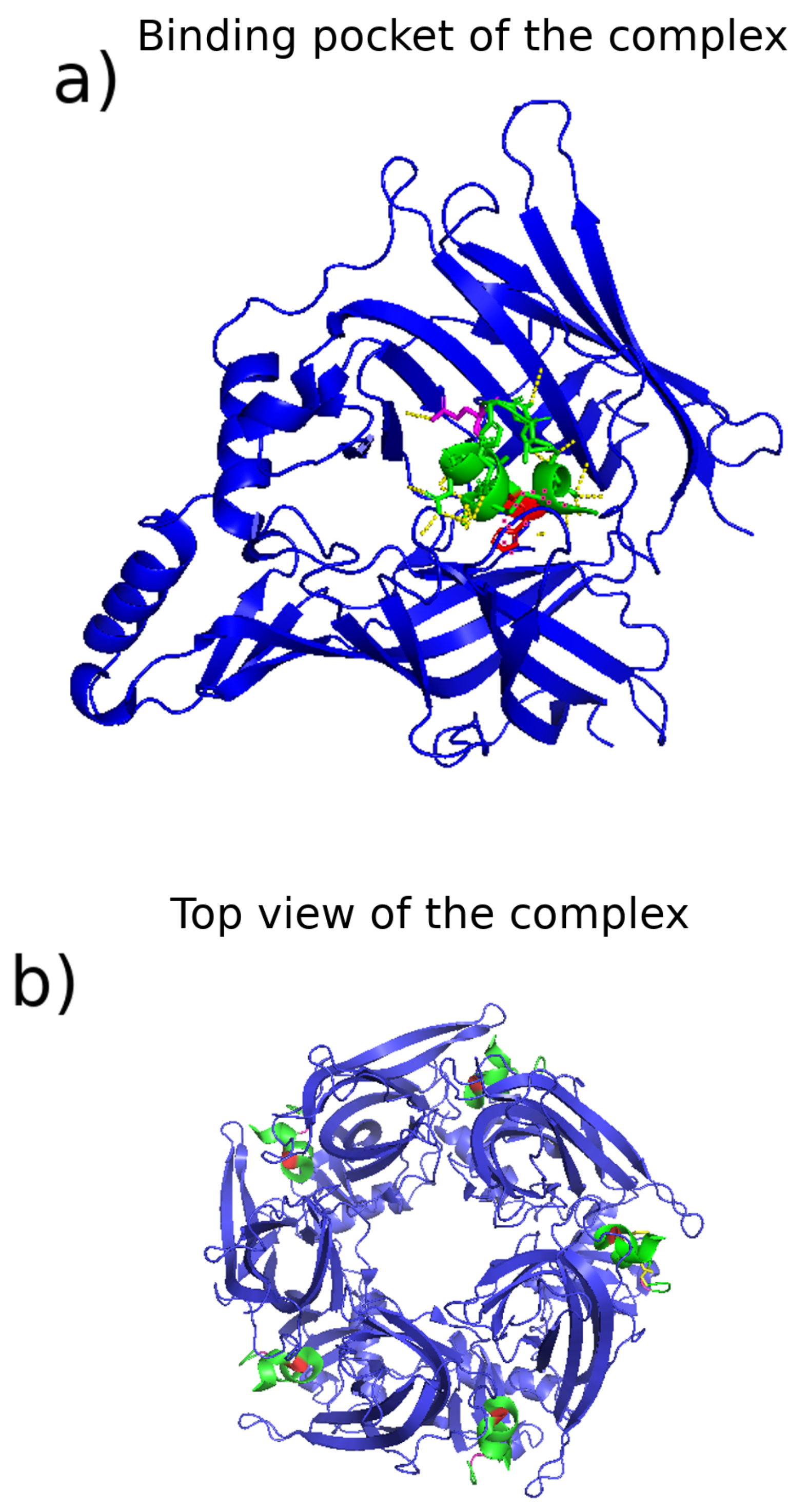
3.2.1. Conopeptide and Receptor Structural Characterization
3.2.2. Molecular Mechanisms of Selectivity and Binding
3.2.3. Identification of Binding Sites, Complexes, and Pathways
3.2.4. Folding Kinetics and Isolated Conformations of Conopeptides in Solution
3.2.5. Summary
3.3. Computational Design of Conopeptides for Specific Applications
4. Future Outlook
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AchBP | Acetylcholine binding protein |
| GPCR | G protein-coupled receptor |
| h-bonding | hydrogen bonding |
| MD | Molecular dynamics |
| ML | Machine learning |
| nAChR | Nicotinic acetylcholine receptor |
| NMR | Nuclear magnetic resonance |
| NMDA | N-methyl-D-aspartate receptor |
| PDB | Protein Data Bank |
| VGCC | Voltage-gated calcium channel |
| VGSC | Voltage-gated sodium channel |
| VGPC | Voltage-gated potassium channel |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Goal | Toxin(s) | Methods | Results/Citations |
|---|---|---|---|
| Train ML classifier (Section 3.1) | toxins targeting VGSC, VGCC, VGPC | Support vector machines (SVMs) | Predictor of sequence to target with average accuracy 90.3% [100] |
| SVMs | Predictor of sequence to target with average accuracy 95.3% [97] | ||
| SVMs | Predictor of sequence to target with average accuracy 94.2% [96] | ||
| Radial basis function network | Predictor of sequence to target with average accuracy 89.7% [98] | ||
| Random forests | Predictor of sequence to target with average accuracy 97.3% [99] | ||
| RNA sequences from ten species | Logit, Label spreading, Perceptron | ConusPipe identifies potential conotoxins from sequence [95] | |
| Structure prediction (Section 3.2.1) | BtIIIA | MD simulation | Structure refinement [105] |
| -MI | MD simulation | Structure refinement [106] | |
| -EIVA | MD simulation | Structure refinement [107] | |
| conantokin G | MD simulation | Structure refinement in complex with calcium [15] | |
| sr11a, -RXIA | MD, homology modeling | Structure refinement of sr11a [108] | |
| Vt3.1 | MD, secondary structure predictors | Structure determination [109] | |
| conantokins conBk-A, conBk-B, conBk-C | MD, secondary structure predictors | Structure determination [110] | |
| -GI | Docking, MD | Validation of in silico predictions [111] | |
| -LvIA | Homology modeling, MD | Revealed molecular interactions between toxin and nAChR [112] | |
| -GIIIA, -PIIIA, -KIIIA | Docking | Validation of homology models for eukaryotic sodium channels [113] | |
| -GIIIA | Docking, biased MD, unbiased MD | Characterization of insertion into mammalian and bacterial channels [114] | |
| -GID and analogues | Docking | Testing of ToxDock algorithm [115] | |
| Molecular mechanisms (Section 3.2.2) | -RIIIK | Docking | Identification of charged ring interaction with Shaker VGPC [116] |
| /-PIXIVA | Docking | Identification of charged ring interaction with Shaker VGPC [77] | |
| -GIIIA | Docking | Identification of charged ring interaction with VGSC [117] | |
| -MII, -TxIA, -[A10L]TxIA | Docking, MD | Charge more important than steric for selectivity for nAChR [118] | |
| -ImI and mutants | Docking, MD | Binding versus selectivity to nAChR subtypes [119] | |
| -MVIIA, -MVIIC, mutants | Quantum and classical MD | Find electronic changes from mutation and relate to h-bonding [121] | |
| -MII | Constant-pH MD | Probe pH effects on protonation [122] | |
| cyclic -PVIIA | MD simulation | Cyclicization effects on interaction with VGPC [120] | |
| - [A10L]PnIA | Docking | Hydrophobic effect on , nAChR selectivity [123] | |
| -RegIIA | MD simulation | Receptor side chain length relation to affinity for , nAChR [124] | |
| -RgIA | MD simulation | Effect of dicarba instead of disulfide bridges on nAChR binding [125] | |
| -PIA | Docking, MD | Tripeptide tail interaction with nAChR [126] | |
| -GIC | Docking, MD | Receptor side chain orientation effects on binding to , nAChR [127] | |
| -GIC | Homology modeling, Docking | His-5, Gln-13 key residues control , nAChR interaction [128] | |
| -BuIA | MD simulation | Mechanism of selectivity for over nAChR [129] | |
| -RegIIA | MD simulation | Residue Glu-198 controls affinity for rat over human nAChR [130] | |
| -MII, -PnIA, -GID | Docking | Conserved proline controls affinity with nAChR [131] | |
| -RgIA | Docking, MD | Arg-7, Arg-9 controls affinity and selectivity for nAChR [132] | |
| -AuIB | Homology modeling, MD | Phe-9 controls binding with nAChR [133] | |
| -TxID | MD simulation | Selectivity for nAChR due to steric changes in binding pocket [134] | |
| -TxID | MD simulation | Selectivity for nAChR due to steric changes in binding pocket, design of mutated analogue [135] | |
| -MVIIA | Docking, MD | Key methionine residue responsible for toxicity [136] | |
| Binding site and pathway characterization (Section 3.2.3) | -ImI, -ImII | Docking | Affinities for different nAChR binding sites [137] |
| -PIIIA | MD simulation | Verified and explained proposed binding orientation in VGSC [138] | |
| -MI | Docking | Identification of two different nAChR binding modes [139] | |
| -AuIB | Docking | Structural isomers bind to different sites on nAChR [71] | |
| -Vc1.1 | MD, binding energy calculations | Identify binding site to nAChR [140] | |
| conantokin-T, conantokin-G | MD simulation | Determined metal-binding models [141] | |
| -LtIA | Docking | Microscopic interactions of rapid unbinding from nAChR [143] | |
| -ImI, -PnIB, -PnIA, -MII | Docking | Differences between conotoxin and snake toxin binding at nAChR [144] | |
| -ImI, analogues | Docking, MD | Binding interaction investigation [142] | |
| -EVIA | Docking, MD | Molecular basis of binding to VGSC [145] | |
| -PIIIA, -KIIIA, -BuIIIB | Docking, MD | Systematic analysis of binding modes to Na1.4 sodium channel [146] | |
| -PIIIA | Docking, MD, umbrella sampling | Predict specificity for 8 different Na subtypes [147] | |
| -GVIA | Umbrella sampling | Predict IC50 values for Ca2.2 channel inhibition [149] | |
| -PIIIA | Umbrella sampling | Dissociation constants from Na1.4 sodium channel [150] | |
| -Imi, -PnIA variants | Umbrella sampling | Binding pathway characterization [151] | |
| -GID mutant | Umbrella sampling | Identification of multiple pathways in binding to and nAChR s [152] | |
| -ImI | Random accelerated MD, Steered MD | Characterize multiple unbinding pathways from nAChR [153] | |
| -PVIIA | MD, coarse-grained Brownian dynamics | Contribution of long-range electrostatics, h-bonding, hydrophobicity to approach and insertion into VGPC [155] | |
| Folding and conformational dynamics (Section 3.2.4) | -GI | Simplified quantum chemical calculations | Rapid simulations of folding/unfolding [156] |
| -AuIB | MD simulation | Size/shape fluctuations, translational and rotational diffusivity determination [157] | |
| MrIIIe | MD simulation | Effects of electric field strength [158] | |
