Bis-Indolyl Benzenoids, Hydroxypyrrolidine Derivatives and Other Constituents from Cultures of the Marine Sponge-Associated Fungus Aspergillus candidus KUFA0062

 , , ,
, , ,  ,
,  ,
,  ,
,  , and
, and 
Abstract
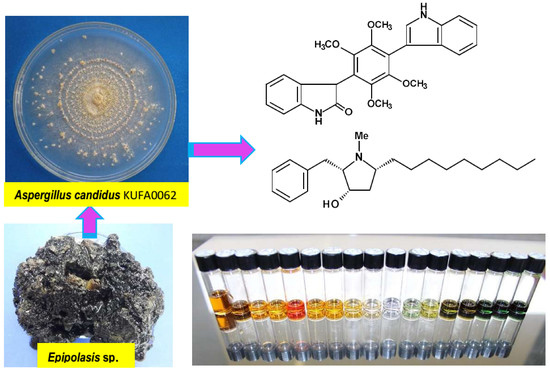
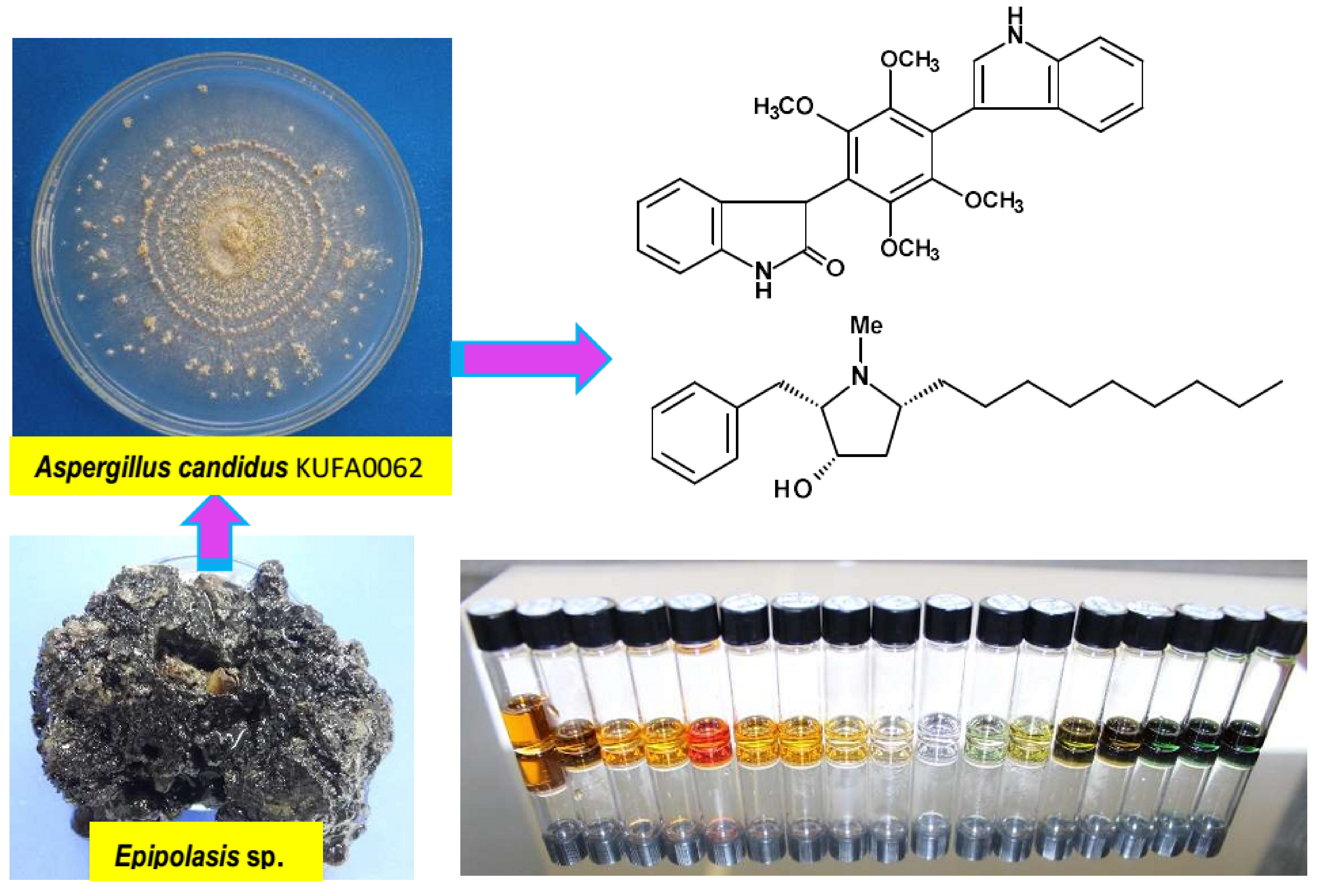
