2. Results and Discussion
The freeze-dried sponge was sequentially extracted with hexane, ethyl acetate, and
n-butanol, and twenty-one secondary metabolites (
Chart 1) were isolated from the ethyl acetate and
n-butanol extracts, including two new compounds, (
Z)-coscinamide D (
1) and (
E)-coscinamide D (
2). Lamellomorphamides A–D (
3–
4) were isolated for the first time as natural products, although they were previously reported as intermediates in the synthesis of 6′,6′′-didebromo-
cis-3,4-dihydrohamacanthin B, 6′-debromo-
cis-3,4-dihydrohamacanthin B, and hamacanthin analogues [
7,
8]. Eight previously reported compounds, (
E)-coscinamide B (
7) [
9], (
Z)-coscinamide B (
8) [
9], deoxytopsentin (
9) [
10], isobromodeoxytopsentin (
10) [
11], bromodeoxytopsentin (
11) [
11], dibromodeoxytopsentin (
12) [
11], 6-bromoindole-3-carboxylic acid (
13) [
12], and (6-bromo-1
H-indol-3-yl) oxoacetamide (
14) [
13], and seven trifluoroacetic acid (TFA)-catalyzed artifacts of the hamacanthin A and B class of compounds, 3,4-seco-(8
R)-6′′-debromohamacanthin A (
15), 3,4-seco-(8
R)-6′,6′′-didebromohamacanthin A (
16), 3,4-seco-(8
S)-hamacanthin A (
17), 3,4-seco-(8
S)-hamacanthin B (
18), 3,4-seco-(8
S)-6′′-debromohamacanthin B (
19), 3,4-seco-(8
R)-6′-debromohamacanthin B (
20), and 3,4-seco-(8
R)-6′,6′′-didebromohamacanthin B (
21), were also isolated. The structures of
1–
21 were determined by analysis of their spectroscopic data, including NMR in DMSO-
d6, UV, IR, ECD, and MS in combination with high-level TDDFT calculations. The structures of the eight previously reported compounds
7–
14 were also confirmed by comparison to literature data [
9,
10,
11,
12,
13].
(
Z)-coscinamide D (
1) was isolated as a yellow amorphous solid. ESIMS analysis revealed a protonated molecular isotopic doublet at
m/z 408/410 in a ratio of 1:1, characteristic of a monobrominated compound. HRESIMS analysis of
1 established a molecular formula of C
20H
14BrN
3O
2, based on the exact mass of the [M + Na]
+ adduct ion (Δmmu −0.6). The presence of a 6-bromoindole moiety was apparent from a comparison of the
1H and
13C NMR (
Table 1) and UV–vis spectroscopic data with those of the previously reported compound coscinamide B (
7) [
9]. Proton signals at δ
H 12.43 (1′-NH) and 8.92 (H-2′) and the ABX spin system comprised of signals at δ
H 7.41 (H-5′, dd,
J = 8.4, 1.8 Hz), 7.76 (H-7′, d,
J = 1.8 Hz), and 8.16 (H-4′, d,
J = 8.4 Hz) indicated the presence of a 3,6-disubstituted indole moiety. The 1′-NH signal showed a COSY correlation with the H-2′ signal at δ
H 8.92, which in turn showed HMBC correlations to C-3′ (δ
C 111.8), C-3a′ (δ
C 125.3), and C-7a′ (δ
C 137.2) (
Figure 1,
Table S1). A COSY correlation was also observed between H-4′ and H-5′. Furthermore, 3-bond HMBC correlations from H-4′ to C-3′, and to the non-protonated carbons C-6′ (δ
C 116.1) and C-7a′ (δ
C 137.2), confirmed the attachment of bromine at C-6′ and established the 3,6-disubstituted bromoindole moiety. Another set of proton signals at δ
H 11.45 (1-NH), 7.64 (H-2), 7.43 (H-4), 7.16 (H-5), 7.07 (H-6), and 7.63 (H-7) was assigned to the 3-substituted indole residue. The 1-NH signal showed a COSY correlation with H-2, which in turn showed HMBC correlations to C-3 (δ
C 109.6), C-3a (δ
C 126.4), and C-7a (δ
C 135.8). COSY correlations defined the spin system from H-4 to H-7, confirming that
1 contained a 3-substituted indole. This interpretation was supported by 3-bond HMBC correlations from H-2 and H-4 to C-7a. IR absorption bands at 1670 and 1623 cm
−1, along with non-protonated
13C NMR resonances at δ
C 159.7 and 180.0, suggest the presence of two carbonyl groups, one being an amide carbonyl. Another set of resonances was assigned to a vinylamide based on COSY correlations from H-9 to H-8 and 10-NH, and an HMBC correlation from 10-NH to the amide carbonyl carbon at C-9′ (δ