| -AuIB, -GI | MD simulation | Ensembles of different isomers thermodynamically favorable under different solvent conditions [159,160] | |
| -GIIIA, -KIIIA, -PIIIA, -SIIIA, -SmIIIA | MD simulation | Effects of removal of successive disulfide bonds and characterization of folding types [161] | |
| cyclic -Vc1.1 | MD simulation | Removal of disulfide bonds does not perturb structure [162] | |
| -Vc1.1, -BuIA, -ImI, -AuIB | MD simulation | Removal of disulfide bonds perturbs non-cyclic structures [163] | |
| Rational design (Section 3.3) | contulakin-G | MD simulation, binding free energy calculations | Design of neurotensin analogues [165] |
| -TxID | MD simulation | Design of methionine-lacking mutant [166] | |
| 148 conopeptides with 3D structures in PDB [56] | Docking, MD | Design of conotoxin targeting LPAR6 [104] | |
| Conus betulinus venoms | Homology search | Find sequences with insecticidal properties [167] | |
| -GVIA and mutants | MD simulation | Find strong copper-binding conopeptides to remove metals from environment [168] | |
| conantokin-G and mutants | Docking | Design of EAR16, EAR18 that reversibly block GluN2B NMDA receptor [169] | |
| -MII and mutants | Docking, genetic algorithms | Design mutant with double the binding affinity for nAChR [170] | |
| -MII and a set of FDA-approved drugs | Docking, genetic algorithms | Drug repurposing algorithm [171] | |
| -PnIA and analogues | Docking, Protein Surface Topography | Design mutant with nanomolar affinity for nAChR [172] | |
| -PIIIA | MD, binding energy calculation | Assess importance of different disulfide bonds to binding with VGSC [164] |
References
- Robinson, S.D.; Norton, R.S. Conotoxin Gene Superfamilies. Mar. Drugs 2014, 12, 6058–6101. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J.; Dutertre, S.; Vetter, I.; Christie, M.J. Conus venom peptide pharmacology. Pharmacol. Rev. 2012, 64, 259–298. [Google Scholar] [CrossRef] [PubMed]
- Akondi, K.B.; Muttenthaler, M.; Dutertre, S.; Kaas, Q.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Discovery, Synthesis, and Structure–Activity Relationships of Conotoxins. Chem. Rev. 2014, 114, 5815–5847. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Jones, A.; Lewis, R.J. Remarkable inter- and intra-species complexity of conotoxins revealed by LC/MS. Peptides 2009, 30, 1222–1227. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.M.; Bulaj, G. Conus peptides—Combinatorial chemistry at a cone snail’s pace. Curr. Opin. Drug Discov. Dev. 2000, 3, 141–154. [Google Scholar]
- Buczek, O.; Bulaj, G.; Olivera, B.M. Conotoxins and the posttranslational modification of secreted gene products. Cell. Mol. Life Sci. 2005, 62, 3067–3079. [Google Scholar] [CrossRef] [PubMed]
- Puillandre, N.; Koua, D.; Favreau, P.; Olivera, B.M.; Stöcklin, R. Molecular Phylogeny, Classification and Evolution of Conopeptides. J. Mol. Evol. 2012, 74, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Lebbe, E.K.M.; Tytgat, J. In the picture: Disulfide-poor conopeptides, a class of pharmacologically interesting compounds. J. Venom. Anim. Toxins Incl. Trop. Dis. 2016, 22, 30. [Google Scholar] [CrossRef] [PubMed]
- Olivera, B.M. Conus Venom Peptides: Reflections from the Biology of Clades and Species. Annu. Rev. Ecol. Evol. Syst. 2002, 33, 25–47. [Google Scholar] [CrossRef]
- Craig, A.G.; Bandyopadhyay, P.; Olivera, B.M. Post-translationally modified neuropeptides from Conus venoms. Eur. J. Biochem. 1999, 264, 271–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, L.J.; Gray, W.R.; Olivera, B.M.; Zeikus, R.D.; Kerr, L.; Yoshikami, D.; Moczydlowski, E. Conus geographus toxins that discriminate between neuronal and muscle sodium channels. J. Biol. Chem. 1985, 260, 9280–9288. [Google Scholar] [PubMed]
- Loughnan, M.; Bond, T.; Atkins, A.; Cuevas, J.; Adams, D.J.; Broxton, N.M.; Livett, B.G.; Down, J.G.; Jones, A.; Alewood, P.F.; et al. alpha-conotoxin EpI, a novel sulfated peptide from Conus episcopatus that selectively targets neuronal nicotinic acetylcholine receptors. J. Biol. Chem. 1998, 273, 15667–15674. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, M.; Cruz, L.; Hunkapiller, M.; Gray, W.; Olivera, B. Isolation and structure of a peptide toxin from the marine snail Conus magus. Arch. Biochem. Biophys. 1982, 218, 329–334. [Google Scholar] [CrossRef]
- Craig, A.G.; Zafaralla, G.; Cruz, L.J.; Santos, A.D.; Hillyard, D.R.; Dykert, J.; Rivier, J.E.; Gray, W.R.; Imperial, J.; DelaCruz, R.G.; et al. An O-Glycosylated Neuroexcitatory Conus Peptide. Biochemistry 1998, 37, 16019–16025. [Google Scholar] [CrossRef] [PubMed]
- Rigby, A.C.; Baleja, J.D.; Li, L.; Pedersen, L.G.; Furie, B.C.; Furie, B. Role of γ-Carboxyglutamic Acid in the Calcium-Induced Structural Transition of Conantokin G, a Conotoxin from the Marine Snail Conus geographus. Biochemistry 1997, 36, 15677–15684. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Peng, C.; Yang, J.; Yi, Y.; Zhang, J.; Shi, Q. Cone snails: A big store of conotoxins for novel drug discovery. Toxins 2017, 9, 397. [Google Scholar] [CrossRef] [PubMed]
- Kaas, Q.; Westermann, J.C.; Halai, R.; Wang, C.K.L.; Craik, D.J. ConoServer, a database for conopeptide sequences and structures. Bioinformatics 2008, 24, 445–446. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.; Terlau, H. Toxins from cone snails: properties, applications and biotechnological production. Appl. Microbiol. Biotechnol. 2008, 79, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mir, R.; Karim, S.; Amjad Kamal, M.; Wilson, C.; Mirza, Z. Conotoxins: Structure, Therapeutic Potential and Pharmacological Applications. Curr. Pharm. Des. 2016, 22, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, S.A.; Christie, M.J. Conotoxin Interactions with α9α10-nAChRs: Is the α9α10-Nicotinic Acetylcholine Receptor an Important Therapeutic Target for Pain Management? Toxins 2015, 7, 3916–3932. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.J.; Yoshikami, D.; Azam, L.; Gajewiak, J.; Olivera, B.M.; Bulaj, G.; Zhang, M.M. μ-Conotoxins that differentially block sodium channels NaV1.1 through 1.8 identify those responsible for action potentials in sciatic nerve. Proc. Natl. Acad. Sci. USA 2011, 108, 10302–10307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, R.; Dai, H.; Mendelman, N.; Cuello, L.G.; Chill, J.H.; Goldstein, S.A.N. Designer and natural peptide toxin blockers of the KcsA potassium channel identified by phage display. Proc. Natl. Acad. Sci. USA 2015, 112, 7013–7021. [Google Scholar] [CrossRef] [PubMed]
