Fusarisolins A–E, Polyketides from the Marine-Derived Fungus Fusarium solani H918




Abstract
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Identification, Fermentation, and Extract
3.3. Isolation and Purification
3.4. Preparation of MPA Esters of 1
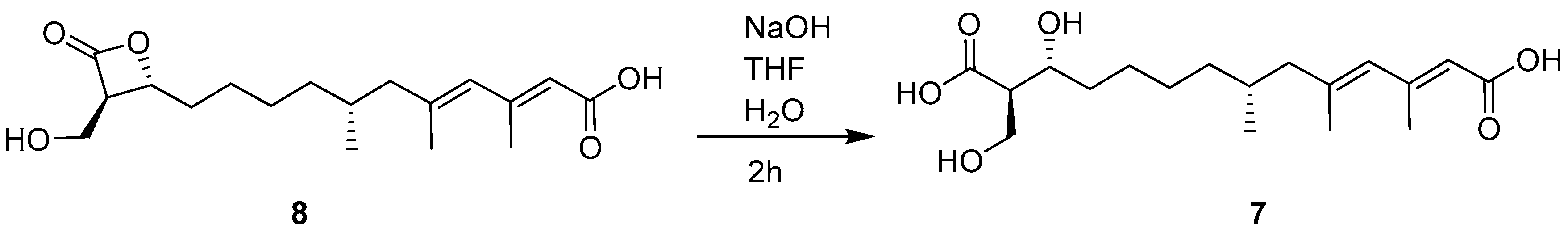
3.5. Alkaline Hydrolysis of 8
3.6. Antifungal Assay
3.7. ED50 Detection
3.8. Total RNA Isolation
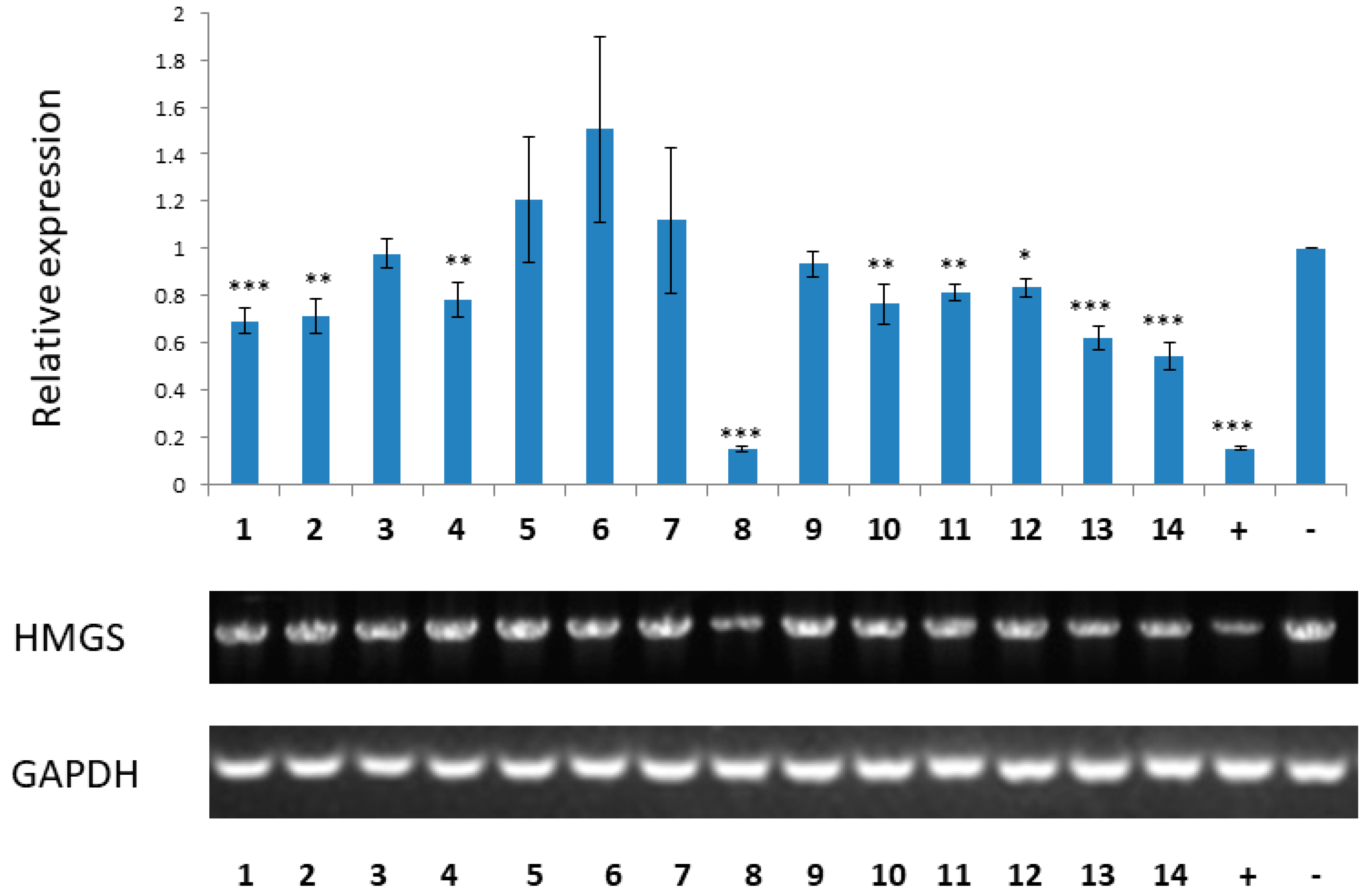
3.9. RT-PCR Analysis of HMG-CoA Synthase Gene Expression
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Rateb, M.E.; Ebel, R. Secondary metabolites of fungi from marine habitats. Nat. Prod. Rep. 2011, 28, 290–344. [Google Scholar] [CrossRef]
- Aldridge, D.C.; Giles, D.; Turner, W.B. Antibiotic 1233A: A fungal β-lactone. J. Chem. Soc. 1971, 23, 3888–3891. [Google Scholar] [CrossRef]
- Greenspan, M.D.; Yudkovitz, J.B.; Lo, C.Y.L.; Chen, J.S.; Alberts, A.W.; Hunt, V.M.; Chang, M.N.; Yang, S.S.; Thompson, K.L.; Chiang, Y.C.P.; et al. Inhibition of hydroxymethylglutaryl-coenzyme A synthase by L-659,699. Proc. Natl. Acad. Sci. USA 1987, 84, 7488–7492. [Google Scholar] [CrossRef]
- Tomoda, H.; Kumagai, H.; Takahashi, Y.; Tanaka, Y.; Iwai, Y.; Omura, S. F-244 (1233A), a specific inhibitor of 3-hydroxy-3-methylglutaryl coenzyme A synthase: Taxonomy of producing strain, fermentation, isolation and biological properties. J. Antibiot. 1988, 41, 247–249. [Google Scholar] [CrossRef]
- Umezawa, H.; Aoyagi, T.; Uotani, K.; Hamada, M.; Takeuchi, T.; Takahashi, S. Ebelactone, an inhibitor of esterase, produced by actinomycetes. J. Antibiot. 1980, 33, 1594–1596. [Google Scholar] [CrossRef]