C 159.7). The observed
3JH-8,H-9 coupling constant (9.2 Hz) established the geometry of the olefinic bond as
Z, which was supported by a strong ROESY correlation between H-8 and H-9. HMBC correlations from H-8 to C-2 (δ
C 123.8) and C-3a (δ
C 126.4), and from H-9 to C-3, indicated direct attachment of this fragment to the unsubstituted indole at C-3 (
Figure 1). The remaining carbonyl substituent (δ
C 180.0) could be placed on C-3′ of the bromoindole moiety. Finally, the two indole-containing fragments were linked through the α-ketovinylamide to give the structure of
1. While no HMBC correlations were observed to C-8′ (δ
C 180.0), the chemical shifts of
1 closely resembled those reported for coscinamides A and B (
7) [
9]. It should also be noted that there are no literature reports of HMBC correlations to C-8′ in analogous known compounds, even for experiments optimized for small range coupling constants [
9].
(
E)-Coscinamide D (
2) was isolated as a yellow amorphous solid. HRESIMS analysis of
2 established the same molecular formula as
1 (C
20H
14BrN
3O
2) based on the deprotonated molecule [M − H]
− (Δmmu +0.2). The NMR data for
2 were very similar to those for
1 (
Table 1), with the only significant differences being in the chemical shifts and coupling constants of the vinyl protons H-8 and H-9. The observed
3JH-8,H-9 coupling constant (14.7 Hz) suggested an
E geometry, which was supported by the absence of a ROESY correlation between H-8 and H-9 (
Figure 1,
Table S2), confirming that
2 is the
E isomer of
1. All other spectroscopic data were consistent with this structure.
Lamellomorphamide A (
3) was isolated as a yellow amorphous solid. HRESIMS analysis revealed a protonated molecule [M + H]
+ consistent with a molecular formula C
20H
15N
3O
3 (Δmmu −0.3). The NMR data for
3 (
Table 2) also showed the presence of two 3-substituted indole residues, which was reminiscent of those of coscinamide B (
7). The main difference from the coscinamide derivatives was the presence of an extra carbonyl carbon. IR absorption bands at 1670 and 1623 cm
−1 along with non-protonated
13C NMR resonances at δ
C 163.8, 181.9, and 189.2, suggest the presence of one amide and two conjugated ketones. HMBC correlations from H-2 (δ
H 8.50, d,
J = 3.2 Hz) to C-3a (δ
C 125.4) and C-7a (δ
C 136.3), and from 1-NH (δ
H 12.07) to C-3 (δ
C 113.9) and C-3a (δ
C 125.4), indicated that the pyrrole 1-NH and H-2 were connected to an ABMX aryl spin system comprised of the signals at δ
H 7.20 (H-5), 7.23 (H-6), 7.49 (H-7), and 8.16 (H-4) (
Figure 1,
Table S3). HMBC correlations from H-2′ (δ
H 8.82, d,
J = 3.2 Hz) and 1′-NH (δ
H 12.26) to C-3a′ (δ
C 126.2) and C-7a′ (δ
C 136.4) indicated that the other pyrrole protons 1′-NH and H-2′ were connected to another ABMX aryl spin system comprised of the signals at δ
H 7.27 (H-5′), 7.28 (H-6′), 7.54 (H-7′) and 8.26 (H-4′). This confirmed the presence of the two 3-substituted indole moieties. An HMBC correlation from H-2 to C-8 (δ
C 189.2) indicated attachment of C-8 at C-3. The amide proton showed an HMBC correlation to its carbonyl C-9′ (δ
C 163.8) and a COSY correlation to methylene H
2-9 (δ
H 4.63, d,
J = 5.9 Hz), which in turn was coupled by HMBC to the carbonyl C-8. ROESY correlations between H-2 and H
2-9, and between H
2-9 and 10-NH (δ
H 8.91) verified the attachment of the fragment RCO–NH–CH
2–CO on C-3 of the indole moiety (
Figure 1). The remaining carbonyl substituent C-8′ (δ
C 181.9) was placed on C-3′ of the additional indole moiety, and the two indole-containing fragments were linked through the ketone (δ
C 181.9) and the amide carbonyl (δ
C 163.8) to give the structure of
3. Analogous to
1, and the literature [
11,
14], no HMBC correlations were observed to C-8′.