- Zamponi, G.W. Targeting voltage-gated calcium channels in neurological and psychiatric diseases. Nat. Rev. Drug. Discov. 2016, 15, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, M.; McArthur, J.R.; Finol-Urdaneta, R.K. Analgesic conopeptides targeting G protein-coupled receptors reduce excitability of sensory neurons. Neuropharmacology 2017, 127, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Olivera, B.M.; Teichert, R.W. Diversity of the neurotoxic Conus peptides: A model for concerted pharmacological discovery. Mol. Interv. 2007, 7, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Terlau, H.; Olivera, B.M. Conus Venoms: A Rich Source of Novel Ion Channel-Targeted Peptides. Physiol. Rev. 2004, 84, 41–68. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.D.; Bokor, G. Conotoxins: Potential Weapons from the Sea. J. Bioterror. Biodef. 2012, 3, 2157–2526. [Google Scholar]
- Dutertre, S.; Jin, A.H.; Alewood, P.F.; Lewis, R.J. Intraspecific variations in Conus geographus defence-evoked venom and estimation of the human lethal dose. Toxicon 2014, 91, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Thapa, P.; Espiritu, M.J.; Cabalteja, C.C.; Bingham, J.P. Conotoxins and their regulatory considerations. Regul. Toxicol. Pharmacol. 2014, 70, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Armishaw, C.; Alewood, P. Conotoxins as Research Tools and Drug Leads. Curr. Protein Pept. Sci. 2005, 6, 221–240. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, D.; Gonzalez, W.; Fissore, R.; Carvacho, I. Conotoxins as Tools to Understand the Physiological Function of Voltage-Gated Calcium (CaV) Channels. Mar. Drugs 2017, 15, 313. [Google Scholar] [CrossRef] [PubMed]
- Netirojjanakul, C.; Miranda, L.P. Progress and challenges in the optimization of toxin peptides for development as pain therapeutics. Curr. Opin. Chem. Biol. 2017, 38, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Olivera, B.M. Conus peptides: biodiversity-based discovery and exogenomics. J. Biol. Chem. 2006, 281, 31173–31177. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.; Jensen, J.; Nevin, S.; Callaghan, B.; Adams, D.; Craik, D. The Engineering of an Orally Active Conotoxin for the Treatment of Neuropathic Pain. Angew. Chem. Int. Ed. 2010, 49, 6545–6548. [Google Scholar] [CrossRef] [PubMed]
- Obata, H.; Conklin, D.; Eisenach, J.C. Spinal noradrenaline transporter inhibition by reboxetine and Xen2174 reduces tactile hypersensitivity after surgery in rats. Pain 2005, 113, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Brust, A.; Palant, E.; Croker, D.E.; Colless, B.; Drinkwater, R.; Patterson, B.; Schroeder, C.I.; Wilson, D.; Nielsen, C.K.; Smith, M.T.; et al. χ-Conopeptide Pharmacophore Development: Toward a Novel Class of Norepinephrine Transporter Inhibitor (Xen2174) for Pain. J. Med. Chem. 2009, 52, 6991–7002. [Google Scholar] [CrossRef] [PubMed]
- Miljanich, G. Ziconotide: Neuronal Calcium Channel Blocker for Treating Severe Chronic Pain. Curr. Med. Chem. 2004, 11, 3029–3040. [Google Scholar] [CrossRef] [PubMed]
- Pope, J.E.; Deer, T.R. Ziconotide: A clinical update and pharmacologic review. Expert Opin. Pharmacother. 2013, 14, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.S.; Boyken, S.E.; Baker, D. The coming of age of de novo protein design. Nature 2016, 537, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, L.; Heyne, M.; Shifman, J.M.; Papo, N. Protein Engineering by Combined Computational and In Vitro Evolution Approaches. Trends Biochem. Sci. 2016, 41, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Hachmann, J.; Afzal, M.A.F.; Haghighatlari, M.; Pal, Y. Building and deploying a cyberinfrastructure for the data-driven design of chemical systems and the exploration of chemical space. Mol. Simul. 2018, 44, 921–929. [Google Scholar] [CrossRef]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Frenkel, D.; Smit, B. Understanding Molecular Simulation: From Algorithms to Applications, 2nd ed.; Academic Press: San Diego, CA, USA, 2001. [Google Scholar]
- Van Gunsteren, W.F.; Daura, X.; Hansen, N.; Mark, A.E.; Oostenbrink, C.; Riniker, S.; Smith, L.J. Validation of Molecular Simulation: An Overview of Issues. Angew. Chem. Int. Ed. 2018, 57, 884–902. [Google Scholar] [CrossRef] [PubMed]
- Moreira, I.S.; Fernandes, P.A.; Ramos, M.J. Protein-protein docking dealing with the unknown. J. Comput. Chem. 2009, 31, 317–342. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.C.; Kuyucak, S. Developing a comparative docking protocol for the prediction of peptide selectivity profiles: investigation of potassium channel toxins. Toxins 2012, 4, 110–138. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.; Hernández-Lobato, J.M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Adams, R.P.; Aspuru-Guzik, A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent. Sci. 2018, 4. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.Y.; Wong, G.C.; Ferguson, A.L. Machine learning-enabled discovery and design of membrane-active peptides. Bioorg. Med. Chem. 2018, 26, 2708–2718. [Google Scholar] [CrossRef] [PubMed]
- Curtarolo, S.; Hart, G.L.W.; Nardelli, M.B.; Mingo, N.; Sanvito, S.; Levy, O. The high-throughput highway to computational materials design. Nat. Mater. 2013, 12, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Kaas, Q.; Westermann, J.C.; Craik, D.J. Conopeptide characterization and classifications: An analysis using ConoServer. Toxicon 2010, 55, 1491–1509. [Google Scholar] [CrossRef] [PubMed]
- Lamthanh, H.; Jegou-Matheron, C.; Servent, D.; Menez, A.; Lancelin, J.M. Minimal conformation of the alpha-conotoxin ImI for the alpha7 neuronal nicotinic acetylcholine receptor recognition: correlated CD, NMR and binding studies. FEBS Lett. 1999, 454, 293–298. [Google Scholar] [PubMed]
- Kavanaugh, J.S.; Rogers, P.H.; Arnone, A. Crystallographic Evidence for a New Ensemble of Ligand-Induced Allosteric Transitions in Hemoglobin: The T-to-THigh Quaternary Transitions. Biochemistry 2005, 44, 6101–6121. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger LLC. The PyMOL Molecular Graphics System, Version 1.8; Technical Report; Schrödinger LLC: New York, NY, USA, 2015. [Google Scholar]
- Kaas, Q.; Yu, R.; Jin, A.H.; Dutertre, S.; Craik, D.J. ConoServer: Updated content, knowledge, and discovery tools in the conopeptide database. Nucleic Acids Res. 2012, 40, D325–D330. [Google Scholar] [CrossRef] [PubMed]
- Cheek, S.; Krishna, S.S.; Grishin, N.V. Structural Classification of Small, Disulfide-rich Protein Domains. J. Mol. Biol. 2006, 359, 215–237. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellison, M.; Feng, Z.P.; Park, A.J.; Zhang, X.; Olivera, B.M.; McIntosh, J.M.; Norton, R.S. α-RgIA, a Novel Conotoxin That Blocks the α9α10 nAChR: Structure and Identification of Key Receptor-Binding Residues. J. Mol. Biol. 2008, 377, 1216–1227. [Google Scholar] [CrossRef] [PubMed]
- Favreau, P.; Benoit, E.; Hocking, H.G.; Carlier, L.; D’hoedt, D.; Leipold, E.; Markgraf, R.; Schlumberger, S.; Córdova, M.A.; Gaertner, H.; et al. A novel μ-conopeptide, CnIIIC, exerts potent and preferential inhibition of NaV1.2/1.4 channels and blocks neuronal nicotinic acetylcholine receptors. Br. J. Pharmacol. 2012, 166, 1654–1668. [Google Scholar] [CrossRef] [PubMed]
