In Vitro Antiproliferative Evaluation of Synthetic Meroterpenes Inspired by Marine Natural Products

 ,
,  ,
,  ,
, 
 ,
, 
Abstract
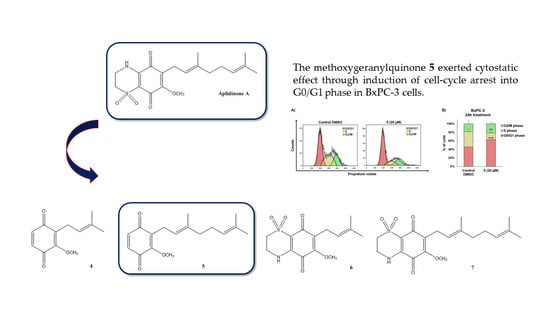
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. In Vitro Evaluation of Antiproliferative Activity of Quinones 4–7 in Cancer Cell Lines
3. Materials and Methods
3.1. Chemistry
3.1.1. Synthesis of 1,2,4-trimethoxy-3-(3-methylbut-2-en-1-yl)benzene (3-R1) and (E)-2-(3,7-dimethylocta-2,6-dien-1-yl)-1,3,4-trimethoxybenzene (3-R2)
3.1.2. Synthesis of 2-methoxy-3-(3-methylbut-2-en-1-yl)cyclohexa-2,5-diene-1,4-dione (4)
3.1.3. Synthesis of (E)-2-(3,7-dimethylocta-2,6-dien-1-yl)-3-methoxycyclohexa-2,5-diene-1,4-dione (5)
3.1.4. Synthesis of 6-methoxy-7-(3-methylbut-2-en-1-yl)-3,4-dihydro-2H-benzo[b][1,4]thiazine-5,8-dione-1,1-dioxide (6) and (E)-7-(3,7-dimethylocta-2,6-dien-1-yl)-6-methoxy-3,4-dihydro-2H-benzo[b][1,4]thiazine-5,8-dione-1,1-dioxide (7)
3.2. In Vitro Evaluation of Antiproliferative Activity of Compounds 4–7 in Cancer Cell Lines
3.2.1. Cell Culture
3.2.2. xCELLigence Assays
3.2.3. Apoptosis Assay and Cell Cycle Analysis
3.2.4. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Polyakov, N.; Leshina, T.; Fedenok, L.; Slepneva, I.; Kirilyuk, I.; Furso, J.; Olchawa, M.; Sarna, T.; Elas, M.; Bilkis, I.; et al. Redox-Active Quinone Chelators: Properties, Mechanisms of Action, Cell Delivery, and Cell Toxicity. Antioxid. Redox Signal. 2018, 28, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Menna, M.; Imperatore, C.; D’Aniello, F.; Aiello, A. Meroterpenes from Marine Invertebrates: Structures, Occurrence, and Ecological Implications. Mar. Drugs 2013, 11, 1602–1643. [Google Scholar] [CrossRef] [PubMed]
- Haque, M.A.; Sailo, B.L.; Padmavathi, G.; Kunnumakkara, A.B.; Jana, C.K. Nature-inspired development of unnatural meroterpenoids as the non-toxic anti-colon cancer agent. Eur. J. Med. Chem. 2018, 160, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Li, G.-Y.; Li, B.-G.; Yang, T.; Yin, J.-H.; Qi, H.-Y.; Liu, G.-Y.; Zhang, G.-L. Sesterterpenoids, terretonins A-D, and an alkaloid, asterrelenin, from Aspergillus terreus. J. Nat. Prod. 2005, 68, 1243–1246. [Google Scholar] [CrossRef] [PubMed]
- García, P.A.; Hernández, Á.P.; San Feliciano, A.; Castro, M.Á. Bioactive prenyl- and terpenyl-quinones/hydroquinones of marine origin. Mar. Drugs 2018, 16, 292. [Google Scholar] [CrossRef]
- Zubia, E.; Ortega, M.J.; Salva, J. Natural products chemistry in marine ascidians of the genus Aplidium. Mini-Rev. Org. Chem. 2005, 2, 389–399. [Google Scholar] [CrossRef]
- Menna, M.; Aiello, A.; D’Aniello, F.; Fattorusso, E.; Imperatore, C.; Luciano, P.; Vitalone, R. Further investigation of the mediterranean sponge Axinella polypoides: Isolation of a new cyclonucleoside and a new betaine. Mar. Drugs 2012, 10, 2509–2518. [Google Scholar] [CrossRef]
- Imperatore, C.; Luciano, P.; Aiello, A.; Vitalone, R.; Irace, C.; Santamaria, R.; Li, J.; Guo, Y.-W.; Menna, M. Structure and Configuration of Phosphoeleganin, a Protein Tyrosine Phosphatase 1B Inhibitor from the Mediterranean Ascidian Sidnyum elegans. J. Nat. Prod. 2016, 79, 1144–1148. [Google Scholar] [CrossRef]
