Jellyfish-Associated Microbiome in the Marine Environment: Exploring Its Biotechnological Potential

 ,
, 
 and
and
Abstract
:1. Introduction
1.1. Biotechnological Potential of Host–Microorganism Systems in the Ocean
1.2. Gelatinous Zooplankton as Host for Specific Microbiome
2. Jellyfish-Associated Microbiome
2.1. Microbiome Associated with Specific Jellyfish Taxa
2.1.1. Medusozoa
2.1.2. Scyphozoa
2.1.3. Semaeostomeae
2.1.4. Ulmaridae
2.1.5. Cyaneidae
2.1.6. Pelagiidae
2.1.7. Rhizostomeae
2.1.8. Cubozoa
2.1.9. Hydrozoa
2.1.10. Ctenophora
2.2. Critical Overview of Methodological Approaches Used to Study Jellyfish Microbiome
3. Characteristics of the Jellyfish-Associated Microbiome
3.1. What is the Degree of Specialization of the Jellyfish-Associated Microbiome?
3.2. Is the Jellyfish-Associated Microbiome Jellyfish Population-Specific?
3.3. Is there a Jellyfish Taxa-Specific Microbiome?
3.4. Is the Jellyfish-Associated Microbiome Specific to Different Life Stages?
3.5. Is the Jellyfish-Associated Microbiome Body Part-Specific?
3.5.1. Microbiome Associated with Outer Body Parts and Its Potential Role
3.5.2. Microbiome Associated with Inner Body Compartments and Its Potential Role
3.6. The Composition, Potential Role, and Biotechnological Potential of the Jellyfish-Associated Microbiome
3.6.1. Gamma- and Alphaproteobacteria
3.6.2. Bacteroidetes, Flavobacteria, Flavobacteriaceae
3.6.3. Tenericutes
3.6.4. Minor Members of the Jellyfish Microbiome
4. Conclusions and Future Research Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bull, A.T. Microbial Diversity and Bioprospecting; American Society for Microbiology (ASM) Press: Washington, DC, USA, 2004. [Google Scholar]
- Azam, F.; Malfatti, F. Microbial structuring of marine ecosystems. Nat. Rev. Microbiol. 2007, 5, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Gulder, T.A.M.; Moore, B.S. Chasing the treasures of the sea—Bacterial marine natural products. Curr. Opin. Microbiol. 2009, 12, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Antranikian, G. Industrial relevance of thermophiles and their enzymes. In Thermophiles: Biology and Technology at High Temperatures; Robb, F., Grogan, D., Eds.; GRC Press: Boca Raton, FL, USA, 2007; pp. 113–189. [Google Scholar]
- Kennedy, J.; Marchesi, J.R.; Dobson, A.D.W. Marine metagenomics: Strategies for discovery of novel enzymes with biotechnological applications from marine environments. Microb. Cell Factories 2008, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Munn, C.B. Marine Microbiology: Ecology and Applications; Garland Science, CRC Press: Abingdon, UK, 2011. [Google Scholar]
- Fenical, W. Marine Pharmaceuticals: Past, Present and Future. Oceanography 2006, 19, 112–119. [Google Scholar] [CrossRef]
- Newman, D.J.; Hill, R.T. New drugs from marine microbes: The tide is running. J. Ind. Microbiol. Biotechnol. 2006, 33, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.G. Panning for chemical gold: Marine bacteria as a source of new therapeutics. Trends Biotechnol. 2009, 27, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, A.; Naughton, L.M.; Montánchez, I.; Dobson, A.D.W.; Rai, D.K. Current Status and Future Prospects of Marine Natural Products (MNPs) as Antimicrobials. Mar. Drugs 2017, 15, 272. [Google Scholar] [CrossRef]
- Taylor, M.W.; Radax, R.; Steger, D.; Wagner, M. Sponge-associated microorganisms: Evolution, ecology, and biotechnological potential. Microbiol. Mol. Mol. Rev. 2007, 71, 295–347. [Google Scholar] [CrossRef]
- Thomas, T.R.A.; Kavlekar, D.P.; LokaBharathi, P.A. Marine drugs from sponge-microbe association—A review. Mar. Drugs 2010, 8, 1417–1468. [Google Scholar] [CrossRef]
- Penesyan, A.; Kjelleberg, S.; Egan, S. Development of novel drugs from marine surface associated microorganisms. Mar. Drugs 2010, 8, 438–459. [Google Scholar] [CrossRef]
- Blockley, A.; Elliott, D.R.; Roberts, A.P.; Sweet, M. Symbiotic Microbes from Marine Invertebrates: Driving a New Era of Natural Product Drug Discovery. Diversity 2017, 9, 49. [Google Scholar] [CrossRef]
- Haygood, M.G.; Schmidt, G.; Davidson, E.W.; Faulkner, D.J. Microbial symbionts of marine invertebrates: Opportunities for microbial biotechnology. J. Mol. Microbiol. Biotechnol. 1999, 1, 33–43. [Google Scholar] [PubMed]
- Brinkmann, C.M.; Marker, A.; Kurtböke, I. An Overview on Marine Sponge-Symbiotic Bacteria as Unexhausted Sources for Natural Product Discovery. Diversity 2017, 9, 40. [Google Scholar] [CrossRef]
- Rizzo, C.; Lo Giudice, A. Marine Invertebrates: Underexplored Sources of Bacteria Producing Biologically Active Molecules. Diversity 2018, 10, 52. [Google Scholar] [CrossRef]