- Wells, J.S.; Trejo, W.H.; Principe, P.A.; Sykes, R.B. Obafluorin, a novel β-lactone produced by Pseudomonas fluorescens. Taxonomy, fermentation and biological properties. J. Antibiot. 1984, 37, 802–803. [Google Scholar] [CrossRef]
- Asai, A.; Hasegawa, A.; Ochiai, K.; Yamashita, Y.; Mizukami, T. Belactosin A, a novel antitumor antibiotic acting on cyclin/CDK mediated cell cycle regulation, produced by Streptomyces sp. J. Antibiot. 2000, 53, 81–83. [Google Scholar] [CrossRef]
- Weibel, E.K.; Hadvary, P.; Hochuli, E.; Kupfer, E.; Lengsfeld, H. Lipstatin, an inhibitor of pancreatic lipase, produced by Streptomyces toxytricini. I. Producing organism, fermentation, isolation and biological activity. J. Antibiot. 1987, 40, 1081–1085. [Google Scholar] [CrossRef]
- Feling, R.H.; Buchanan, G.O.; Mincer, T.J.; Kauffman, C.A.; Jensen, P.R.; Fenical, W. Salinosporamide A: A highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus Salinospora. Angew. Chem. Int. Ed. 2003, 42, 355–357. [Google Scholar] [CrossRef]
- Liu, D.Z.; Wang, F.; Liao, T.G.; Tang, J.G.; Steglich, W.; Zhu, H.J.; Liu, J.K. Vibralactone: A lipase inhibitor with an unusual fused β-lactone produced by cultures of the basidiomycete Boreostereum vibrans. Org. Lett. 2006, 8, 5749–5752. [Google Scholar] [CrossRef]
- Morris, B.D.; Smyth, R.R.; Foster, S.P.; Hoffmann, M.P.; Roelofs, W.L.; Franke, S.; Francke, W. Vittatalactone, a β-lactone from the striped cucumber beetle, Acalymma vittatum. J. Nat. Prod. 2005, 68, 26–30. [Google Scholar] [CrossRef]