Lamellomorphamide B (
4) was also isolated as a yellow, amorphous solid. ESIMS analysis revealed a protonated molecule isotopic cluster [M + H]
+ at
m/z 424/426 in the ratio 1:1, suggesting the presence of one bromine atom. HRESIMS of
4 established a molecular formula of C
20H
14BrN
3O
3, based on the [M + Na]
+ adduct ion (Δmmu −0.3). The
1H NMR data for
4 were similar to
3, indicating the same basic scaffold. The NMR data for
4 (
Table 2) also showed the presence of one 3-substituted indole and one 3,6-disubstituted bromoindole. The main difference from
3 was the presence of a bromine atom, and its attachment on the indole ring of the “left hand” of the compound was confirmed by HMBC and ROESY NMR experiments (
Figure 1,
Table S4). The 1-NH signal (δ
H 12.17), the methine H-2 (δ
H 8.53), and the AMX spin system comprised of the signals at δ
H 7.35 (H-5, dd,
J = 8.5, 1.8 Hz), 7.69 (H-7, d,
J =1.8 Hz), 8.09 (H-4, d,
J = 8.5 Hz), indicated the presence of a 3,6-disubstituted indole moiety with the bromine atom located on C-6 (δ
C 115.0). HMBC correlations were observed from 1-NH, H-2, H-5, and H-7 to C-3a (δ
C 124.5) and from H-2 and H-4 to C-7a (δ
C 137.4). The connectivity between 3,6-disubstituted bromoindole to the carbonyl C-8 (δ
C 189.4) was established by a ROESY correlation between H-2 and H
2-9 (δ
H 4.62). The proton signals at δ
H 12.25 (1′-NH), 8.80 (H-2′), 8.25 (H-4′), 7.27 (H-5′), 7.28 (H-6′), and 7.53 (H-7′) suggested the presence of a 3-substituted indole moiety. Attaching this second indole to the ketone C-8′ (δ
C 181.8) gave
4 as the only possible structure for lamellomorphamide B.
Lamellomorphamide C (
5) was isolated as a yellow, amorphous solid. ESIMS analysis revealed a protonated molecule isotopic cluster [M + H]
+ at
m/z 424/426 in the ratio 1:1, indicating the presence of one bromine atom. HRESIMS of
5 established the same molecular formula as
4 (C
20H
14BrN
3O
3) based on the [M + Na]
+ adduct ion (Δmmu −0.3). This immediately suggested that
5 is a regioisomer of
4, with the bromine on the other (“right hand”) indole ring (
Table 3). This interpretation was supported by ROESY correlations between 1′-NH (δ
H 12.32) and H-7′ (δ
H 7.75, d,
J = 1.8 Hz) (
Figure 1,
Table S5). The presence of a small meta coupling between H-7′ and H-5′ (δ
H 7.42), and the absence of an ortho coupling revealed the attachment of the bromine atom at C-6′ (δ
C 116.0). The observed proton signals and coupling constants at δ
H 8.18 (H-4′, d,
J = 8.4 Hz), 7.42 (H-5′, dd,
J = 1.8,
J = 8.4 Hz), and 7.75 (H-7′, d,
J = 1.8 Hz), together with HMBC correlations from 1′-NH and H-2′ to C-3a′ (δ
C 125.3, is characteristic of a 3,6-disubstituted indole moiety with the bromine atom located on the indole ring of the “right hand” of
5. The proton signals at δ
H 12.07 (1-NH), 8.50 (H-2), 8.16 (H-4), 7.20 (H-5), 7.23 (H-6), and 7.49 (H-7) suggest the presence of the 3-substituted indole moiety, which was confirmed by HMBC correlations from H-2 to C-3, C-3a, and C-7a, and from H-4 to C-3, C-6, and C-7a. The connection of 3-indole to C-8 was confirmed by a strong ROESY correlation between H-2 and H-9. The amide proton showed an HMBC correlation to its carbonyl C-9′ (δ
C 163.4) and a COSY correlation to methylene H