- Stabili, L.; Parisi, M.G.; Parrinello, D.; Cammarata, M. Cnidarian Interaction with Microbial Communities: From Aid to Animal’s Health to Rejection Responses. Mar Drugs 2018, 16, 296. [Google Scholar] [CrossRef] [PubMed]
- Bosch, T.C.G. Cnidarian-Microbe Interactions and the Origin of Innate Immunity in Metazoans. Ann. Rev. Microbiol. 2013, 67, 499–518. [Google Scholar] [CrossRef] [PubMed]
- Condon, R.H.; Duarte, C.M.; Pitt, K.A.; Robinson, K.L.; Lucas, C.H.; Sutherland, K.R.; Mianzan, H.W.; Bogeberg, M.; Purcell, J.E.; Decker, M.B.; et al. Recurrent jellyfish blooms are a consequence of global oscillations. Proc. Natl. Acad. Sci. USA 2013, 110, 1000–1005. [Google Scholar] [CrossRef]
- Condon, R.H.; Steinberg, D.K.; del Giorgio, P.A.; Bouvier, T.C.; Bronk, D.A.; Graham, W.M.; Hugh, W.; Ducklow, H.W. Jellyfish blooms result in a major microbial respiratory sink of carbon in marine systems. Proc. Natl. Acad. Sci. USA 2011, 108, 10225–10230. [Google Scholar] [CrossRef]
- Riemann, L.; Titelman, J.; Båmstedt, U. Links between jellyfish and microbes in a jellyfish dominated fjord. Mar. Ecol. Prog. Ser. 2006, 325, 29–42. [Google Scholar] [CrossRef]
- Dinasquet, J.; Granhag, L.; Riemann, L. Stimulated bacterioplankton growth and selection for certain bacterial taxa in the vicinity of the ctenophore Mnemiopsis leidyi. Front. Microbiol. 2012, 3, 302. [Google Scholar] [CrossRef]
- Manzari, C.; Fosso, B.; Marzano, M.; Annese, A.; Caprioli, R.; D’Erchia, A.M.; Gissi, C.; Intranuovo, M.; Picardi, E.; Santamaria, M.; et al. The influence of invasive jellyfish blooms on the aquatic microbiome in a coastal lagoon (Varano, SE Italy) detected by an Illumina-based deep sequencing strategy. Biol. Invas. 2015, 17, 923–940. [Google Scholar] [CrossRef]
- Blanchet, M.; Pringault, O.; Bouvy, M.; Catala, P.; Oriol, L.; Caparros, J.; Ortega-Retuerta, E.; Intertaglia, L.; West, N.; Agis, M.; et al. Changes in bacterial community metabolism and composition during the degradation of dissolved organic matter from the jellyfish Aurelia aurita in a Medeterranean coastal lagoon. Environ. Sci. Pollut. Res. Int. 2014, 22, 13638–13653. [Google Scholar] [CrossRef] [PubMed]
- Dinasquet, J.; Kragh, T.; Schroter, M.L.; Sondergard, M.; Riemann, L. Functional and compositional succession of bacterioplankton in response to a gradient in bioavailable dissolved organic carbon. Environ. Microbiol. 2013, 15, 2616–2628. [Google Scholar] [CrossRef] [PubMed]
- Tinta, T.; Kogovšek, T.; Malej, A.; Turk, V. Jellyfish Modulate Bacterial Dynamic and Community Structure. PLoS ONE 2012, 7, e39274. [Google Scholar] [CrossRef] [PubMed]
- Tinta, T.; Kogovšek, T.; Turk, V.; Shiganova, T.A.; Mikaelyan, A.S.; Malej, A. Microbial transformation of jellyfish organic matter affects the nitrogen cycle in the marine water column—A Black Sea case study. J. Exp. Mar. Biol. Ecol. 2016, 475, 19–30. [Google Scholar] [CrossRef]
- Tinta, T.; Malej, A.; Kos, M.; Turk, V. Degradation of the Adriatic medusa Aurelia sp. by ambient bacteria. Hydrobiologia 2010, 645, 179–191. [Google Scholar] [CrossRef]
- Bruno, D.W.; Ellis, A.E. Mortalities in farmed Atlantic salmon associated with the jellyfish Phialella quadrata. Bull. Eur. Ass. Fish Pathol. 1985, 5, 64. [Google Scholar]
- Margulis, L.; Thorington, G.; Berger, B.; Stolz, J. Endosymbiotic bacteria associated with the intracellular green algae of Hydria Viridis. Curr. Microbiol. 1987, 1, 227–232. [Google Scholar] [CrossRef]
- Ferguson, H.W.; Delannoy, C.M.J.; Hay, S.; Nicolson, J.; Sutherland, D.; Crumlish, M. Jellyfish as vectors of bacterial disease for farmed salmon (Salmo salar). J. Vet. Diagn. Investig. 2010, 22, 376–382. [Google Scholar] [CrossRef]
- Delannoy, C.M.J.; Houghton, J.D.R.; Fleming, N.E.C.; Ferguson, H.W. Mauve Stingers (Pelagia noctiluca) as carriers of the bacterial fish pathogen Tenacibaculum maritimum. Aquaculture 2011, 311, 255–257. [Google Scholar] [CrossRef]
- Fringuelli, E.; Savage, P.D.; Gordon, A.; Baxter, E.J.; Rodger, H.D.; Graham, D.A. Development of a quantitative real-time PCR for the detection of Tenacibaculum maritimum and its application to field samples. J. Fish Dis. 2012, 35, 579–590. [Google Scholar] [CrossRef]
- Weiland-Bräuer, N.; Neulinger, S.C.; Pinnow, N.; Künzel, S.; Baines, J.F.; Schmitz, R.A. Composition of bacterial communities associated with Aurelia aurita changes with compartment, life stage, and population. Appl. Environ. Microbiol. 2015, 81, 6038–6052. [Google Scholar] [CrossRef] [PubMed]