- Gill, K.A.; Berrue, F.; Arens, J.C.; Carr, G.; Kerr, R.G. Cystargolides, 20S Proteasome Inhibitors Isolated from Kitasatospora cystarginea. J. Nat. Prod. 2015, 78, 822–826. [Google Scholar] [CrossRef]
- Böttcher, T.; Sieber, S.A. β-Lactams and β-lactones as activity-based probes in chemical biology. Med. Chem. Commun. 2012, 3, 408–417. [Google Scholar] [CrossRef]
- De Pascale, G.; Nazi, I.; Harrison, P.H.M.; Wright, G.D. β-lactone natural products and derivatives inactivate homoserine transacetylase, a target for antimicrobial agents. J. Antibiot. 2011, 64, 483–487. [Google Scholar] [CrossRef]
- Pojer, F.; Ferrer, J.L.; Richard, S.B.; Nagegowda, D.A.; Chye, M.L.; Bach, T.J.; Noel, J.P. Structural basis for the design of potent and species-specific inhibitors of 3-hydroxy-3-methylglutaryl CoA synthases. Proc. Natl. Acad. Sci. USA 2006, 103, 11491–11496. [Google Scholar] [CrossRef]
- Guerciolini, R. Mode of action of orlistat. Int. J. Obes. Relat. Metab. Disord. 1997, 21 (Suppl. 3), S12–S23. [Google Scholar]
- Yang, X.W.; Peng, K.; Liu, Z.; Zhang, G.Y.; Li, J.; Wang, N.; Steinmetz, A.; Liu, Y. Strepsesquitriol, a rearranged zizaane-type sesquiterpenoid from the deep-sea-derived actinomycete Streptomyces sp. SCSIO 10355. J. Nat. Prod. 2013, 76, 2360–2363. [Google Scholar] [CrossRef]
- Xie, C.L.; Liu, Q.; Xia, J.M.; Gao, Y.; Yang, Q.; Shao, Z.Z.; Liu, G.; Yang, X.W. Anti-allergic compounds from the deep-sea-derived actinomycete Nesterenkonia flava MCCC 1K00610. Mar. Drugs 2017, 15, 71. [Google Scholar] [CrossRef]