2-9 (δ
H 4.63,
J = 5.9 Hz), which in turn was coupled by HMBC to the carbonyl C-8. Comparison of the chemical shifts of the indole protons with those of
4, together with HMBC correlations from H-2′ to C-3′, C-3a′, and C-7a′, and from H-4′ to C-3′, C-6′, and C-7a′, confirmed the connection of the 3′-bromoindolyl residue to C-8′, even though this carbon, like all others in this series, showed no correlations (
Table S5). This left
5 as the only possible structure of lamellomorphamide C that matched the observed spectroscopic data.
Lamellomorphamide D (
6) was isolated as a yellow, amorphous solid. ESIMS analysis revealed a protonated molecule isotopic cluster [M + H]
+ at
m/z 502/504/506 in the ratio 1:2:1, indicative of two bromine atoms. HRESIMS of
6 established a molecular formula of C
20H
13Br
2N
3O
3, based on the [M + H]
+ protonated molecule (Δmmu 0.0). The similarity in
1H NMR spectra between
6 and
3–
5 suggested
6 had both indoles substituted with bromine. This was confirmed by the presence of two aromatic ABX systems; δ
H 7.35 (H-5, dd,
J = 8.5, 1.8 Hz), 7.69 (H-7, d,
J = 1.8 Hz), and 8.09 (H-4, d,
J = 8.5 Hz); and δ
H 7.42 (H-5′, dd,
J = 8.4, 1.8 Hz), 7.75 (H-7′, d,
J = 1.8 Hz), and 8.18 (H-4′, d,
J = 8.4 Hz). ROESY correlations between 1-NH (δ
H 12.17) and H-7 (δ
H 7.69, d,
J = 1.8 Hz) and from 1′-NH (δ
H 12.32) and H-7′ (δ
H 7.75, d,
J = 1.8 Hz) were observed (
Figure 1,
Table S6). The small meta coupling constants of H-7 and H-7′ to H-5 and H-5′, respectively, revealed the attachment of the bromine atoms at C-6 (δ
C 115.5) and C-6′ (δ
C 116.0), consistent with the previously identified structures. The remaining spectroscopic data (
Table 3) were consistent with
6 being 6,6′-dibromolamellomorphamide A.
Compounds
3 and
4 have previously been reported as intermediates in the synthesis of
cis-3,4-dihydrohamacanthin B [
7], and compounds 5 and 6 have previously been reported as intermediates in the synthesis of hamacanthin B anologues [
8] but to date have not been reported as natural products. The reported
1H and
13C NMR data for
4 matched our data exactly. Although no HMBC correlations to C-8′ were detected for any of the compounds
1–
6, the connectivity between the keto enamide and the indole moieties was determined by comparison of our NMR data with those of coscinamide B. The absence of any correlations to C-8′ is also consistent with the topsentins [
11] and spongotines [
14]. Coscinamides are the only known natural products that contain the unusual α-ketovinylamide functionality, and this is the first example of naturally occurring
E isomers, although both coscinamide B and its
Z isomer have been synthesized as intermediates in the synthesis of dihydrohamacanthins [
15].
Deoxytopsentin (
9) [
10] was the major metabolite of
L. strongylata and was obtained pure after Sephadex LH-20 gel permeation chromatography. As previously reported for topsentins [
16], the doubling of
1H NMR signals was observed in DMSO-
d6 due to slow tautomerization of the imidazole. The addition of a trace amount of TFA to the NMR sample resulted in one set of signals (
Figure S47), but with broadening of the imidazolium methine. In methanol-
d4 (
Figure S48), all peaks were sharp and could be easily assigned.