- Daley, M.C.; Urban-Rich, J.; Moisander, P.H. Bacterial associations with the hydromedusa Nemopsis bachei and scyphomedusa Aurelia aurita from the North Atlantic Ocean. Mar. Biol. Res. 2016, 12, 1088–1100. [Google Scholar] [CrossRef]
- Kos Kramar, M.; Tinta, T.; Lučić, D.; Malej, A.; Turk, V. Bacteria Associated with Moon Jellyfish during Bloom and Post-bloom Periods in the Gulf of Trieste (northern Adriatic). PLoS ONE 2019, 14, e0198056. [Google Scholar] [CrossRef] [PubMed]
- Schuett, C.; Doepke, H. Endobiotic bacteria and their pathogenic potential in cnidarian tentacles. Helgol. Mar. Res. 2010, 64, 205–212. [Google Scholar] [CrossRef]
- Hao, W.; Gerdts, G.; Holst, S.; Wichels, A. Bacterial communities associated with scyphomedusae at Helgoland Roads. Mar. Biodiv. 2018. [Google Scholar] [CrossRef]
- Lee, D.L.; Kling, J.D.; Araya, R.; Ceh, J. Jellyfish life stages shape associated microbial communities, while a core microbiome is maintained across all. Front. Microbiol. 2018, 9, 1534. [Google Scholar] [CrossRef]
- Cleary, D.F.R.; Becking, L.E.; Polónia, A.R.M.; Freitas, R.M.; Gomes, N.C.M. Jellyfish-associated bacterial communities and bacterioplankton in Indonesian Marine lakes. FEMS Microbiol. Ecol. 2016, 92, 1–14. [Google Scholar] [CrossRef]
- Cortés-Lara, S.; Urdiain, M.; Mora-Ruiz, M.; Prieto, L.; Rosselló-Móra, R. Prokaryotic microbiota in the digestive cavity of the jellyfish Cotylorhiza tuberculata. Syst. Appl. Microbiol. 2015, 38, 494–500. [Google Scholar] [CrossRef]
- Viver, T.; Orellana, L.H.; Hatt, J.K.; Urdiain, M.; Diaz, S.; Richter, M.; Anton, J.; Avian, M.; Amann, R.; Konstantinidis, K.T.; et al. The low diverse gastric microbiome of the jellyfish Cotylorhiza tuberculata is dominated by four novel taxa. Environ. Microbiol. 2017, 19, 3039–3058. [Google Scholar] [CrossRef]
- Daniels, C.; Breitbart, M. Bacterial communities associated with the ctenophores Mnemiopsis leidyi and Beroe ovata. FEMS Microbiol. Ecol. 2012, 82, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Hao, W.; Gerdts, G.; Peplies, J.; Wichels, A. Bacterial communities associated with four ctenophore genera from the German Bight (North Sea). FEMS Microbiol. Ecol. 2015, 91, 1–11. [Google Scholar] [CrossRef]
- Mayer, A.G. Medusae of the World. Volume III. The Scyphomedusae; Carnegie Institution of Washington: Washington, DC, USA, 1910; 735p. [Google Scholar]
- Kramp, P.L. Synopsis of the Medusae of the World. J. Mar. Biol. Assoc. UK 1961, 40, 7–382. [Google Scholar] [CrossRef]
- Russell, F.S. Medusae of the British Isles. Vol II. Pelagic Scyphozoa, with a Supplement to Vol. 1; Cambridge University Press: Cambridge, UK, 1970. [Google Scholar]
- Gambill, M.; Jarms, G. Can Aurelia (Cnidaria, Scyphozoa) species be differentiated by comparing their scyphistomae and ephyrae. Eur. J. Taxon. 2014, 107, 1–23. [Google Scholar]
- Dawson, M.N.; Jacobs, D.K. Molecular Evidence for Cryptic Species of Aurelia aurita (Cnidaria, Scyphozoa). Biol. Bull. 2001, 200, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.N. Macro-morphological variation among cryptic species of the moon jellyfish, Aurelia (Cnidaria: Scyphozoa). Mar. Biol. 2003, 143, 369–379. [Google Scholar] [CrossRef]
- Ryan, R.P.; Monchy, S.; Cardinale, M.; Taghavi, S.; Crossman, L.; Avison, M.B.; Berg, G.; Van der Lelie, D.; Dow, J.M. The versatility and adaptation of bacteria from the genus Stenotrophomonas. Nat. Rev. Microbiol. 2009, 7, 514–525. [Google Scholar] [CrossRef]
- Hong, Y.H.; Ye, C.C.; Zhou, Q.Z.; Wu, X.Y.; Yuan, J.P.; Peng, J.; Deng, H.; Wang, J.H. Genome sequencing reveals the potential of Achromobacter sp. HZ01 for bioremediation. Front. Microbiol. 2017, 8, 1507. [Google Scholar] [CrossRef]
- Maravić, A.; Skočibušić, M.; Šprung, M.; Šamanić, I.; Puizina, J.; Pavela-Vrančić, M. Occurrence and antibiotic susceptibility profiles of Burkholderia cepacia complex in coastal marine environment. Int. J. Environ. Health Res. 2012, 22, 531–542. [Google Scholar] [CrossRef]
- Deng, M.C.; Li, J.; Liang, F.R.; Yi, M.; Xu, X.M.; Yuan, J.P.; Peng, J.; Wu, C.F.; Wang, J.H. Isolation and characterization of a novel hydrocarbon-degrading bacterium Achromobacter sp. HZ01 from the crude oil-contaminated seawater at the Daya Bay, southern China. Mar. Pollut. Bull. 2014, 83, 79–86. [Google Scholar] [CrossRef]
- Harayama, S.; Kishira, H.; Kasai, Y.; Shutsubo, K. Petroleum biodegradation in marine environments. J. Mol. Microbol. Biotechnol. 1999, 1, 63–70. [Google Scholar]
- Dang, H.; Lovell, C.R. Microbial Surface Colonization and Biofilm Development in Marine Environments. Microbiol. Mol. Biol. Rev. 2016, 80, 91–138. [Google Scholar] [CrossRef] [PubMed]