- Volpon, L.; Lamthanh, H.; Barbier, J.; Gilles, N.; Molgó, J.; Ménez, A.; Lancelin, J.M. NMR solution structures of δ-conotoxin EVIA from Conus ermineus that selectively acts on vertebrate neuronal Na+ channels. J. Biol. Chem. 2004, 279, 21356–21366. [Google Scholar] [CrossRef] [PubMed]
- Gehrmann, J.; Alewood, P.F.; Craik, D.J. Structure determination of the three disulfide bond isomers of α-conotoxin GI: A model for the role of disulfide bonds in structural stability. J. Mol. Biol. 1998, 278, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.S.; Jois, S.D.; Kini, R.M. Solution structures of two structural isoforms of CMrVIA χ/λ-conotoxin. Biomacromolecules 2006, 7, 2337–2346. [Google Scholar] [CrossRef] [PubMed]
- Imperial, J.S.; Bansal, P.S.; Alewood, P.F.; Daly, N.L.; Craik, D.J.; Sporning, A.; Terlau, H.; López-Vera, E.; Bandyopadhyay, P.K.; Olivera, B.M. A novel conotoxin inhibitor of Kv1.6 channel and nAChR subtypes defines a new superfamily of conotoxins. Biochemistry 2006, 45, 8331–8340. [Google Scholar] [CrossRef] [PubMed]
- Korukottu, J.; Bayrhuber, M.; Montaville, P.; Vijayan, V.; Jung, Y.S.; Becker, S.; Zweckstetter, M. Fast High-Resolution Protein Structure Determination by Using Unassigned NMR Data. Angew. Chem. Int. Ed. 2007, 46, 1176–1179. [Google Scholar] [CrossRef] [PubMed]
- Du, W.H.; Han, Y.H.; Huang, F.J.; Li, J.; Chi, C.W.; Fang, W.H. Solution structure of an M-1 conotoxin with a novel disulfide linkage. FEBS J. 2007, 274, 2596–2602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, K.H.; Hwang, K.J.; Kim, S.M.; Kim, S.K.; Gray, W.R.; Olivera, B.M.; Rivier, J.; Shon, K.J. NMR structure determination of a novel conotoxin, [Pro 7, 13] αA-conotoxin PIVA. Biochemistry 1997, 36, 1669–1677. [Google Scholar] [CrossRef] [PubMed]
- Eliseo, T.; Cicero, D.O.; Romeo, C.; Schininà, M.E.; Massilia, G.R.; Polticelli, F.; Ascenzi, P.; Paci, M. Solution structure of the cyclic peptide contryphan-Vn, a Ca2+-dependent K+ channel modulator. Biopolymers 2004, 74, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Huang, F.; Jiang, H.; Liu, L.; Wang, Q.; Wang, Y.; Shao, X.; Chi, C.; Du, W.; Wang, C. Purification and structural characterization of a d-amino acid-containing conopeptide, conomarphin, from Conus marmoreus. FEBS J. 2008, 275, 1976–1987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Huang, F.; Du, W. Solution structure of a novel α-conotoxin with a distinctive loop spacing pattern. Amino Acids 2012, 43, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Daly, N.L.; Craik, D.J. Structural studies of conotoxins. IUBMB Life 2009, 61, 144–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, T.; Zhang, M.M.; Walewska, A.; Gruszczynski, P.; Robertson, C.; Cheatham, T.; Yoshikami, D.; Olivera, B.; Bulaj, G. Structurally Minimized μ-Conotoxin Analogues as Sodium Channel Blockers: Implications for Designing Conopeptide-Based Therapeutics. ChemMedChem 2009, 4, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Grishin, A.A.; Wang, C.I.A.; Muttenthaler, M.; Alewood, P.F.; Lewis, R.J.; Adams, D.J. Alpha-conotoxin AuIB isomers exhibit distinct inhibitory mechanisms and differential sensitivity to stoichiometry of alpha3beta4 nicotinic acetylcholine receptors. J. Biol. Chem. 2010, 285, 22254–22263. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.H.; Gehrmann, J.; W Guddat, L.; Alewood, P.F.; Craik, D.J.; Martin, J.L. The 1.1 å crystal structure of the neuronal acetylcholine receptor antagonist, α-conotoxin PnIA from Conus pennaceus. Structure 1996, 4, 417–423. [Google Scholar] [CrossRef]
- Nilsson, K.P.R.; Lovelace, E.S.; Caesar, C.E.; Tynngård, N.; Alewood, P.F.; Johansson, H.M.; Sharpe, I.A.; Lewis, R.J.; Daly, N.L.; Craik, D.J. Solution structure of χ-conopeptide MrIA, a modulator of the human norepinephrine transporter. Biopolymers 2005, 80, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.M.; Alewood, P.F.; Craik, D.J. Solution structure of the sodium channel antagonist conotoxin GS: A new molecular caliper for probing sodium channel geometry. Structure 1997, 5, 571–583. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef] [PubMed]
- Kudryavtsev, D.S.; Shelukhina, I.V.; Son, L.V.; Ojomoko, L.O.; Kryukova, E.V.; Lyukmanova, E.N.; Zhmak, M.N.; Dolgikh, D.A.; Ivanov, I.A.; Kasheverov, I.E.; et al. Neurotoxins from snake venoms and α-conotoxin ImI inhibit functionally active ionotropic γ-aminobutyric acid (GABA) receptors. J. Biol. Chem. 2015, 290, 22747–22758. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Babu, R.M.; Bhavna, R.; Ramakumar, S. In silico detection of binding mode of J-superfamily conotoxin pl14a with Kv1.6 channel. In Silico Biol. 2007, 7, 175–186. [Google Scholar] [PubMed]
- Turner, M.W.; Cort, J.R.; McDougal, O.M. α-Conotoxin Decontamination Protocol Evaluation: What Works and What Doesn’t. Toxins 2017, 9, 281. [Google Scholar] [CrossRef] [PubMed]
- Maaten, L.v.d.; Hinton, G. Visualizing Data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Guddat, L.W.; Martin, J.A.; Shan, L.; Edmundson, A.B.; Gray, W.R. Three-Dimensional Structure of the α-Conotoxin GI at 1.2 Å Resolution. Biochemistry 1996, 35, 11329–11335. [Google Scholar] [CrossRef] [PubMed]
- Chi, S.W.; Kim, D.H.; Olivera, B.M.; McIntosh, J.M.; Han, K.H. NMR structure determination of α-conotoxin BuIA, a novel neuronal nicotinic acetylcholine receptor antagonist with an unusual 4/4 disulfide scaffold. Biochem. Biophys. Res. Commun. 2006, 349, 1228–1234. [Google Scholar] [CrossRef] [PubMed]
- Jin, A.H.; Brandstaetter, H.; Nevin, S.T.; Tan, C.; Clark, R.J.; Adams, D.J.; Alewood, P.F.; Craik, D.J.; Daly, N.L. Structure of α-conotoxin BuIA: influences of disulfide connectivity on structural dynamics. BMC Struct. Biol. 2007, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Park, K.H.; Suk, J.E.; Jacobsen, R.; Gray, W.R.; McIntosh, J.M.; Han, K.H. Solution conformation of alpha-conotoxin EI, a neuromuscular toxin specific for the alpha 1/delta subunit interface of torpedo nicotinic acetylcholine receptor. J. Biol. Chem. 2001, 276, 49028–49033. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, K.J.; Thomas, L.; Lewis, R.J.; Alewood, P.F.; Craik, D.J. A Consensus Structure for ω-Conotoxins with Different Selectivities for Voltage-sensitive Calcium Channel Subtypes: Comparison of MVIIA, SVIB and SNX-202. J. Mol. Biol. 1996, 263, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Buczek, O.; Wei, D.; Babon, J.J.; Yang, X.; Fiedler, B.; Chen, P.; Yoshikami, D.; Olivera, B.M.; Bulaj, G.; Norton, R.S. Structure and Sodium Channel Activity of an Excitatory I1-Superfamily Conotoxin. Biochemistry 2007, 46, 9929–9940. [Google Scholar] [CrossRef] [PubMed]
- Norton, R.S.; Pallaghy, P.K. The cystine knot structure of ion channel toxins and related polypeptides. Toxicon 1998, 36, 1573–1583. [Google Scholar] [CrossRef]
- Xie, B.; Huang, Y.; Baumann, K.; Fry, B.; Shi, Q. From Marine Venoms to Drugs: Efficiently Supported by a Combination of Transcriptomics and Proteomics. Mar. Drugs 2017, 15, 103. [Google Scholar] [CrossRef] [PubMed]