- Niu, S.; Zhou, T.T.; Xie, C.L.; Zhang, G.Y.; Yang, X.W. Microindolinone A, a novel 4,5,6,7-tetrahydroindole, from the deep-sea-derived actinomycete Microbacterium sp. MCCC 1A11207. Mar. Drugs 2017, 15, 230. [Google Scholar] [CrossRef]
- Niu, S.; Liu, Q.; Xia, J.; Xie, C.; Luo, Z.; Shao, Z.; Liu, G.; Yang, X. Polyketides from the deep-sea-derived fungus Graphostroma sp. MCCC 3A00421 showed potent antifood allergic activities. J. Agric. Food Chem. 2018, 66, 1369–1376. [Google Scholar] [CrossRef]
- Chiang, Y.C.P.; Yang, S.S.; Heck, J.V.; Chabala, J.C.; Chang, M.N. Total synthesis of L-659,699, a novel inhibitor of cholesterol biosynthesis. J. Org. Chem. 1989, 54, 5708–5712. [Google Scholar] [CrossRef]
- Yasuhara, F.; Yamaguchi, S. Use of shift reagent with MTPA derivatives in 1H NMR spectroscopy. III. Determination of absolute configuration and enantiomeric purity of primary carbinols with chiral center at the C-2 position. Tetrahedron Lett. 1977, 18, 4085–4088. [Google Scholar] [CrossRef]
- Sugimoto, Y.; Tsuyuki, T.; Moriyama, Y.; Takahashi, T. Application of the MTPA method to determination of abusolute stereochemistry. The hydroxymethyl-substituted chiral carbon of carbocycles. Bull. Chem. Soc. Jpn. 1980, 53, 3723–3724. [Google Scholar] [CrossRef]
- Wovkulich, P.M.; Shankaran, K.; Kiegiel, J.; Uskokovic, M.R. Total synthesis of 1233A. J. Org. Chem. 1993, 58, 832–839. [Google Scholar] [CrossRef]