- Hay, S.J.; Hislop, J.R.G.; Shanks, A.M. North Sea Scyphomedusae; summer distribution, estimated biomass and significance particularly for 0-group gadoid fish. Neth. J. Sea Res. 1990, 25, 113–130. [Google Scholar] [CrossRef]
- Fehr, A.; Walther, E.; Schmidt-Posthaus, H.; Nufer, L.; Wilson, A.; Svercel, M.; Richter, D.; Segner, H.; Pospischil, A.; Vaughan, L. Candidatus Syngnamydia venezia, a novel member of the phylum Chlamydiae from the broad nosed pipefish Syngnathus typhle. PLoS ONE 2013, 8, e70853. [Google Scholar] [CrossRef] [PubMed]
- Nylund, S.; Steigen, A.; Karlsbakk, E.; Plarre, H.; Andersen, L.; Karlsen, M.; Watanabe, K.; Nylund, A. Characterization of “Candidatus Syngnamydia salmonis” (Chlamydiales, Simkaniaceae), a bacterium associated with epitheoliocystis in Atlantic salmon (Salmo salar). Arch. Microbiol. 2015, 197, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Moss, A.G.; Estes, A.M.; Muellner, L.A.; Morgan, D.D. Protistan epibionts of the ctenophore Mnemiopsis mccradyi Mayer. Hydrobiologia 2001, 451, 295–304. [Google Scholar] [CrossRef]
- Hay, S. Marine Ecology: Gelatinous Bells May Ring Change in Marine Ecosystems. Curr. Biol. 2006, 16, 679–682. [Google Scholar] [CrossRef]
- Reitzel, A.M.; Sullivan, J.C.; Brown, B.K.; Chin, D.W.; Cira, E.K.; Edquist, S.K.; Genco, B.M.; Joseph, O.C.; Kaufman, C.A.; Kovitvongsa, K.; et al. Ecological and developmental dynamics of a host-parasite system involving a sea anemone and two ctenophores. J. Parasitol. 2007, 93, 1392–1402. [Google Scholar] [CrossRef]
- Smith, K.; Dodson, M.; Santos, S.; Gast, R.; Rogerson, A.; Sullivan, B.; Moss, A.G. Pentapharsodinium tyrrhenicum is a parasitic dinoflagellate of the ctenophore Mnemiopsis leidyi. J. Phycol. 2007, 43, 119. [Google Scholar]
- Kogovšek, T.; Vodopivec, M.; Raicich, F.; Uye, S.; Malej, A. Comparative analysis of the ecosystems in the northern Adriatic Sea and the Inland Sea of Japan: Can anthropogenic pressures disclose jellyfish outbreaks? Sci. Total Environ. 2018, 626, 982–994. [Google Scholar] [CrossRef]
- Stewart, E.J. Growing Unculturable Bacteria. J. Bacteriol. 2012, 194, 4151–4160. [Google Scholar] [CrossRef] [PubMed]
- Sogin, M.L.; Morrison, H.G.; Huber, J.A.; Welch, D.M.; Huse, S.M.; Neal, P.R.; Arrieta, J.M.; Herndl, G.J. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc. Natl. Acad. Sci. USA 2006, 103, 12115–12120. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, F.; Seguritan, V.; Azam, F.; Knowlton, N. Diversity and distribution of coral-associated bacteria. Mar. Ecol. Prog. Ser. 2002, 243, 1–10. [Google Scholar] [CrossRef]
- Webster, N.S.; Taylor, M.W.; Behnam, F.; Lücker, S.; Rattei, T.; Whalan, S.; Horn, M.; Wagner, M. Deep sequencing reveals exceptional diversity and modes of transmission for bacterial sponge symbionts. Environ. Microbiol. 2010, 12, 2070–2082. [Google Scholar] [CrossRef]
- Lee, O.O.; Wang, Y.; Yang, J.; Lafi, F.F.; Al-Suwailem, A.; Qian, P.Y. Pyrosequencing reveals highly diverse and species-specific microbial communities in sponges from the Red Sea. ISME J. 2011, 5, 650–664. [Google Scholar] [CrossRef] [PubMed]
- Ovchinnikova, T.V.; Balandin, S.V.; Aleshina, G.M.; Tagaev, A.A.; Leonova, Y.F.; Krasnodembsky, E.D.; Men’shenin, A.V.; Kokryakov, V.N. Aurelin, a novel antimicrobial peptide from jellyfish Aurelia aurita with structural features of defensins and channel-blocking toxins. Biochem. Biophys. Res. Commun. 2006, 348, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Bhosale, S.H.; Nagle, V.L.; Jagtap, T.G. Antifouling potential of some marine organisms from India against species of Bacillus and Pseudomonas. Mar. Biotechnol. 2002, 4, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, F.; Kim, E.L.; Li, J.L.; Hong, J.; Bae, K.S.; Chung, H.Y.; Kim, H.S.; Jung, J.H. Antibacterial polyketides from the jellyfish-derived fungus Paecilomyces variotii. J. Nat. Prod. 2011, 74, 1826–1829. [Google Scholar] [CrossRef]
- Mills, C.E. Jellyfish blooms: Are populations increasing globally in response to changing ocean conditions? Hydrobiologia 2001, 451, 55–68. [Google Scholar] [CrossRef]
- Lucas, C.H. Reproduction and life history strategies of the common jellyfish, Aurelia aurita, in relation to its ambient environment. Hydrobiologia 2001, 451, 229–246. [Google Scholar] [CrossRef]
- Purcell, J.E.; Arai, M.N. Interactions of pelagic cnidarians and ctenophores with fish: A review. Hydrobiologia 2001, 451, 27–44. [Google Scholar] [CrossRef]
- Vodopivec, M.; Peliz, A.J.; Malej, A. Offshore marine constructions as propagators of moon jellyfish dispersal. Environ. Res. Lett. 2017, 12, 084003. [Google Scholar] [CrossRef]