- Imperatore, C.; D’Aniello, F.; Aiello, A.; Fiorucci, S.; D’Amore, C.; Sepe, V.; Menna, M. Phallusiasterols A and B: Two new sulfated sterols from the mediterranean tunicate Phallusia fumigata and their effects as modulators of the PXR receptor. Mar. Drugs 2014, 12, 2066–2078. [Google Scholar] [CrossRef]
- Luciano, P.; Imperatore, C.; Senese, M.; Aiello, A.; Casertano, M.; Guo, Y.-W.; Menna, M. Assignment of the Absolute Configuration of Phosphoeleganin via synthesis of Model Compounds. J. Nat. Prod. 2017, 80, 2118–2123. [Google Scholar] [CrossRef]
- Casertano, M.; Imperatore, C.; Luciano, P.; Aiello, A.; Menna, M.; Putra, M.Y.; Gimmelli, R.; Ruberti, G. Chemical Investigation of the Indonesian Tunicate Polycarpa aurata and Evaluation of the Effects Against Schistosoma mansoni of the Novel Alkaloids Polyaurines A and B. Mar. Drugs 2019, 17, 278. [Google Scholar] [CrossRef] [PubMed]
- Menna, M.; Aiello, A.; D’Aniello, F.; Imperatore, C.; Luciano, P.; Vitalone, R.; Irace, C.; Santamaria, R. Conithiaquinones A and B, Tetracyclic Cytotoxic Meroterpenes from the Mediterranean Ascidian Aplidium conicum. Eur. J. Org. Chem. 2013, 3241–3246. [Google Scholar] [CrossRef]
- Aiello, A.; Fattorusso, E.; Luciano, P.; Macho, A.; Menna, M.; Munoz, E. Antitumor Effects of Two Novel Naturally Occurring Terpene Quinones Isolated from the Mediterranean Ascidian Aplidium conicum. J. Med. Chem. 2005, 48, 3410–3416. [Google Scholar] [CrossRef] [PubMed]
- Aiello, A.; Fattorusso, E.; Luciano, P.; Mangoni, A.; Menna, M. Isolation and structure determination of aplidinones A-C from the Mediterranean ascidian Aplidium conicum: A successful regiochemistry assignment by quantum mechanical 13C NMR chemical shift calculations. Eur. J. Org. Chem. 2005, 2005, 5024–5030. [Google Scholar] [CrossRef]
- Imperatore, C.; Cimino, P.; Cebrián-Torrejón, G.; Persico, M.; Aiello, A.; Senese, M.; Fattorusso, C.; Menna, M.; Doménech-Carbó, A. Insight into the mechanism of action of marine cytotoxic thiazinoquinones. Mar. Drugs 2017, 15, 335. [Google Scholar] [CrossRef] [PubMed]
- Imperatore, C.; Persico, M.; Aiello, A.; Luciano, P.; Guiso, M.; Sanasi, M.F.; Taramelli, D.; Parapini, S.; Cebrián-Torrejón, G.; Doménech-Carbó, A.; et al. Marine inspired antiplasmodial thiazinoquinones: Synthesis, computational studies and electrochemical assays. RSC Adv. 2015, 5, 70689–70702. [Google Scholar] [CrossRef]
- Imperatore, C.; Persico, M.; Senese, M.; Aiello, A.; Casertano, M.; Luciano, P.; Basilico, N.; Parapini, S.; Paladino, A.; Fattorusso, C.; et al. Exploring the antimalarial potential of the methoxy-thiazinoquinone scaffold: Identification of a new lead candidate. Bioorg. Chem. 2019, 85, 240–252. [Google Scholar] [CrossRef]
- Gimmelli, R.; Persico, M.; Imperatore, C.; Saccoccia, F.; Guidi, A.; Casertano, M.; Luciano, P.; Pietrantoni, A.; Bertuccini, L.; Paladino, A.; et al. Thiazinoquinones as new promising multi-stage schistosomicidal compounds impacting Schistosoma mansoni and egg viability. ACS Infect. Dis. 2019. [Google Scholar] [CrossRef]