- Franot, C.; Benezra, C.; Lepoittevin, J.P. Synthesis and interaction studies of 13C labeled lactone derivatives with a model protein using 13C NMR. Bioorg. Med. Chem. 1993, 1, 389–397. [Google Scholar] [CrossRef]
- White, J.D.; Johnson, A.T. Synthesis of the aliphatic depside (+)-bourgeanic acid. J. Org. Chem. 1994, 59, 3347–3358. [Google Scholar] [CrossRef]
- Yoshida, W.Y.; Bryan, P.J.; Baker, B.J.; McClintock, J.B. Pteroenone: A defensive metabolite of the abducted Antarctic Pteropod Clione antarctica. J. Org. Chem. 1995, 60, 780–782. [Google Scholar] [CrossRef]
- Nanda, S.; Scott, A.I. Asymmetric synthesis of (E)- and (Z)-3,7-dimethyl-2-octene-1,8-diol and callosobruchusic acid. Tetrahedron Asymmetry 2004, 15, 963–970. [Google Scholar] [CrossRef]
- Kornsakulkarn, J.; Dolsophon, K.; Boonyuen, N.; Boonruangprapa, T.; Rachtawee, P.; Prabpai, S.; Kongsaeree, P.; Thongpanchang, C. Dihydronaphthalenones from endophytic fungus Fusarium sp. BCC14842. Tetrahedron 2011, 67, 7540–7547. [Google Scholar] [CrossRef]
- Kimura, Y.; Shimada, A.; Nakajima, H.; Hamasaki, T. Structures of naphthoquinones produced by the fungus, Fusarium sp., and their biological activity toward pollen germination. Agric. Biol. Chem. 1988, 52, 1253–1259. [Google Scholar] [CrossRef]
- Chilton, W.S. Isolation and structure of norjavanicin. J. Org. Chem. 1968, 33, 4299–4301. [Google Scholar] [CrossRef]
- Tatum, J.H.; Baker, R.A. Naphthoquinones produced by Fusarium solani isolated from citrus. Phytochemistry 1983, 22, 543–547. [Google Scholar] [CrossRef]