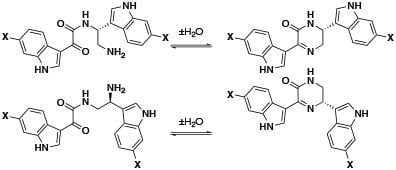
Hamacanthins A (
24) and B (
25) [
14,
17] are part of a series of marine alkaloids that contain a six-membered nitrogen heterocycle (5,6-dihydro-2-(1
H)-pyrazinone) as a linker unit between two variously substituted indole groups. Seven putative acid-catalyzed artifacts of the hamacanthin A and B class of compounds were isolated from the ethyl acetate and
n-butanol extracts: 3,4-seco-6′′-debromohamacanthin A (
15), 3,4-seco-6′,6′′-didebromohamacanthin A (
16), 3,4-seco-hamacanthin A (
17), 3,4-seco-hamacanthin B (
18), 3,4-seco-6′′-debromohamacanthin B (
19), 3,4-seco-6′-debromohamacanthin B (
20), and seco-6′,6′′-didebromohamacanthin B (
21). NMR spectroscopy of these HPLC purified compounds (containing traces of TFA) showed only one set of peaks corresponding to the acyclic form (
Figures S56–S69, Tables S7–S9), while LC-MS analysis (with or without formic acid) showed two peaks (
Figure S1). The first peak (
m/z 425/427; λ
max 214, 262, 278, 329 nm) corresponded to the acyclic form and the second peak (
m/z 407/409,
Figure S75; λ
max 216, 280, 385 nm;) corresponded to the cyclic form (
Figure S2). Compounds
15–
21 were isolated by HPLC purification with 0.05% TFA in the mobile phase. An LC-MS analysis of crude
n-butanol and ethyl acetate fractions did not reveal masses corresponding to the acyclic compounds (
Figures S3 and S4), but rather the cyclic hamacanthin A and B class of compounds. We therefore propose that the TFA-catalyzed ring-opening occurred during HPLC purification of
15–
21. A similar mechanism has been proposed by Kuoko et al. [
18] in the total synthesis of optically active hamacanthins A and B from the acyclic intermediates. Alternatively, the previously isolated hamacanthin A and B type alkaloids could be artifacts of isolation as previous reports all used silica gel chromatography, which we found efficiently cyclizes compounds
15–
21.
Given there are no specific rotation data reported for any compounds related to
15–
21, our ORD data could not be compared to any literature values. Therefore,
15–
21 were passed through a silica SPE column, eluting with dry 20% ethyl acetate in hexane, to facilitate dehydration and ring closure (
Scheme 1). The specific rotations of the resulting cyclized compounds
22–
28 were measured for comparison to the hamacantins (
Table 4). The specific rotation of
22 indicated it was (
R)-6′′-debromohamacanthin A and that
15 must therefore also have an
R configuration at C-8. Compound
23 had a specific rotation suggesting it was (
R)-didebromohamacanthin A, the enantiomer of natural (
S)-bisdebromohamacanthin A. Compound
24 had a specific rotation similar to synthetic and natural (
S)-hamacanthin A (
Table 4) and opposite to synthetic (
R)-hamacanthin A (−79 deg cm
2 g
–1 [
19]). The specific rotations of natural and synthetic hamacanthin B (
25) matched the sign but not the amplitude of the specific rotation for cyclized
18. Similarly,
28 had a negative specific rotation but much smaller than expected from the literature. In contrast, the specific rotation of
26 was a reasonable match for the published specific rotation of (
S)-6′′-debromohamacanthin B. Compound
27, did not match the amplitude and was opposite in sign, suggesting
27 (and
20) had the opposite configurations to the previously isolated compounds. Kuoko et al. [
18] in the total synthesis (+)-hamacanthins A and B, reported the specific rotations of the Boc-protected acyclic amide intermediates as [α]
D −2.6, +9.8, and −5.0 deg.cm
2.g
–1, which are very small compared to the specific rotations of the cyclic (
S)-hamacanthins A and B (+83.7 and +170.1 deg.cm
2.g
–1, respectively) and generally consistent with the fact that specific rotation is not a useful indicator of absolute configuration in this series of closely related compounds.