- Schmahl, G. Induction of stolon settlement in the scyphopolyps of Aurelia aurita (Cnidaria, Scyphozoa, Semaeostomeae) by glycolipids of marine bacteria. Helgol. Meeresunters. 1985, 39, 117–127. [Google Scholar] [CrossRef]
- Schmahl, G. Bacterially induced stolon settlement in the scyphopolyp of Aurelia aurita (Cnidaria, Scyphozoa). Helgol. Meeresunters. 1985, 39, 33–42. [Google Scholar] [CrossRef]
- Hofmann, D.K.; Neumann, R.; Henne, K. Strobilation, budding and initiation of scyphistoma morphogenesis in the rhizostome Cassiopea andromeda (Cnidaria, Scyphozoa). Mar. Biol. 1978, 47, 161–176. [Google Scholar] [CrossRef]
- Hofmann, D.K.; William, K.F.; Fleck, J. Checkpoints in the life-cycle of Cassiopea spp.: Control of metagenesis and metamorphosis in a tropical jellyfish. Int. J. Dev. Biol. 1996, 40, 331–338. [Google Scholar] [PubMed]
- Neumann, A. Bacterial induction of settlement and metamorphosis in the planula larvae of Cassiopea andromeda (Cnidaria: Scyphozoa, Rhizostomeae). Mar. Ecol. Prog. Ser. 1979, 1, 21–28. [Google Scholar] [CrossRef]
- Neumann, R.; Schmahl, G.; Hofmann, D.K. Bud formation and control of polyp morphogenesis in Cassiopea andromeda (Scyphozoa). In Developmental and Cellular Biology of Coelenterates; Tardent, P., Tardent, R., Eds.; Elsevier, North-Holland Biomedical Press: Amsterdam, The Netherlands, 1980; pp. 217–223. [Google Scholar]
- Brekhman, V.; Malik, A.; Haas, B.; Sher, N.; Lotan, T. Transcriptome profiling of the dynamic life cycle of the scypohozoan jellyfish Aurelia aurita. BMC Genom. 2015, 16, 74. [Google Scholar] [CrossRef]
- Fuchs, B.; Wang, W.; Graspeuntner, S.; Li, Y.; Insua, S.; Herbst, E.M.; Dirksen, P.; Marei Bohm, A.M.; Hemmrich, G.; Sommer, F.; et al. Regulation of Polyp-to-Jellyfish Transition in Aurelia aurita. Curr. Biol. 2014, 24, 263–273. [Google Scholar] [CrossRef]
- Gold, D.A.; Katsuki, T.; Li, Y.; Yan, X.; Regulski, M.; Ibberson, D.; Holstein, T.; Steele, R.E.; Jacobs, D.K.; Greenspan, R.J. The genome of the jellyfish Aurelia and the evolution of animal complexity. Nat. Ecol. Evol. 2018, 3, 96–104. [Google Scholar] [CrossRef]
- Freckelton, M.L.; Nedved, B.T.; Hadfield, M.G. Induction of Invertebrate Larval Settlement; Different Bacteria, Different Mechanisms? Sci. Rep. 2017, 7, 42557. [Google Scholar] [CrossRef] [PubMed]
- Dobretsov, S.; Qian, P.Y. The role of epibotic bacteria from the surface of the soft coral Dendronephthya sp. in the inhibition of larval settlement. J. Exp. Mar. Biol. Ecol. 2004, 299, 35–50. [Google Scholar] [CrossRef]
- Dang, H.; Li, T.; Chen, M.; Huang, G. Cross-ocean distribution of Rhodobacterales bacteria as primary surface colonizers in temperate coastal marine waters. Appl. Environ. Microbiol. 2008, 74, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Porsby, C.H.; Nielsen, K.F.; Gram, L. Phaeobacter and Ruegeria Species of the Roseobacter Clade Colonize Separate Niches in a Danish Turbot (Scophthalmus maximus)-rearing farm and antagonize Vibrio anguillarum under different growth conditions. Appl. Environ. Microbiol. 2008, 74, 7356–7364. [Google Scholar] [CrossRef] [PubMed]
- Elsayed, Y.; Refaat, J.; Abdelmohsen, U.R.; Fouad, M.A. The Genus Rhodococcus as a source of novel bioactive substances: A review. J. Pharma. Phytochem. 2017, 6, 83–92. [Google Scholar]
- Shnit-Orland, M.; Kushmaro, A. Coral mucus-associated bacteria: A possible first line of defense. FEMS Microbiol. Ecol. 2009, 67, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Lesh-Laurie, G.E.; Suchy, P.E. Scyphozoa and Cubozoa. In Placozoa, Porifera, Cnidaria, and Ctenophora, Volume II; Harrison, F.W., Westfall, J.A., Eds.; Wiley-Liss: New York, NY, USA, 1991; pp. 185–266. [Google Scholar]
- Heeger, T.; Möller, H. Ultrastructural observations on prey capture and digestion in the scyphomedusa Aurelia aurita. Mar. Biol. 1987, 96, 391–400. [Google Scholar] [CrossRef]
- Patwa, A.; Thiéry, A.; Lombard, F.; Lilley, M.K.S.; Boisset, C.; Bramard, J.; Bottero, J.Y.; Philippe Barthélémy, P. Accumulation of nanoparticles in “jellyfish” mucus: A bio-inspired route to decontamination of nano-waste. Sci. Rep. 2015, 5, 11387. [Google Scholar] [CrossRef]
- Shanks, A.; Graham, W. Chemical defense in a scyphomedusa. Mar. Ecol. Prog. Ser. 1988, 45, 81–86. [Google Scholar] [CrossRef]
- Shaposhnikovaa, T.; Matveevb, I.; Naparac, T.; Podgornayab, O. Mesogleal cells of the jellyfish Aurelia aurita are involved in the formation of mesogleal fibres. Cell. Biol. Int. 2005, 29, 952–958. [Google Scholar] [CrossRef]