- Kurobane, I.; Zaita, N.; Fukuda, A. New metabolites of Fusarium martii related to dihydrofusarubin. J. Antibiot. 1986, 39, 205–214. [Google Scholar] [CrossRef]
- Arsenault, G.P. Fungal metabolites. III. Quinones from Fusarium solani D2 purple and structure of (+)-solaniol. Tetrahedron 1968, 24, 4745–4749. [Google Scholar] [CrossRef]
- Hashimoto, J.; Motohashi, K.; Sakamoto, K.; Hashimoto, S.; Yamanouchi, M.; Tanaka, H.; Takahashi, T.; Takagi, M.; Shin-ya, K. Screening and evaluation of new inhibitors of hepatic glucose production. J. Antibiot. 2009, 62, 625–629. [Google Scholar] [CrossRef]
- Alex, D.; Bach, T.J.; Chye, M.L. Expression of Brassica juncea 3-hydroxy-3-methylglutaryl CoA synthase is developmentally regulated and stress-responsive. Plant J. 2001, 22, 415–426. [Google Scholar] [CrossRef]
- Woo, J.H.; Kitamura, E.; Myouga, H.; Kamei, Y. An antifungal protein from the marine bacterium Streptomyces sp. Strain AP77 is specific for pythium porphyrae, a causative agent of red rot disease in Porphyra spp. Appl. Environ. Microb. 2002, 68, 2666–2675. [Google Scholar] [CrossRef]
- Kundu, A.; Saha, S.; Walia, S.; Shakil, N.A.; Kumar, J.; Annapurna, K. Cadinene sesquiterpenes from Eupatorium adenophorum and their antifungal activity. J. Environ. Sci. Health B 2013, 48, 516–522. [Google Scholar] [CrossRef]

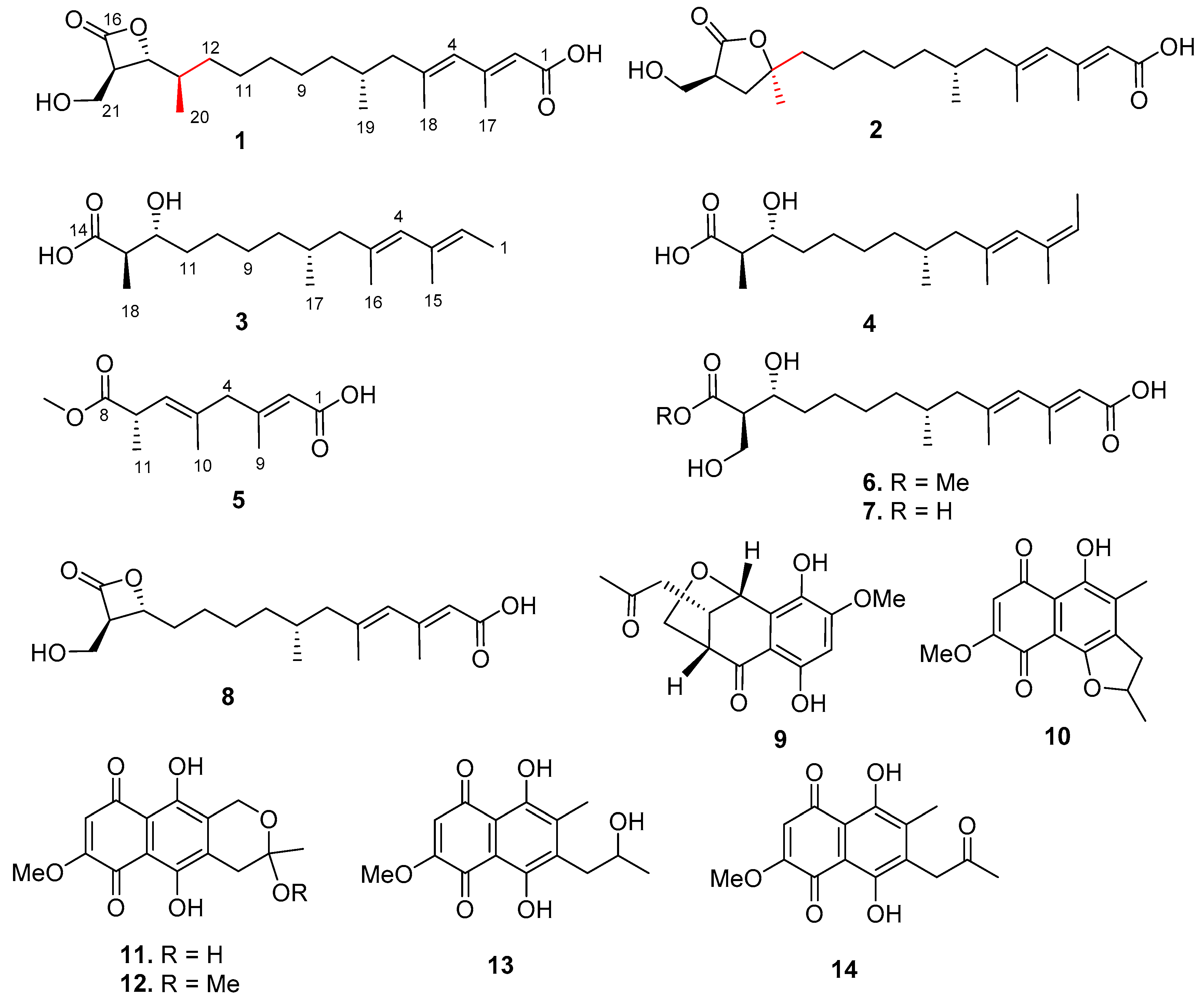
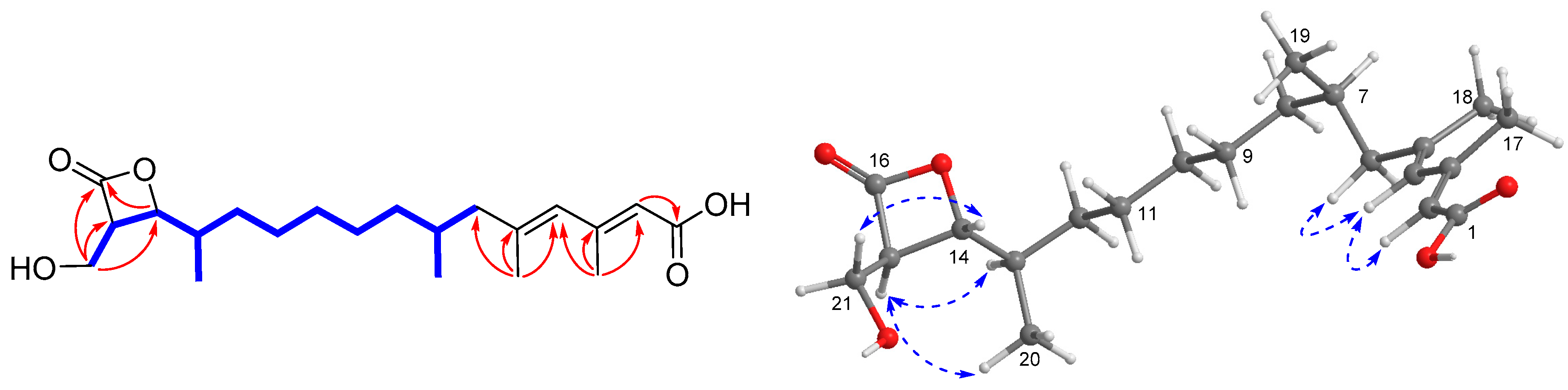
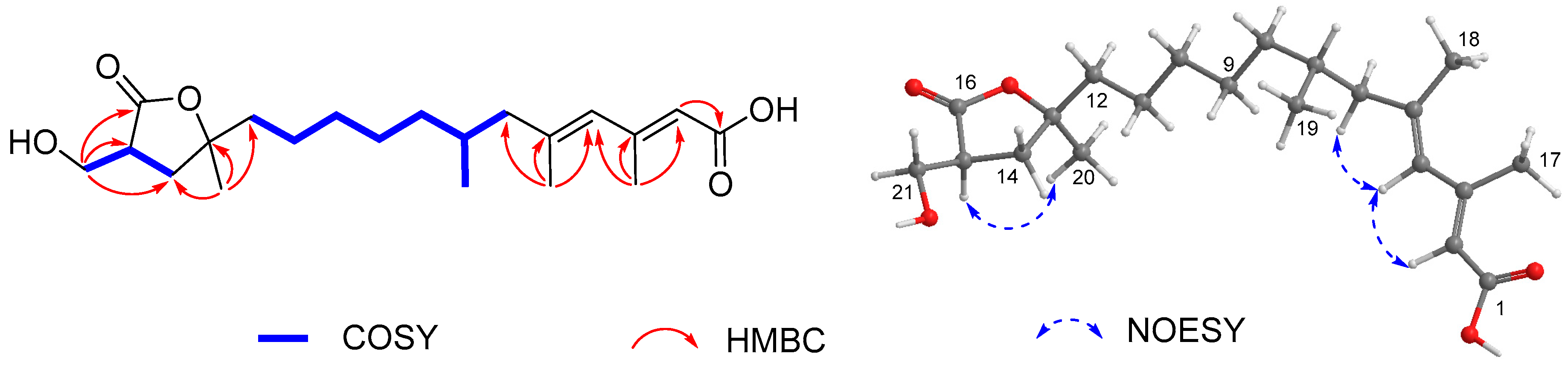
 ), HMBC (
), HMBC ( ), and NOESY (
), and NOESY ( ) correlations of 1.