- Kaas, Q.; Craik, D.J. Bioinformatics-Aided Venomics. Toxins 2015, 7, 2159–2187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prashanth, J.R.; Lewis, R.J.; Dutertre, S. Towards an integrated venomics approach for accelerated conopeptide discovery. Toxicon 2012, 60, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Kamilaris, A.; Prenafeta-Boldú, F.X. Deep learning in agriculture: A survey. Comput. Electron. Agric. 2018, 147, 70–90. [Google Scholar] [CrossRef]
- Shen, D.; Wu, G.; Suk, H.I. Deep Learning in Medical Image Analysis. Annu. Rev. Biomed. Eng. 2017, 19, 221–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, S.; Lee, B.; Yoon, S. Deep learning in bioinformatics. Brief. Bioinform. 2016, 18, bbw068. [Google Scholar] [CrossRef] [PubMed]
- Dao, F.Y.; Yang, H.; Su, Z.D.; Yang, W.; Wu, Y.; Hui, D.; Chen, W.; Tang, H.; Lin, H. Recent Advances in Conotoxin Classification by Using Machine Learning Methods. Molecules 2017, 22, 1057. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Watkins, M.; Robinson, S.; Safavi-Hemami, H.; Yandell, M. Discovery of Novel Conotoxin Candidates Using Machine Learning. Toxins 2018, 10, 503. [Google Scholar] [CrossRef] [PubMed]
- Xianfang, W.; Junmei, W.; Xiaolei, W.; Yue, Z. Predicting the Types of Ion Channel-Targeted Conotoxins Based on AVC-SVM Model. BioMed. Res. Int. 2017, 2017, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zheng, Y.; Tang, H. Identifying the Types of Ion Channel-Targeted Conotoxins by Incorporating New Properties of Residues into Pseudo Amino Acid Composition. BioMed. Res. Int. 2016, 2016, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.F.; Ding, C.; Guo, S.H.; Ding, H.; Chen, W.; Lin, H. Prediction of the types of ion channel-targeted conotoxins based on radial basis function network. Toxicol. In Vitro 2013, 27, 852–856. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, C.; Gao, R.; Yang, R.; Song, Q. Using the SMOTE technique and hybrid features to predict the types of ion channel-targeted conotoxins. J. Theor. Biol. 2016, 403, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Deng, E.Z.; Yuan, L.F.; Liu, L.; Lin, H.; Chen, W.; Chou, K.C. iCTX-type: A sequence-based predictor for identifying the types of conotoxins in targeting ion channels. BioMed. Res. Int. 2014, 2014, 286419. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.X.; Song, J.; Kong, X.; Shen, H.B. PredCSF: An Integrated Feature-Based Approach for Predicting Conotoxin Superfamily. Protein Pept. Lett. 2011, 18, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Śledź, P.; Caflisch, A. Protein structure-based drug design: from docking to molecular dynamics. Curr. Opin. Struct. Biol. 2018, 48, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Younis, S.; Rashid, S. Alpha conotoxin-BuIA globular isomer is a competitive antagonist for oleoyl-L-alpha-lysophosphatidic acid binding to LPAR6; A molecular dynamics study. PLoS ONE 2017, 12, e0189154. [Google Scholar] [CrossRef] [PubMed]
- Akcan, M.; Cao, Y.; Chongxu, F.; Craik, D.J. The three-dimensional solution structure of mini-M conotoxin BtIIIA reveals a disconnection between disulfide connectivity and peptide fold. Bioorg. Med. Chem. 2013, 21, 3590–3596. [Google Scholar] [CrossRef] [PubMed]
- Gouda, H.; Yamazaki, K.i.; Hasegawa, J.; Kobayashi, Y.; Nishiuchi, Y.; Sakakibara, S.; Hirono, S. Solution structure of α-conotoxin MI determined by 1H-NMR spectroscopy and molecular dynamics simulation with the explicit solvent water. BBA Protein Struct. Mol. Enzymol. 1997, 1343, 327–334. [Google Scholar] [CrossRef]
- Chi, S.W.; Park, K.H.; Suk, J.E.; Olivera, B.M.; McIntosh, J.M.; Han, K.H. Solution conformation of alphaA-conotoxin EIVA, a potent neuromuscular nicotinic acetylcholine receptor antagonist from Conus ermineus. J. Biol. Chem. 2003, 278, 42208–42213. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, M.B.; Pérez-Reyes, L.I.; López, Z.; de la Cotera, E.P.H.; Falcón, A.; Ayala, C.; Galván, M.; Salvador, C.; Escobar, L.I. Peptide sr11a from Conus spurius is a novel peptide blocker for Kv1 potassium channels. Peptides 2010, 31, 1287–1291. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chang, S.; Yang, L.; Shi, J.; McFarland, K.; Yang, X.; Moller, A.; Wang, C.; Zou, X.; Chi, C.; et al. Conopeptide Vt3.1 preferentially inhibits BK potassium channels containing β4 subunits via electrostatic interactions. J. Biol. Chem. 2014, 289, 4735–4742. [Google Scholar] [CrossRef] [PubMed]
- Platt, R.J.; Curtice, K.J.; Twede, V.D.; Watkins, M.; Gruszczyński, P.; Bulaj, G.; Horvath, M.P.; Olivera, B.M. From molecular phylogeny towards differentiating pharmacology for NMDA receptor subtypes. Toxicon 2014, 81, 67–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasiripourdori, A.; Ranjbar, B.; Naderi-Manesh, H. Binding of long-chain alpha-neurotoxin would stabilize the resting state of nAChR: a comparative study with alpha-conotoxin. Theor. Biol. Med. Model. 2009, 6, 3. [Google Scholar] [CrossRef] [PubMed]
- Zhangsun, D.; Zhu, X.; Wu, Y.; Hu, Y.; Kaas, Q.; Craik, D.J.; McIntosh, J.M.; Luo, S. Key residues in the nicotinic acetylcholine receptor β2 subunit contribute to α-conotoxin LvIA binding. J. Biol. Chem. 2015, 290, 9855–9862. [Google Scholar] [CrossRef] [PubMed]
- Korkosh, V.S.; Zhorov, B.S.; Tikhonov, D.B. Folding similarity of the outer pore region in prokaryotic and eukaryotic sodium channels revealed by docking of conotoxins GIIIA, PIIIA, and KIIIA in a NavAb-based model of Nav1.4. J. Gen. Physiol. 2014, 144, 231–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, D.; Mahdavi, S.; Kuyucak, S. Computational Study of Binding of μ-Conotoxin GIIIA to Bacterial Sodium Channels NaVAb and NaVRh. Biochemistry 2016, 55, 1929–1938. [Google Scholar] [CrossRef] [PubMed]
- Leffler, A.E.; Kuryatov, A.; Zebroski, H.A.; Powell, S.R.; Filipenko, P.; Hussein, A.K.; Gorson, J.; Heizmann, A.; Lyskov, S.; Tsien, R.W.; et al. Discovery of peptide ligands through docking and virtual screening at nicotinic acetylcholine receptor homology models. Proc. Natl. Acad. Sci. USA 2017, 114, E8100–E8109. [Google Scholar] [CrossRef] [PubMed]
- Verdier, L.; Al-Sabi, A.; Rivier, J.E.F.; Olivera, B.M.; Terlau, H.; Carlomagno, T. Identification of a novel pharmacophore for peptide toxins interacting with K+ channels. J. Biol. Chem. 2005, 280, 21246–21255. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, G.; Aliste, M.P.; Tieleman, D.P.; French, R.J.; Dudley, S.C., Jr. Docking of μ-Conotoxin GIIIA in the Sodium Channel Outer Vestibule. Channels 2007, 1, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Beissner, M.; Dutertre, S.; Schemm, R.; Danker, T.; Sporning, A.; Grubmuller, H.; Nicke, A. Efficient Binding of 4/7 α-Conotoxins to Nicotinic α4β2 Receptors Is Prevented by Arg185 and Pro195 in the α4 Subunit. Mol. Pharmacol. 2012, 82, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Craik, D.J.; Kaas, Q. Blockade of Neuronal α7-nAChR by α-Conotoxin ImI Explained by Computational Scanning and Energy Calculations. PLoS Comput. Biol. 2011, 7, e1002011. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.; Bosmans, F.; Kaas, Q.; Cheneval, O.; Conibear, A.C.; Rosengren, K.J.; Wang, C.K.; Schroeder, C.I.; Craik, D.J. Efficient enzymatic cyclization of an inhibitory cystine knot-containing peptide. Biotechnol. Bioeng. 2016, 113, 2202–2212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lúcio, A.D.; Mazzoni, M.S. Toxins by first-principles: Electronic structure mapping structural changes. J. Mol. Struc-Theochem 2008, 853, 58–61. [Google Scholar] [CrossRef]