- Hoeger, U. Biochemical composition of ctenophores. J. Exp. Mar. Biol. Ecol. 1983, 72, 251–261. [Google Scholar] [CrossRef]
- Hsieh, Y.H.P.; Rudloe, J. Potential of utilizing jellyfish as food in Western countries. Trends Food Sci. Technol. 1994, 5, 225–229. [Google Scholar] [CrossRef]
- Ducklow, H.W.; Mitchell, R. Composition of mucus released by coral reef coelenterates. Limnol. Oceanogr. 1979, 24, 706–714. [Google Scholar] [CrossRef]
- Stabili, L.; Schirosi, R.; Parisi, M.G.; Piraino, S.; Cammarata, M. The Mucus of Actinia equina (Anthozoa, Cnidaria): An Unexplored Resource for Potential Applicative Purposes. Mar. Drugs 2015, 13, 5276–5296. [Google Scholar] [CrossRef] [PubMed]
- Wahl, M.; Goecke, F.; Labes, A.; Dobretsov, S.; Weinberger, F. The second skin: Ecological role of epibiotic biofilms on marine organisms. Front. Microbiol. 2012, 3, 292. [Google Scholar] [CrossRef] [PubMed]
- Harder, T.; Lau, S.C.K.; Dobretsov, S.; Fang, T.K.; Qian, P.Y. A distinctive epibiotic bacterial community on the soft coral Dendronephthya sp. and antibacterial activity of coral tissue extracts suggest a chemical mechanism against bacterial epibiosis. FEMS Microbiol. Ecol. 2003, 43, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Holmström, C.; Egan, S.; Franks, A.; McCloy, S.; Kjelleberg, S. Antifouling activities expressed by marine surface associated Pseudoalteromonas species. FEMS Microbiol. Ecol. 2002, 41, 47–58. [Google Scholar] [CrossRef]
- Long, R.A.; Azam, F. Antagonistic Interactions among Marine Pelagic Bacteria. Appl. Environ. Microbiol. 2001, 67, 4975–4983. [Google Scholar] [CrossRef]
- Bruhn, J.B.; Nielsen, K.F.; Hjelm, M.; Hansen, M.; Bresciani, J.; Schulz, S.; Gram, L. Ecology, Inhibitory Activity, and Morphogenesis of a Marine Antagonistic Bacterium Belonging to the Roseobacter Clade. Appl. Environ. Microbiol. 2005, 71, 7263–7270. [Google Scholar] [CrossRef]
- Gram, L.; Melchiorsen, J.; Bruhn, J.B. Antibacterial Activity of Marine Culturable Bacteria Collected from a Global Sampling of Ocean Surface Waters and Surface Swabs of Marine Organisms. Mar. Biotechnol. 2010, 12, 439–451. [Google Scholar] [CrossRef]
- Bishop, C.D.; Brandhorst, B.P. On nitric oxide signalling, metamorphosis, and the evolution of biphasic life cycles. Evol. Dev. 2003, 5, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Moroz, L.L.; Meech, R.W.; Sweedler, J.V.; Mackie, G.O. Nitric oxide regulates swimming in the jellyfish Aglantha digitale. J. Comp. Neurol. 2004, 471, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Salleo, A.; Musci, G.; Barra, P.; Calabrese, L. The discharge mechanism of acontial nematocytes involves the release of nitric oxide. J. Exp. Biol. 1996, 199 Pt. 6, 1261–1267. [Google Scholar]
- Almeda, R.; Wambaugh, Z.; Chai, C.; Wang, Z.; Liu, Z.; Buskey, E.J. Effects of crude oil exposure on bioaccumulation of polycyclic aromatic hydrocarbons and survival of adult and larval stages of gelatinous zooplankton. PLoS ONE 2013, 8, e74476. [Google Scholar] [CrossRef] [PubMed]
- Schiraldi, C.; Giuliano, M.; De Rosa, M. Perspectives on biotechnological applications of archaea. Archaea 2002, 1, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Egorova, K.; Antranikian, G. Industrial relevance of thermophilic Archaea. Curr. Opin. Microbiol. 2005, 8, 649–655. [Google Scholar] [CrossRef]
- Ainsworth, T.D.; Krause, L.; Bridge, T.; Torda, G.; Raina, J.B.; Zakrzewski, M.; Gates, R.D.; Padilla-Gamiño, J.L.; Spalding, H.L.; Smith, C.; et al. The coral core microbiome identifies rare bacterial taxa as ubiquitous endosymbionts. ISME J. 2015, 9, 2261–2274. [Google Scholar] [CrossRef]
- Giovannoni, S.J.; Rappe, M. Evolution, diveristy and molecular ecology of marine prokaryotes. In Microbial Ecology of the Ocean; Kirchman, D.L., Ed.; Wiley-Liss: New York, NY, USA, 2000; pp. 47–84. [Google Scholar]
- Solano, F.; Sanchez-Amat, A. Studies on the phylogenetic relationships of melanogenic marine bacteria: Proposal of Marinomonas mediterranea sp. nov. Int. J. Syst. Bacteriol. 1999, 49, 1241–1246. [Google Scholar] [CrossRef]
- Sanchez-Amat, A.; Lucas-Elo, P.; Fernández, E.; García-Borrón, J.C.; Solano, F. Molecular cloning and functional characterization of a unique multipotent polyphenol oxidase from Marinomonas mediterranea. Biochim. Biophys. Acta 2001, 1547, 104–116. [Google Scholar] [CrossRef]
- Bowman, J.P. Bioactive compound synthetic capacity and ecological significance of marine bacterial genus pseudoalteromonas. Mar. Drugs 2007, 5, 220–241. [Google Scholar] [CrossRef]
- Thompson, F.L.; Iida, T.; Swings, J. Biodiversity of Vibrios. Microbiol. Mol. Biol. Rev. 2004, 68, 403–431. [Google Scholar] [CrossRef] [PubMed]