) correlations of 1.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 a | 2 a | 3 a | 4 a | 5 b | 6 a |
|---|---|---|---|---|---|---|
| 1 | 1.67, d (7.0) | 1.50, dm (6.8) | ||||
| 2 | 5.65, br s | 5.65, br s | 5.29, q (7.0) | 5.30, qq (6.8, 1.2) | 5.74, br s | 5.65, br s |
| 4 | 5.76, br s | 5.76, br s | 5.59, br s | 5.53, br s | 2.85, s | 5.76, br s |
| 6 | 2.13, dd (13.2, 6.2) | 2.14, dd (13.2, 7.2) | 2.03, dd (12.8, 6.4) | 2.09, dd (13.2, 6.3) | 5.32, d (8.7) | 2.14, dd (13.1, 6.4) |
| 1.88, dd (13.2, 8.1) | 1.89, dd (13.2, 8.1) | 1.79, m | 1.86, ddd (13.2, 8.3, 0.8) | 1.89, dd (13.1, 8.2) | ||
| 7 | 1.70, m | 1.71, m | 1.65, m | 1.66, m | 3.39, m | 1.71, m |
| 8 | 1.35, m; 1.15, m | 1.34, m; 1.15, m | 1.36, m; 1.12, m | 1.38, m; 1.15, m | 1.36, m; 1.16, m | |
| 9 | 1.44, m | 1.34, m | 1.36, m | 1.38, m | 2.12, s | 1.37, m |
| 10 | 1.33, m | 1.34, m | 1.49, m; 1.37, m | 1.50, m; 1.38, m | 1.63, s | 1.48, m; 1.37, m |
| 11 | 1.33, m | 1.42, m | 1.52, m; 1.40, m | 1.53, m; 1.40, m | 1.26, d (6.8) | 1.53, m; 1.45, m |
| 12 | 1.44, m; 1.18, m | 1.72, m | 3.71, t (6.9) | 3.70, t (6.9) | 3.79, m | |
| 13 | 1.82, m | 2.48, m | 2.48, m | 2.67, ddd (8.5, 6.3, 5.4) | ||
| 14 | 4.31, dd (8.4, 4.2) | 2.16, br d (10.3) | ||||
| 15 | 3.53, q (4.2) | 3.02, m | 1.69, s | 1.70, m | 2.21, d (1.2) | |
| 16 | 1.71, s | 1.54, d (1.3) | 1.82, d (1.2) | |||
| 17 | 2.20, s | 2.21, s | 0.84, d (6.6) | 0.88, d (6.6) | 0.87, d (6.6) | |
| 18 | 1.82, s | 1.82, s | 1.13, d (6.9) | 1.13, d (7.0) | 3.82, dd (10.9, 8.7) | |
| 3.73, dd (10.9, 5.3) | ||||||
| 19 | 0.87, d (6.6) | 0.87, d (6.5) | ||||
| 20 | 1.04, d (6.5) | 1.38, s | ||||
| 21 | 3.91, dd (11.9, 4.5) | 3.87, dd (11.0, 4.6) | ||||
| 3.77, dd (11.9, 3.8) | 3.72, dd (11.0, 3.6) | |||||
| OMe | 3.70, s | 3.71, s |
| No. | 1 a | 2 a | 3 a | 4 a | 5 b | 6 a |
|---|---|---|---|---|---|---|
| 1 | 171.0, C | 172.1, C | 13.7, CH3 | 15.3, CH3 | 171.7, C | 170.6, C |
| 2 | 119.1, CH | 119.1, CH | 123.7, CH | 121.8, CH | 116.6, CH | 118.8, CH |
| 3 | 155.2, C | 155.0, C | 134.9, C | 135.3, C | 160.1, C | 155.5, C |
| 4 | 130.6, CH | 130.6, CH | 131.6, CH | 126.8, CH | 51.1, CH2 | 130.6, CH |