- McDougal, O.M.; Granum, D.M.; Swartz, M.; Rohleder, C.; Maupin, C.M. pKa Determination of Histidine Residues in α-Conotoxin MII Peptides by 1H NMR and Constant pH Molecular Dynamics Simulation. J. Phys. Chem. B 2013, 117, 2653–2661. [Google Scholar] [CrossRef] [PubMed]
- Hopping, G.; Wang, C.I.A.; Hogg, R.C.; Nevin, S.T.; Lewis, R.J.; Adams, D.J.; Alewood, P.F. Hydrophobic residues at position 10 of α-conotoxin PnIA influence subtype selectivity between α7 and α3β2 neuronal nicotinic acetylcholine receptors. Biochem. Pharmacol. 2014, 91, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Cuny, H.; Kompella, S.N.; Tae, H.S.; Yu, R.; Adams, D.J. Key Structural Determinants in the Agonist Binding Loops of Human β2 and β4 Nicotinic Acetylcholine Receptor Subunits Contribute to α3β4 Subtype Selectivity of α-Conotoxins. J. Biol. Chem. 2016, 291, 23779–23792. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, S.; Belgi, A.; Bartels, P.; van Lierop, B.J.; Robinson, S.D.; Kompella, S.N.; Hung, A.; Callaghan, B.P.; Adams, D.J.; Robinson, A.J.; et al. Dicarba Analogues of α-Conotoxin RgIA. Structure, Stability, and Activity at Potential Pain Targets. J. Med. Chem. 2014, 57, 9933–9944. [Google Scholar] [CrossRef] [PubMed]
- Pucci, L.; Grazioso, G.; Dallanoce, C.; Rizzi, L.; De Micheli, C.; Clementi, F.; Bertrand, S.; Bertrand, D.; Longhi, R.; De Amici, M.; et al. Engineering of α-conotoxin MII-derived peptides with increased selectivity for native α6β2* nicotinic acetylcholine receptors. FASEB J. 2011, 25, 3775–3789. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Lee, S.H.; Kim, D.H.; Han, K.H. Molecular docking study on the α3β2 neuronal nicotinic acetylcholine receptor complexed with α-Conotoxin GIC. BMB Rep. 2012, 45, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Xu, M.; Zhu, X.; Wu, Y.; Liu, X.; Zhangsun, D.; Hu, Y.; Xiang, S.H.; Kasheverov, I.E.; Tsetlin, V.I.; et al. From crystal structure of α-conotoxin GIC in complex with Ac-AChBP to molecular determinants of its high selectivity for α3β2 nAChR. Sci. Rep. 2016, 6, 22349. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.W.; McIntosh, J.M. α6 nAChR subunit residues that confer α-conotoxin BuIA selectivity. FASEB J. 2012, 26, 4102–4110. [Google Scholar] [CrossRef] [PubMed]
- Kompella, S.N.; Cuny, H.; Hung, A.; Adams, D.J. Molecular Basis for Differential Sensitivity of α-Conotoxin RegIIA at Rat and Human Neuronal Nicotinic Acetylcholine Receptors. Mol. Pharmacol. 2015, 88, 993–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutertre, S.; Nicke, A.; Lewis, R.J. Beta2 subunit contribution to 4/7 alpha-conotoxin binding to the nicotinic acetylcholine receptor. J. Biol. Chem. 2005, 280, 30460–30468. [Google Scholar] [CrossRef] [PubMed]
- Pérez, E.G.; Cassels, B.K.; Zapata-Torres, G. Molecular modeling of the α9α10 nicotinic acetylcholine receptor subtype. Bioorg. Med. Chem. Lett. 2009, 19, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Grishin, A.A.; Cuny, H.; Hung, A.; Clark, R.J.; Brust, A.; Akondi, K.; Alewood, P.F.; Craik, D.J.; Adams, D.J. Identifying key amino acid residues that affect α-conotoxin AuIB inhibition of α3β4 nicotinic acetylcholine receptors. J. Biol. Chem. 2013, 288, 34428–34442. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhu, X.; Harvey, P.J.; Kaas, Q.; Zhangsun, D.; Craik, D.J.; Luo, S. Single Amino Acid Substitution in α-Conotoxin TxID Reveals a Specific α3β4 Nicotinic Acetylcholine Receptor Antagonist. J. Med. Chem. 2018, 61, 9256–9265. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhangsun, D.; Zhu, X.; Kaas, Q.; Zhangsun, M.; Harvey, P.J.; Craik, D.J.; McIntosh, J.M.; Luo, S. α-Conotoxin [S9A]TxID Potently Discriminates between α3β4 and α6/α3β4 Nicotinic Acetylcholine Receptors. J. Med. Chem. 2017, 60, 5826–5833. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Yan, Z.; Liu, Z.; Wang, S.; Wu, Q.; Yu, S.; Ding, J.; Dai, Q. Molecular basis of toxicity of N-type calcium channel inhibitor MVIIA. Neuropharmacology 2016, 101, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Ellison, M.; Gao, F.; Wang, H.L.W.; Sine, S.M.; McIntosh, J.M.; Olivera, B.M. α-Conotoxins ImI and ImII Target Distinct Regions of the Human α7 Nicotinic Acetylcholine Receptor and Distinguish Human Nicotinic Receptor Subtypes. Biochemistry 2004, 43, 16019–16026. [Google Scholar] [CrossRef] [PubMed]
- McArthur, J.R.; Singh, G.; O’Mara, M.L.; McMaster, D.; Ostroumov, V.; Tieleman, D.P.; French, R.J. Orientation of μ-Conotoxin PIIIA in a Sodium Channel Vestibule, Based on Voltage Dependence of Its Binding. Mol. Pharmacol. 2011, 80, 219–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortez, L.; Marino-Buslje, C.; de Jiménez Bonino, M.B.; Hellman, U. Interactions between α-conotoxin MI and the Torpedo marmorata receptor α–δ interface. Biochem. Biophys. Res. Commun. 2007, 355, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Kompella, S.N.; Adams, D.J.; Craik, D.J.; Kaas, Q. Determination of the α-Conotoxin Vc1.1 Binding Site on the α9α10 Nicotinic Acetylcholine Receptor. J. Med. Chem. 2013, 56, 3557–3567. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Chan, F.; Hwang, J.; Lyu, P. Calcium binding mode of γ-carboxyglutamic acids in conantokins. Protein Eng. Des. Sel. 1999, 12, 589–595. [Google Scholar] [CrossRef]
- Armishaw, C.J.; Singh, N.; Medina-Franco, J.L.; Clark, R.J.; Scott, K.C.M.; Houghten, R.A.; Jensen, A.A. A synthetic combinatorial strategy for developing alpha-conotoxin analogs as potent alpha7 nicotinic acetylcholine receptor antagonists. J. Biol. Chem. 2010, 285, 1809–1821. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Akondi, K.B.; Zhangsun, D.; Wu, Y.; Zhu, X.; Hu, Y.; Christensen, S.; Dowell, C.; Daly, N.L.; Craik, D.J.; Wang, C.I.A.; Lewis, R.J.; Alewood, P.F.; McIntosh, J.M. Atypical alpha-conotoxin LtIA from Conus litteratus targets a novel microsite of the alpha3beta2 nicotinic receptor. J. Biol. Chem. 2010, 285, 12355–12366. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, S.; Nicke, A.; Tyndall, J.D.A.; Lewis, R.J. Determination of α-conotoxin binding modes on neuronal nicotinic acetylcholine receptors. J. Mol. Recognit. 2004, 17, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Tietze, D.; Leipold, E.; Heimer, P.; Böhm, M.; Winschel, W.; Imhof, D.; Heinemann, S.H.; Tietze, A.A. Molecular interaction of δ-conopeptide EVIA with voltage-gated Na+ channels. BBA—Gen. Subjects 2016, 1860, 2053–2063. [Google Scholar] [CrossRef] [PubMed]