- Kodama, Y.; Stiknowati, L.I.; Ueki, A.; Ueki, K.; Watanabe, K. Thalassospira tepidiphila sp. nov., a polycyclic aromatic hydrocarbon-degrading bacterium isolated from seawater. Int. J. Syst. Evol. Microbiol. 2008, 58, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Gallego, S.; Vila, J.; Tauler, M.; Nieto, J.M.; Breugelmans, P.; Springael, D.; Gri-foll, M. Community structure and PAH ring-hydroxylating dioxygenasegenes of a marine pyrene-degrading microbial consortium. Biodegradation 2014, 25, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, C.; Michaud, L.; Hörmann, B.; Gerçe, B.; Syldatk, C.; Hausmann, R.; DeDomenico, E.; Lo Giudice, A. Bacteria associated with sabellids (Polychaeta: Annelida; as a novel source of surface active compounds. Mar. Pollut. Bull. 2013, 70, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Wiese, J.; Thiel, V.; Gartner, A.; Schmaljohann, R.; Imhoff, J.F. Kiloniella laminariae gen. nov., sp. nov., an Alphaproteobacterium from the marine macroalga Laminaria saccharina. Int. J. Syst. Evol. Micrrobiol. 2009, 59, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Converse, R.R.; Blackwood, A.D.; Kirs, M.; Griffith, J.F.; Noble, R.T. Rapid Q-PCR-based assay for fecal Bacteroidetes spp. as tool for assessing fecal contamination in recreational water. Water Res. 2009, 43, 4828–4837. [Google Scholar] [CrossRef] [PubMed]
- Toranzo, A.E.; Magarinos, B.; Romalde, J.L. A review of the main bacterial fish diseases in mariculture systems. Aquaculture 2005, 246, 37–61. [Google Scholar] [CrossRef]
- Handlinger, J.; Soltani, M.; Percival, S. The pathology of Flexibacter maritimus in aquaculture species in Tasmania, Australia. J. Fish. Dis. 1997, 20, 159–168. [Google Scholar] [CrossRef]
- Roberts, R.J. Miscellaneous non-infectious diseases. In Fish Pathology; Roberts, R.J., Ed.; WB Saunders: London, UK, 2001; pp. 367–379. [Google Scholar]
- Queruel, P. Envenimations par la méduse Pelagia noctiluca sur nos côtes méditerranéennes. Presse Méd. 2000, 29, 188. [Google Scholar]
- Mitchell, S.O.; Rodger, H.D. A review of infectious gill disease in marine salmonid fish. J. Fish. Dis. 2011, 34, 411–432. [Google Scholar] [CrossRef]
- Avendanõ-Herrera, R.; Toranzo, A.E.; Magarinõs, B. Tenacibaculosis infection in marine fish caused by Tenacibaculum maritimum: A review. Dis. Aquat. Organ. 2006, 71, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Kellogg, C.A.; Lisle, J.T.; Galkiewicz, J.P. Culture-independent characterization of bacterial communities associated with the cold-water coral Lophelia pertusa in the northeastern Gulf of Mexico. Appl. Environ. Microbiol. 2009, 75, 2294–2303. [Google Scholar] [CrossRef] [PubMed]
- Neulinger, S.C.; Gartner, A.; Jarnegren, J.; Ludvigsen, M.; Lochte, K.; Dullo, W.C. Tissue-associated “Candidatus Mycoplasma corallicola” and filamentous bacteria on the cold-water coral Lophelia pertusa (Scleractinia). Appl. Environ. Microbiol. 2009, 75, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Bull, A.T.; Stach, J.E. Marine actinobacteria: New opportunities for natural product search and discovery. Trends Microbiol. 2007, 11, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Bouillon, J.; Boero, F. Synopsis of the families and genera of the Hydromedusae of the world, with a list of the worldwide species. Thalassia Salentina 2000, 24, 47–296. [Google Scholar]
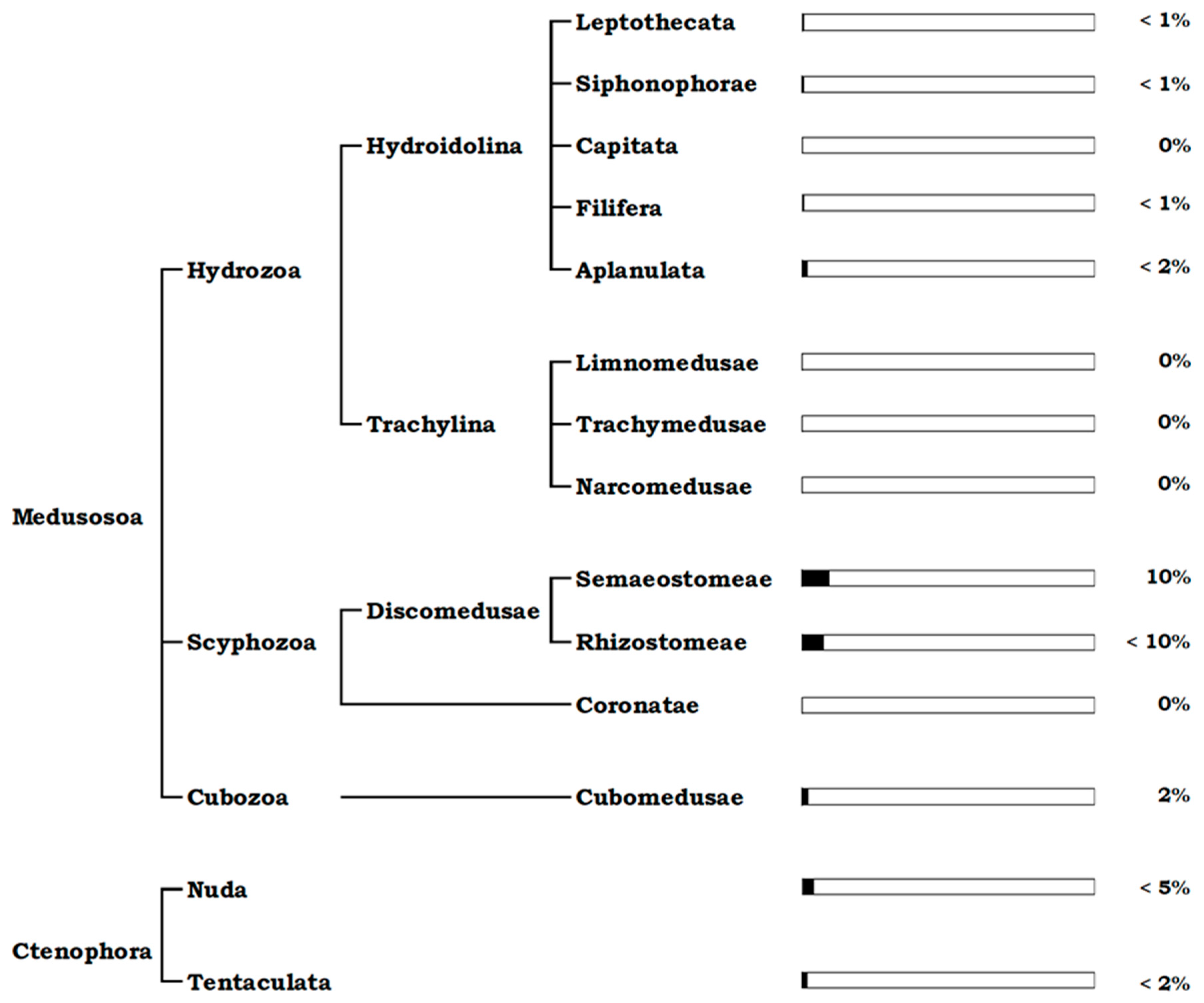

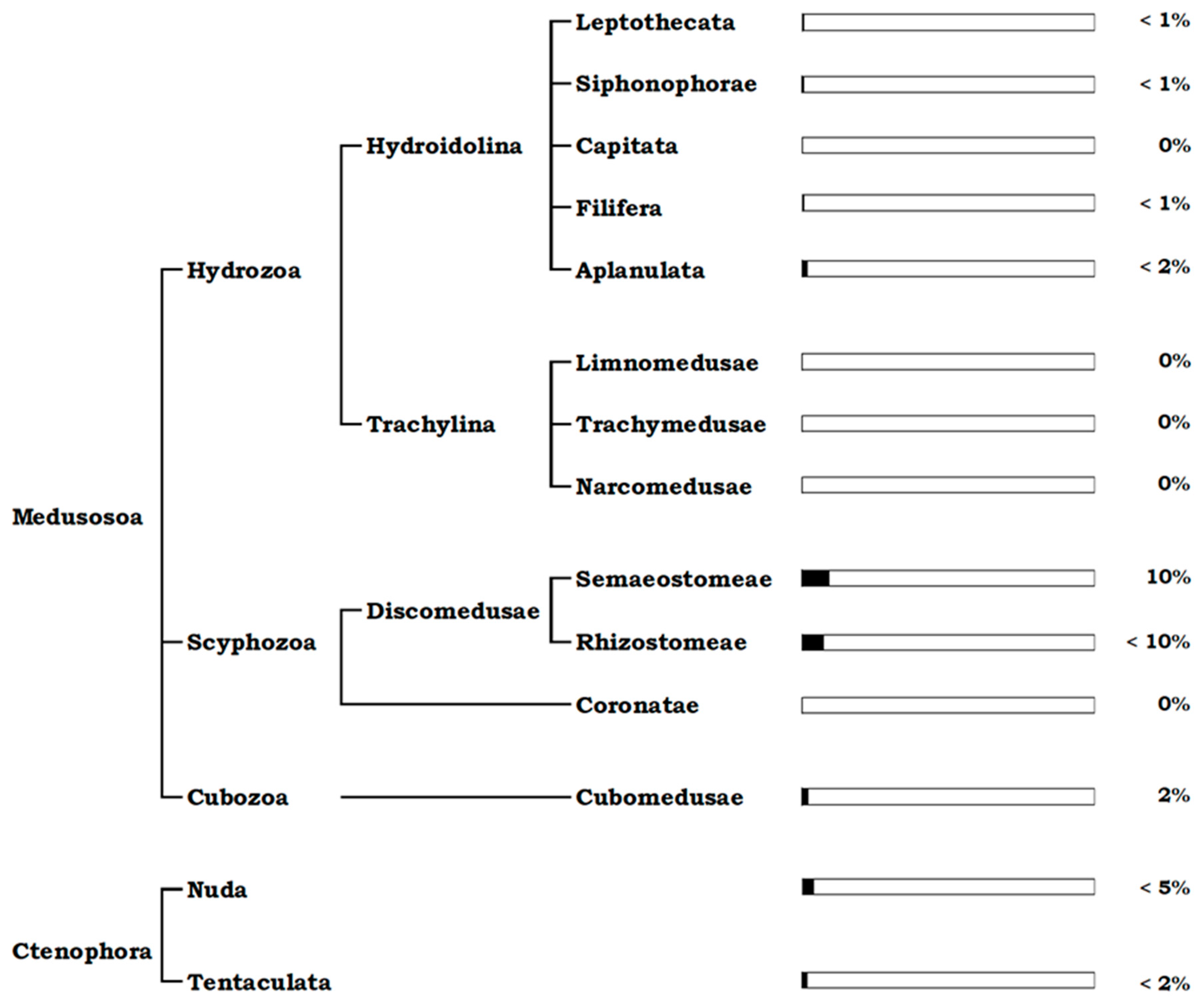{kind=link}
| Jellyfish Taxonomy | Study Design | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Phylum/Subphylum | Class | Order | Family | Species | Life Stage | Body Part (Adult Medusae) | Sampling Location | Methodology to Study Associated Microbiota | Publication |
| Medusozoa | Scyphozoa | Semaeostomeae | Ulmaridae | Aurelia aurita | Adult medusae | Whole body | North Atlantic coastal waters | 16S rRNA gene clone libraries | [36] |
| Polyps, strobila, ephyra, juvenile adult medusae | Mucus, gastric cavity | Kiel Bight, Baltic Sea, Southern English Channel, North Sea | Confocal laser scanning microscopy FISH NGS 454 technology V1–V2 16S rRNA region | [35] | |||||
| Adult medusae | Oral arms umbrella gastric cavity | Northern Adriatic | Culturing, DGGE, 16S rRNA gene clone libraries | [37] | |||||
| Cyaneidae | Cyanea capillata | Adult medusae | Tentacles | Scottish waters (Orkney) | Culturing DGGE, sequencing bands | [38] | |||
| Cyanea lamarckii | Adult medusae | Tentacles | Scottish waters (Orkney) | Culturing DGGE, sequencing bands | [38] | ||||