- Aiello, A.; Fattorusso, E.; Luciano, P.; Menna, M.; Calzado, M.A.; Muñoz, E.; Bonadies, F.; Guiso, M.; Sanasi, M.F.; Cocco, G.; et al. Synthesis of structurally simplified analogues of aplidinone A, a pro-apoptotic marine thiazinoquinone. Bioorg. Med. Chem. 2010, 18, 719–727. [Google Scholar] [CrossRef]
- Fedorov, S.N.; Radchenko, O.S.; Shubina, L.K.; Balaneva, N.N.; Bode, A.M.; Stonik, V.A.; Dong, Z. Evaluation of Cancer-Preventive Activity and Structure–Activity Relationships of 3-Demethylubiquinone Q2, Isolated from the Ascidian Aplidium glabrum, and its Synthetic Analogs. Pharm. Res. 2006, 23, 70–81. [Google Scholar] [CrossRef]
- Della Sala, G.; Agriesti, F.; Mazzoccoli, C.; Tataranni, T.; Costantino, V.; Piccoli, C. Clogging the Ubiquitin-Proteasome Machinery with Marine Natural Products: Last Decade Update. Mar. Drugs 2018, 16, 467. [Google Scholar] [CrossRef] [PubMed]
- Teta, R.; Della Sala, G.; Esposito, G.; Via, C.W.; Mazzoccoli, C.; Piccoli, C.; Bertin, M.J.; Costantino, V.; Mangoni, A. A joint molecular networking study of a Smenospongia sponge and a cyanobacterial bloom revealed new antiproliferative chlorinated polyketides. Org. Chem. Front. 2019, 6, 1762–1774. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 6 a | 7 a | |||
|---|---|---|---|---|
| Pos. | δC | δH, mult. (J in Hz) | δC | δH, mult. (J in Hz) |
| 1 | - | - | - | - |
| 2 | 49.0 | 3.29, m | 49.0 | 3.28, m |
| 3 | 39.9 | 4.04, m | 39.7 | 4.04, m |
| 4 | - | - | - | - |
| 4a | 142.9 | - | 143.2 | - |
| 5 | 176.8 | - | 176.5 | - |
| 6 | 152.7 | - | 153.1 | - |
| 7 | 136.9 | - | 137.0 | - |
| 8 | 178.1 | - | 178.8 | - |
| 8a | 110.2 | - | 109.8 | - |
| 9 | 60.8 | 3.91, s | 61.0 | 3.89, s |
| 1′ | 22.9 | 3.20, d (7.3) | 22.9 | 3.20, d (5.5) |
| 2′ | 119.1 | 5.04, t (7.6) | 119.5 | 5.04, m b |
| 3′ | 134.5 | - | 138.5 | - |
| 4′ | 17.8 | 1.72, s | 39.9 | 1.93, m |
| 5′ | 25.7 | 1.65, s | 26.5 | 2.02, m |
| 6′ | - | - | 124.5 | 5.04, m b |
| 7′ | - | - | 131.8 | - |
| 8′ | - | - | 17.8 | 1.56, s |
| 9′ | - | - | 16.9 | 1.71, s |
| 10′ | - | - | 25.3 | 1.64, s |
| -NH | 6.57, brs | - | 6.84, brs | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imperatore, C.; Della Sala, G.; Casertano, M.; Luciano, P.; Aiello, A.; Laurenzana, I.; Piccoli, C.; Menna, M. In Vitro Antiproliferative Evaluation of Synthetic Meroterpenes Inspired by Marine Natural Products. Mar. Drugs 2019, 17, 684. https://doi.org/10.3390/md17120684
Imperatore C, Della Sala G, Casertano M, Luciano P, Aiello A, Laurenzana I, Piccoli C, Menna M. In Vitro Antiproliferative Evaluation of Synthetic Meroterpenes Inspired by Marine Natural Products. Marine Drugs. 2019; 17(12):684. https://doi.org/10.3390/md17120684
Chicago/Turabian StyleImperatore, Concetta, Gerardo Della Sala, Marcello Casertano, Paolo Luciano, Anna Aiello, Ilaria Laurenzana, Claudia Piccoli, and Marialuisa Menna. 2019. "In Vitro Antiproliferative Evaluation of Synthetic Meroterpenes Inspired by Marine Natural Products" Marine Drugs 17, no. 12: 684. https://doi.org/10.3390/md17120684
APA StyleImperatore, C., Della Sala, G., Casertano, M., Luciano, P., Aiello, A., Laurenzana, I., Piccoli, C., & Menna, M. (2019). In Vitro Antiproliferative Evaluation of Synthetic Meroterpenes Inspired by Marine Natural Products. Marine Drugs, 17(12), 684. https://doi.org/10.3390/md17120684