- Mahdavi, S.; Kuyucak, S. Systematic study of binding of μ-conotoxins to the sodium channel NaV1.4. Toxins 2014, 6, 3454–3470. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Huang, W.; Jiang, T.; Yu, R. Determination of the μ-Conotoxin PIIIA Specificity Against Voltage-Gated Sodium Channels from Binding Energy Calculations. Mar. Drugs 2018, 16, 153. [Google Scholar] [CrossRef] [PubMed]
- Torrie, G.; Valleau, J. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Chen, R.; Chung, S.H. Complex Structures between the N-Type Calcium Channel (CaV 2.2) and ω-Conotoxin GVIA Predicted via Molecular Dynamics. Biochemistry 2013, 52, 3765–3772. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Robinson, A.; Chung, S.H. Mechanism of μ-Conotoxin PIIIA Binding to the Voltage-Gated Na+ Channel NaV1.4. PLoS ONE 2014, 9, e93267. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Tabassum, N.; Jiang, T. Investigation of α-conotoxin unbinding using umbrella sampling. Bioorg. Med. Chem. Lett. 2016, 26, 1296–1300. [Google Scholar] [CrossRef] [PubMed]
- Suresh, A.; Hung, A. Molecular Simulation study of the unbinding of α-conotoxin [Υ4E]GID at the α7 and α4β2 neuronal nicotinic acetylcholine receptors. J. Mol. Graph. Model. 2016, 70, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Kaas, Q.; Craik, D.J. Delineation of the Unbinding Pathway of α-Conotoxin ImI from the α7 Nicotinic Acetylcholine Receptor. J. Phys. Chem. B 2012, 116, 6097–6105. [Google Scholar] [CrossRef] [PubMed]
- Jarzynski, C. Nonequilibrium equality for free energy differences. Phys. Rev. Lett. 1997, 78, 2690. [Google Scholar] [CrossRef]
- Huang, X.; Dong, F.; Zhou, H.X. Electrostatic Recognition and Induced Fit in the κ-PVIIA Toxin Binding to Shaker Potassium Channel. J. Am. Chem. Soc. 2005, 127, 6836–6849. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Ma, J. Conformational Simulations of Aqueous Solvated α-Conotoxin GI and Its Single Disulfide Analogues Using a Polarizable Force Field Model. J. Phys. Chem. A 2008, 112, 9854–9867. [Google Scholar] [CrossRef] [PubMed]
- Karayiannis, N.C.; Laso, M.; Kroger, M. Detailed Atomistic Molecular Dynamics Simulations of α-Conotoxin AuIB in Water. J. Phys. Chem. B 2009, 113, 5016–5024. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Pirogova, E. Static electric fields induce conformational changes in alpha conotoxin: A molecular dyanamics simulation study. In Proceedings of the 2017 Progress in Electromagnetics Research Symposium—Fall (PIERS— FALL), Singapore, 19–22 November 2017; pp. 1273–1277. [Google Scholar]
- Sajeevan, K.A.; Roy, D. Aqueous ionic liquids influence the disulfide bond isoform equilibrium in conotoxin AuIB: A consequence of the Hofmeister effect? Biophys. Rev. 2018, 10, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Sajeevan, K.A.; Roy, D. Peptide Sequence and Solvent as Levers to Control Disulfide Connectivity in Multiple Cysteine Containing Venom Toxins. J. Phys. Chem. B 2018, 122, 5776–5789. [Google Scholar] [CrossRef] [PubMed]
- Paul George, A.A.; Heimer, P.; Maaß, A.; Hamaekers, J.; Hofmann-Apitius, M.; Biswas, A.; Imhof, D. Insights into the Folding of Disulfide-Rich μ-Conotoxins. ACS Omega 2018, 3, 12330–12340. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Seymour, V.A.L.; Berecki, G.; Jia, X.; Akcan, M.; Adams, D.J.; Kaas, Q.; Craik, D.J. Less is More: Design of a Highly Stable Disulfide-Deleted Mutant of Analgesic Cyclic α-Conotoxin Vc1.1. Sci. Rep. 2015, 5, 13264. [Google Scholar] [CrossRef] [PubMed]
- Tabassum, N.; Tae, H.S.; Jia, X.; Kaas, Q.; Jiang, T.; Adams, D.J.; Yu, R. Role of Cys I–Cys III Disulfide Bond on the Structure and Activity of α-Conotoxins at Human Neuronal Nicotinic Acetylcholine Receptors. ACS Omega 2017, 2, 4621–4631. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Xu, Q.; Chen, F.; Shi, J.; Liu, Y.; Chu, Y.; Wan, S.; Jiang, T.; Yu, R. Role of the disulfide bond on the structure and activity of μ-conotoxin PIIIA in the inhibition of NaV1.4. RSC Adv. 2019, 9, 668–674. [Google Scholar] [CrossRef]
- Lee, H.K.; Zhang, L.; Smith, M.D.; Walewska, A.; Vellore, N.A.; Baron, R.; McIntosh, J.M.; White, H.S.; Olivera, B.M.; Bulaj, G. A marine analgesic peptide, Contulakin-G, and neurotensin are distinct agonists for neurotensin receptors: uncovering structural determinants of desensitization properties. Front. Pharmacol. 2015, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Li, R.; Ning, J.; Zhu, X.; Zhangsun, D.; Wu, Y.; Luo, S. Effect of Methionine Oxidation and Substitution of α-Conotoxin TxID on α3β4 Nicotinic Acetylcholine Receptor. Mar. Drugs 2018, 16, 215. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Peng, C.; Lin, B.; Chen, Q.; Zhang, J.; Shi, Q. Screening and Validation of Highly-Efficient Insecticidal Conotoxins from a Transcriptome-Based Dataset of Chinese Tubular Cone Snail. Toxins 2017, 9, 214. [Google Scholar] [CrossRef] [PubMed]
- Barba, M.; Sobolev, A.P.; Zobnina, V.; Bonaccorsi di Patti, M.C.; Cervoni, L.; Spiezia, M.C.; Schininà, M.E.; Pietraforte, D.; Mannina, L.; Musci, G.; et al. Cupricyclins, Novel Redox-Active Metallopeptides Based on Conotoxins Scaffold. PLoS ONE 2012, 7, e30739. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Guzman, E.A.; Vega-Castro, N.; Reyes-Montaño, E.A.; Recio-Pinto, E. Antagonistic action on NMDA/GluN2B mediated currents of two peptides that were conantokin-G structure-based designed. BMC Neurosci. 2017, 18, 44. [Google Scholar] [CrossRef] [PubMed]
- King, M.D.; Long, T.; Andersen, T.; McDougal, O.M. Genetic Algorithm Managed Peptide Mutant Screening: Optimizing Peptide Ligands for Targeted Receptor Binding. J. Chem. Inf. Model. 2016, 56, 2378–2387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, M.D.; Long, T.; Pfalmer, D.L.; Andersen, T.L.; McDougal, O.M. SPIDR: small-molecule peptide-influenced drug repurposing. BMC Bioinform. 2018, 19, 138. [Google Scholar] [CrossRef] [PubMed]
- Kasheverov, I.E.; Chugunov, A.O.; Kudryavtsev, D.S.; Ivanov, I.A.; Zhmak, M.N.; Shelukhina, I.V.; Spirova, E.N.; Tabakmakher, V.M.; Zelepuga, E.A.; Efremov, R.G.; et al. High-Affinity α-Conotoxin PnIA Analogs Designed on the Basis of the Protein Surface Topography Method. Sci. Rep. 2016, 6, 36848. [Google Scholar] [CrossRef] [PubMed]
- Pitera, J.W.; Swope, W. Understanding folding and design: replica-exchange simulations of “Trp-cage” miniproteins. Proc. Natl. Acad. Sci. USA 2003, 100, 7587–7592. [Google Scholar] [CrossRef] [PubMed]
- Ensign, D.L.; Kasson, P.M.; Pande, V.S. Heterogeneity Even at the Speed Limit of Folding: Large-scale Molecular Dynamics Study of a Fast-folding Variant of the Villin Headpiece. J. Mol. Biol. 2007, 374, 806–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voelz, V.A.; Bowman, G.R.; Beauchamp, K.; Pande, V.S. Molecular Simulation of ab Initio Protein Folding for a Millisecond Folder NTL9(1-39). J. Am. Chem. Soc. 2010, 132, 1526–1528. [Google Scholar] [CrossRef] [PubMed]