| Larvae polyps adult medusae | Tentacles, umbrella, mouth arm, gonads | German Bight | ARISA of ITS region | [39] | |||||
| Pelagiidae | Pelagia noctiluca | Adult medusae | Mouth | Ireland | Sequencing of specific bacterial 16S rRNA gene | [33] | |||
| Chrysaora plocamia | Polyps podocyst excyst | Whole body | Northern Chile | NGS Illumina MiSeq platform 2 × 300 bp paired end, V1–V2 16S rRNA region | [40] | ||||
| Chrysaora hysoscella | Larvae polyps adult medusae | Tentacles, umbrella, mouth arm, gonads | German Bight | ARISA of ITS region | [39] | ||||
| Rhizostomeae | Mastigiidae | Mastigias cf. papua | Adult medusae | Dome, tentacles | Indonesian marine lakes | NGS 454 technology, V3–V4 region | [41] | ||
| Cepheidae | Cotylorhiza tuberculata | Adult medusae | Gastric cavity | Alcudia Bay, Balearic Sea | Culturing, NGS—454 pyrosequencing | [42] | |||
| Adult medusae | Gastric cavity | Alcudia Bay, Balearic Sea | NGS—Illumina MiSeq platform, 2 × 250 bp, paired end | [43] | |||||
| Cubozoa | Carybdeida | Tripedaliidae | Tripedalia cf. cystophora | Adult medusae | Whole body | Indonesian marine lakes | NGS—454 technology V3-V4 16S rRNA region | [41] | |
| Hydrozoa | Anthoathecata | Bougainvilliidae | Nemopsis bachei | Adult medusae | Whole body | North Atlantic coastal waters | 16S rRNA gene clone libraries | [36] | |
| Tubulariidae | Tubularia indivisa | Adult medusae | Tentacles | Scottish waters (Orkney) | Culturing DGGE, sequencing bands | [38] | |||
| Leptothecata | Phialellidae | Phialella quadrata | Adult medusae | Whole body | Shetland Isles | Nested PCR with specific bacterial primers | [32] | ||
| Adult medusae | Whole body | Ireland | RT PCR with specific bacterial primers | [34] | |||||
| Siphonophorae | Diphyidae | Muggiaea atlantica | Adult medusae | Whole body | Ireland | RT PCR with specific bacterial primers | [34] | ||
| Ctenophora | Tentaculata | Lobata | Bolinopsidae | Mnemiopsis leidyi | Adult specimen | Whole body | Tampa Bay, Florida, USA | 16S rRNA gene clone libraries, T-RFLP | [44] |
| Adult specimen | Whole body guts | Gullmar fjord, west coast of Sweden | NGS—454 pyrosequencing | [23] | |||||
| Adult specimen | Whole body | Helgoland roads, German Bight | ARISA of ITS region | [45] | |||||
| Bolinopsis infundibulum | Adult specimen | Whole body | Helgoland roads, German Bight | ARISA of ITS region | [45] | ||||
| Cydippida | Pleurobrachiidae | Pleurobrachia pileus | Adult specimen | Whole body | Helgoland roads, German Bight | ARISA of ITS region | [45] | ||
| Nuda | Beroida | Beroidae | Beroe ovata | Adult specimen | Whole body | Helgoland roads, German Bight | ARISA of ITS region | [45] | |
| Adult specimen | Whole body | Tampa Bay, Florida, USA | 16S rRNA gene clone libraries, T-RFLP | [44] | |||||
| Bacteria Associated with Jellyfish | Features with Biotechnological Potential | Jellyfish | |
|---|---|---|---|
| Class | Representative Families | ||
| Gammaproteobacteria | Vibrionaceae Pseudoalteromonadaceae Alteromonadaceae Oceanospirillaceae Shewanellaceae Crenotrichaceae Methylococcalaceae Endozoicimonadaceae Moraxellaceae Legionelaceae |
|
|
| Alphaproteobacteria | Rhodospirillaceae Rhodobacteriaceae Kiloniellaceae |
|
|
| Flavobacteria | Flavobacteriaceae |
|
|
| Mollicutes | Spiroplasmataceae Mycoplasmataceae |
|
|
| Spirochaetes |
|
| |
| Actinobacteria |
|
| |
| Firmicutes |
|
| |
| Chlamydiae | Simkania-like bacteria |
|
|
| Nitrospirae and Nitrospinae |
|
| |
| Betaproteobacteria | Burkholderia Achromobacter Cupriavidus |
|
|
| Cyanobacteria |
|
| |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tinta, T.; Kogovšek, T.; Klun, K.; Malej, A.; Herndl, G.J.; Turk, V. Jellyfish-Associated Microbiome in the Marine Environment: Exploring Its Biotechnological Potential. Mar. Drugs 2019, 17, 94. https://doi.org/10.3390/md17020094
Tinta T, Kogovšek T, Klun K, Malej A, Herndl GJ, Turk V. Jellyfish-Associated Microbiome in the Marine Environment: Exploring Its Biotechnological Potential. Marine Drugs. 2019; 17(2):94. https://doi.org/10.3390/md17020094
Chicago/Turabian StyleTinta, Tinkara, Tjaša Kogovšek, Katja Klun, Alenka Malej, Gerhard J. Herndl, and Valentina Turk. 2019. "Jellyfish-Associated Microbiome in the Marine Environment: Exploring Its Biotechnological Potential" Marine Drugs 17, no. 2: 94. https://doi.org/10.3390/md17020094
APA StyleTinta, T., Kogovšek, T., Klun, K., Malej, A., Herndl, G. J., & Turk, V. (2019). Jellyfish-Associated Microbiome in the Marine Environment: Exploring Its Biotechnological Potential. Marine Drugs, 17(2), 94. https://doi.org/10.3390/md17020094