Turning to electronic circular dichroism (ECD;
Figure 2 and
Figures S5–S11), it is clear that
15,
16,
20, and
21 have the same configurations, while
17–
19 have the opposite configurations. The weak ECD for
17,
18 and
20,
21 suggests that they are partial racemates, supporting the conclusions above (
Table 4). The ECD spectra of
15–
21 showed negative Cotton effects at 280 and 330 nm and positive Cotton effects at 360 nm for all the compounds except
17–
19, which were the opposite. This is consistent with the specific rotations found for
27 and
28 (derived from
20 and
21 respectively) supporting the assignment of
15 and
16 as
R, and
17–
19 as
S. Based on the ECD data,
20 and
21 should have
R configurations, consistent with a negative specific rotation, but not consistent with the magnitude of the published specific rotations. Taken together, these data suggest that
20 and
21 are partial racemates with the
R form predominating and that
18 and
19 are partial racemates with the
S form predominating. This result is intriguing for a number of reasons. Firstly, the one sponge produces enantiomerically pure but opposite configurations of compounds that only differ in the number of bromines, suggesting parallel rather than sequential biosynthesis. Secondly, four compounds are partial racemates, which (if the above is true) suggests these compounds are biosynthesized via two routes in non-equivalent amounts. This type of enantiodivergent biosynthesis has previously been observed in related bromotyrosine and bromopyrrole alkaloids [
3,
23,
24,
25]. While it is possible that this chiral center could undergo epimerization during handling, this is unlikely as partial racemization was seen in only four out of the seven compounds isolated from the same extract, and all compounds were handled identically. Finally, our results explain the range of optical rotations published for these compounds. For example, synthetic (
S)-hamacanthin B (
25) has been reported with a specific rotation (methanol) of +170–183 deg.cm
2.g
–1 [
18,
22] whereas natural
25 has been reported with +172 deg.cm
2.g
–1 [
17] or +56 deg.cm
2.g
–1 [
20]. The latter is close to the value we obtained (+46 deg.cm
2.g
–1) for the partial racemate, suggesting that the compound isolated by Bao et al. was also a partial racemate. Enantiodivergent biosynthesis of natural products may be more common than currently recognized.
Finally, turning to time-dependent density functional theory (TDDFT) calculations, we were able to confirm the
R configuration for
16 and
21. TDDFT-D3//pbe0/TZVPP-COSMO(methanol) was able to qualitatively reproduce the UV (
Figure S12) and ECD (
Figure 3) spectra for
16 and
21. Specifically, the positive Cotton effect at 360 nm and negative Cotton effect at 330 nm were reproduced, albeit with differing oscillator strengths. These results add weight to the interpretation that
17–
19 have
S configurations while the others have
R configurations.
Bisindole alkaloids are a rapidly growing group of sponge metabolites that exhibit potent and diverse bioactivities, including cytotoxicity, antiviral, antifungal, and anti-inflammatory activities [
20]. For example, the bisindole alkaloids vinblastine and vincristine have been developed as effective anticancer drugs [
26]. Examples of bisindole alkaloids reported from marine sponges include the topsentins [
16], which have a ketone and an imidazole moiety as a linker between two indole rings; the nortopsentins [
27], which lack the central ketone; and dragmacidins [
17] and the hamacanthins (
22–
28) [
17,
18], which have a piperazine–piperazinone linker. Compounds
1–
9 and
13–
21 were screened against Methicillin-resistant
Staphylococcus aureus (MRSA, ATTC 43300),
Escherichia coli (ATCC 25922),
Klebsiella pneumoniae (MDR, ATCC 700603),
Acinetobacter baumannii (ATCC 19606),
Pseudomonas aeruginosa (ATCC 27853),
Candida albicans (ATCC 90028), and
Cryptococcus neoformans var.
grubii (ATCC 208821). Compounds
2,
3,
6,
13, and
18 showed some activity against MRSA (
Table S10).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}