- Sborgi, L.; Verma, A.; Piana, S.; Lindorff-Larsen, K.; Cerminara, M.; Santiveri, C.M.; Shaw, D.E.; de Alba, E.; Muñoz, V. Interaction Networks in Protein Folding via Atomic-Resolution Experiments and Long-Time-Scale Molecular Dynamics Simulations. J. Am. Chem. Soc. 2015, 137, 6506–6516. [Google Scholar] [CrossRef] [PubMed]
- Rohl, C.A.; Strauss, C.E.; Misura, K.M.; Baker, D. Protein structure prediction using Rosetta. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2004; Volume 383, pp. 66–93. [Google Scholar]
- Dill, K.A.; MacCallum, J.L. The protein-folding problem, 50 years on. Science 2012, 338, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.; Sali, A. Protein structure prediction and structural genomics. Science 2001, 294, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z. Advances in Homology Protein Structure Modeling. Curr. Protein Pept. Sci. 2006, 7, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Nabuurs, S.B.; Vriend, G. Homology modeling. Methods Biochem. Anal. 2003, 44, 509–523. [Google Scholar] [PubMed]
- Rost, B. Twilight zone of protein sequence alignments. Protein Eng. Des. Sel. 1999, 12, 85–94. [Google Scholar] [CrossRef] [Green Version]
- Everhart, D.; Cartier, G.E.; Malhotra, A.; Gomes, A.V.; McIntosh, J.M.; Luetje, C.W. Determinants of Potency on α-Conotoxin MII, a Peptide Antagonist of Neuronal Nicotinic Receptors. Biochemistry 2004, 43, 2732–2737. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Vijayan, R.; Shichina, K.; Babu, R.M.; Ramakumar, S. I-Superfamily Conotoxins: Sequence and Structure Analysis. In Silico Biol. 2005, 5, 557–571. [Google Scholar] [PubMed]
- Twede, V.D.; Teichert, R.W.; Walker, C.S.; Gruszczynski, P.; Kazmierkiewicz, R.; Bulaj, G.; Olivera, B.M. Conantokin-Br from Conus brettinghami and Selectivity Determinants for the NR2D Subunit of the NMDA Receptor. Biochemistry 2009, 48, 4063–4073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdes, A.; Anand, P.; Gorson, J.; Jannetti, S.; Kelly, P.; Leffler, A.; Simpson, D.; Ramrattan, G.; Holford, M. From Mollusks to Medicine: A Venomics Approach for the Discovery and Characterization of Therapeutics from Terebridae Peptide Toxins. Toxins 2016, 8, 117. [Google Scholar] [CrossRef] [PubMed]
- Heimer, P.; Tietze, A.A.; Bäuml, C.A.; Resemann, A.; Mayer, F.J.; Suckau, D.; Ohlenschläger, O.; Tietze, D.; Imhof, D. Conformational μ-Conotoxin PIIIA Isomers Revisited: Impact of Cysteine Pairing on Disulfide-Bond Assignment and Structure Elucidation. Anal. Chem. 2018, 90, 3321–3327. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.H.; Tseng, L.Y. DBCP: A web server for disulfide bonding connectivity pattern prediction without the prior knowledge of the bonding state of cysteines. Nucleic Acids Res. 2010, 38, W503–W507. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; He, B.J.; Jang, R.; Zhang, Y.; Shen, H.B. Accurate disulfide-bonding network predictions improve ab initio structure prediction of cysteine-rich proteins. Bioinformatics 2015, 31, btv459. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Zou, S.; Sun, Y.; Zhang, S. GL-BLSTM: A novel structure of bidirectional long-short term memory for disulfide bonding state prediction. arXiv, 2018; arXiv:1808.03745. [Google Scholar]
- Espiritu, M.J. Disulfide Bond and Topological Isomerization of the Conopeptide PnID: Disulfide Bonds with a Twist. Ph.D. Thesis, University of Hawai’i at Manoa, Honolulu, HI, USA, 2017. [Google Scholar]
- Steiner, A.M.; Bulaj, G. Optimization of oxidative folding methods for cysteine-rich peptides: A study of conotoxins containing three disulfide bridges. J. Pept. Sci. 2011, 17, 1–7. [Google Scholar] [CrossRef] [PubMed]



| Category | Type | Description |
|---|---|---|
| Gene superfamily | sequence | Clustering of precursor region |
| Cysteine framework | sequence | Arrangement of cysteines |
| Loop class | sequence | Number of amino acids between cysteines |
| Disulfide connectivity | structure | Pattern of disulfide bond formation |
| Fold | structure | General three-dimensional structure |
| Subfold | structure | More specific three-dimensional structure |
| Pharmacological family | action | Target and mode of action (agonist, antagonist, etc.) |
| Framework Name | Pattern | No. Cysteines |
|---|---|---|
| I | CC-C-C | 4 |
| II | CCC-C-C-C | 6 |
| III | CC-C-C-CC | 6 |
| IV | CC-C-C-C-C | 6 |
| V | CC-CC | 4 |
| VI/VII | C-C-CC-C-C | 6 |
| VIII | C-C-C-C-C-C-C-C-C-C | 10 |
| IX | C-C-C-C-C-C | 6 |
| X | CC-C.[PO]C | 4 |
| XI | C-C-CC-CC-C-C | 8 |
| XII | C-C-C-C-CC-C-C | 8 |
| XIII | C-C-C-CC-C-C-C | 8 |
| XIV | C-C-C-C | 4 |
| XV | C-C-CC-C-C-C-C | 8 |
| XVI | C-C-CC | 4 |
| XVII | C-C-CC-C-CC-C | 8 |
| XVIII | C-C-CC-CC | 6 |
| XIX | C-C-C-CCC-C-C-C-C | 10 |
| XX | C-CC-C-CC-C-C-C-C | 10 |
| XXI | CC-C-C-C-CC-C-C-C | 10 |
| XXII | C-C-C-C-C-C-C-C | 8 |
| XXIII | C-C-C-CC-C | 6 |
| XXIV | C-CC-C | 4 |
| XXV | C-C-C-C-CC | 6 |
| XXVI | C-C-C-C-CC-CC | 8 |
| XXVII | C-CC-C-C-C | 6 |
| Family | Target | Mode of Action |
|---|---|---|
| (alpha) | Nicotinic acetylcholine receptors (nAChRs) | orthosteric, allosteric inhibition |
| (gamma) | Neuronal pacemaker cation currents | increase calcium current |
| (delta) | Voltage-gated sodium channels (VGSCs) | agonist, delayed inactivation |
| (epsilon) | Presynaptic calcium channels or G protein-coupled presynaptic receptors (GPCRs) | blocker |
| (iota) | VGSC | agonist, no delayed inactivation |
| (kappa) | Voltage-gated potassium channels (VGPCs) | blocker |
| (mu) | VGSC | antagonist, blocker |
| (rho) | Alpha-1 adrenergic receptors | allosteric inhibitor |
| (sigma) | Serotonin-gated ion channels | antagonist |
| (tau) | Somatostatin receptor | antagonist |
| (chi) | Neuronal noradrenaline transporter | unknown |
| (omega) | Voltage-gated calcium channels (VGCCs) | blocker |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mansbach, R.A.; Travers, T.; McMahon, B.H.; Fair, J.M.; Gnanakaran, S. Snails In Silico: A Review of Computational Studies on the Conopeptides. Mar. Drugs 2019, 17, 145. https://doi.org/10.3390/md17030145
Mansbach RA, Travers T, McMahon BH, Fair JM, Gnanakaran S. Snails In Silico: A Review of Computational Studies on the Conopeptides. Marine Drugs. 2019; 17(3):145. https://doi.org/10.3390/md17030145
Chicago/Turabian StyleMansbach, Rachael A., Timothy Travers, Benjamin H. McMahon, Jeanne M. Fair, and S. Gnanakaran. 2019. "Snails In Silico: A Review of Computational Studies on the Conopeptides" Marine Drugs 17, no. 3: 145. https://doi.org/10.3390/md17030145
APA StyleMansbach, R. A., Travers, T., McMahon, B. H., Fair, J. M., & Gnanakaran, S. (2019). Snails In Silico: A Review of Computational Studies on the Conopeptides. Marine Drugs, 17(3), 145. https://doi.org/10.3390/md17030145