| 5 | 142.2, C | 142.1, C | 135.1, C | 137.2, C | 133.5, C | 142.3, C |
| 6 | 49.9, CH2 | 49.9, CH2 | 49.8, CH2 | 48.8, CH2 | 128.0, CH | 50.0, CH2 |
| 7 | 32.1, CH | 32.2, CH | 32.1, CH | 32.1, CH | 38.9, CH | 32.1, CH |
| 8 | 37.8, CH2 | 37.9, CH2 | 38.0, CH2 | 38.0, CH2 | 175.7, C | 37.9, CH2 |
| 9 | 27.9, CH2 | 28.9, CH2 | 28.2, CH2 | 28.1, CH2 | 17.9, CH3 | 32.1, CH2 |
| 10 | 27.8, CH2 | 31.2, CH2 | 27.0, CH2 | 27.0, CH2 | 15.9, CH3 | 26.9, CH2 |
| 11 | 31.0, CH2 | 24.8, CH2 | 35.0, CH2 | 35.0, CH2 | 18.4, CH3 | 36.1, CH2 |
| 12 | 32.4, CH2 | 42.6, CH2 | 74.1, CH | 74.1, CH | 71.0, CH | |
| 13 | 38.1, CH | 86.5, C | 47.5, CH | 47.5, CH | 56.2, CH | |
| 14 | 80.2, CH | 36.5, CH2 | 179.7, C | 179.5, C | 175.1, C | |
| 15 | 58.2, CH | 44.6, CH | 17.0, CH3 | 24.2, CH3 | 19.9, CH3 | |
| 16 | 172.1, C | 179.5, C | 17.9, CH3 | 17.8, CH3 | 18.5, CH3 | |
| 17 | 19.8, CH3 | 19.8, CH3 | 20.0, CH3 | 19.9, CH3 | 19.8, CH3 | |
| 18 | 18.5, CH3 | 18.5, CH3 | 13.9, CH3 | 13.8, CH3 | 61.6, CH2 | |
| 19 | 19.9, CH3 | 19.9, CH3 | ||||
| 20 | 15.3, CH3 | 25.5, CH3 | ||||
| 21 | 58.3, CH2 | 61.0, CH2 | ||||
| OMe | 51.9, CH3 | 52.0, CH3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niu, S.; Tang, X.-X.; Fan, Z.; Xia, J.-M.; Xie, C.-L.; Yang, X.-W. Fusarisolins A–E, Polyketides from the Marine-Derived Fungus Fusarium solani H918. Mar. Drugs 2019, 17, 125. https://doi.org/10.3390/md17020125
Niu S, Tang X-X, Fan Z, Xia J-M, Xie C-L, Yang X-W. Fusarisolins A–E, Polyketides from the Marine-Derived Fungus Fusarium solani H918. Marine Drugs. 2019; 17(2):125. https://doi.org/10.3390/md17020125
Chicago/Turabian StyleNiu, Siwen, Xi-Xiang Tang, Zuowang Fan, Jin-Mei Xia, Chun-Lan Xie, and Xian-Wen Yang. 2019. "Fusarisolins A–E, Polyketides from the Marine-Derived Fungus Fusarium solani H918" Marine Drugs 17, no. 2: 125. https://doi.org/10.3390/md17020125
APA StyleNiu, S., Tang, X.-X., Fan, Z., Xia, J.-M., Xie, C.-L., & Yang, X.-W. (2019). Fusarisolins A–E, Polyketides from the Marine-Derived Fungus Fusarium solani H918. Marine Drugs, 17(2), 125. https://doi.org/10.3390/md17020125