1. Introduction
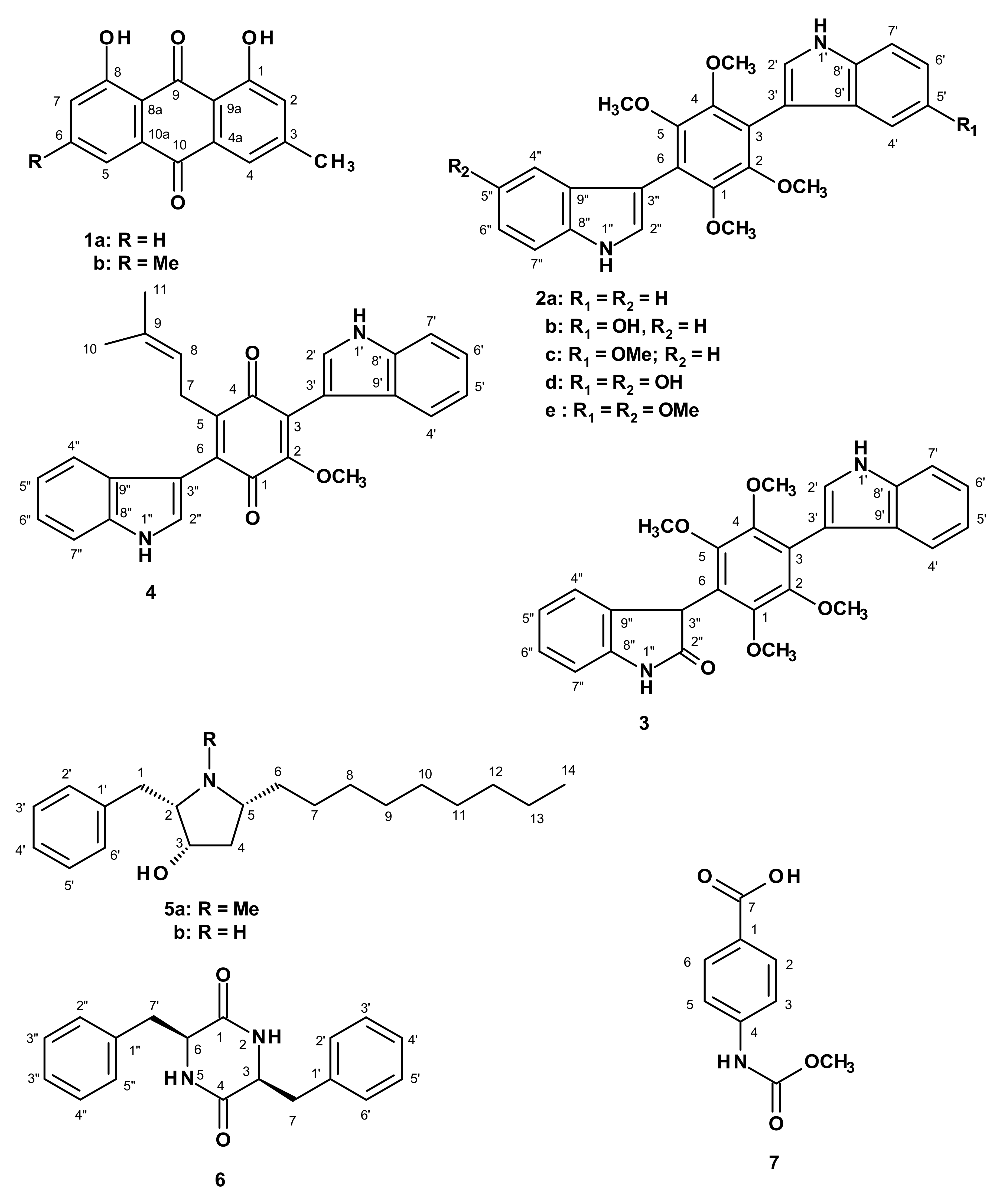
2. Results and Discussion
3. Experimental Section
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Extraction and Isolation
3.3.1. Candidusin D (2e)
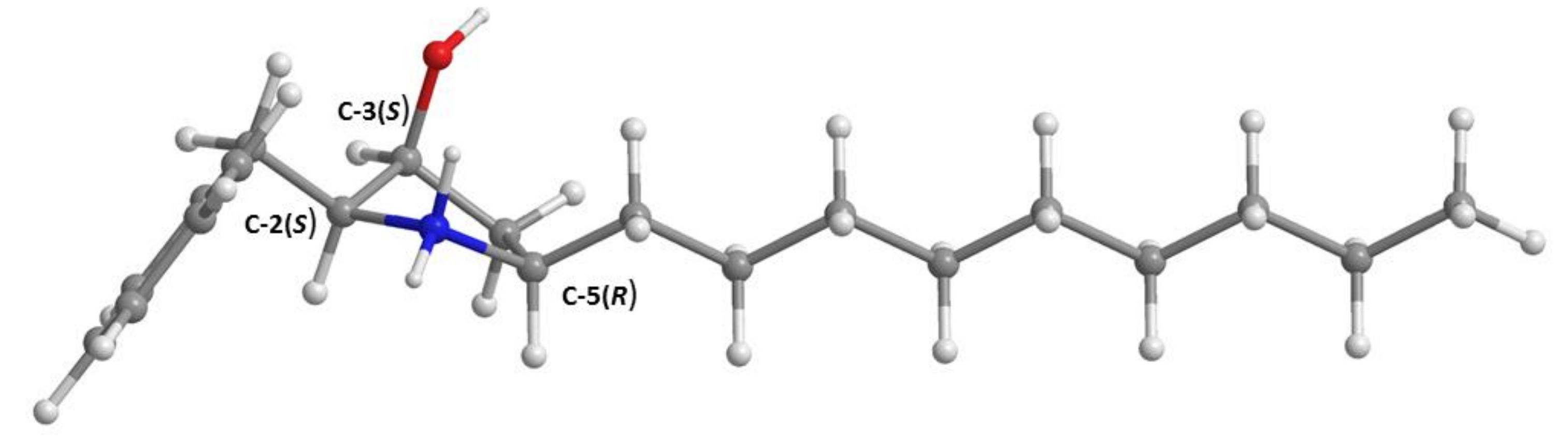
3.3.2. Preussin C (5b)
3.4. Electronic Circular Dichroism
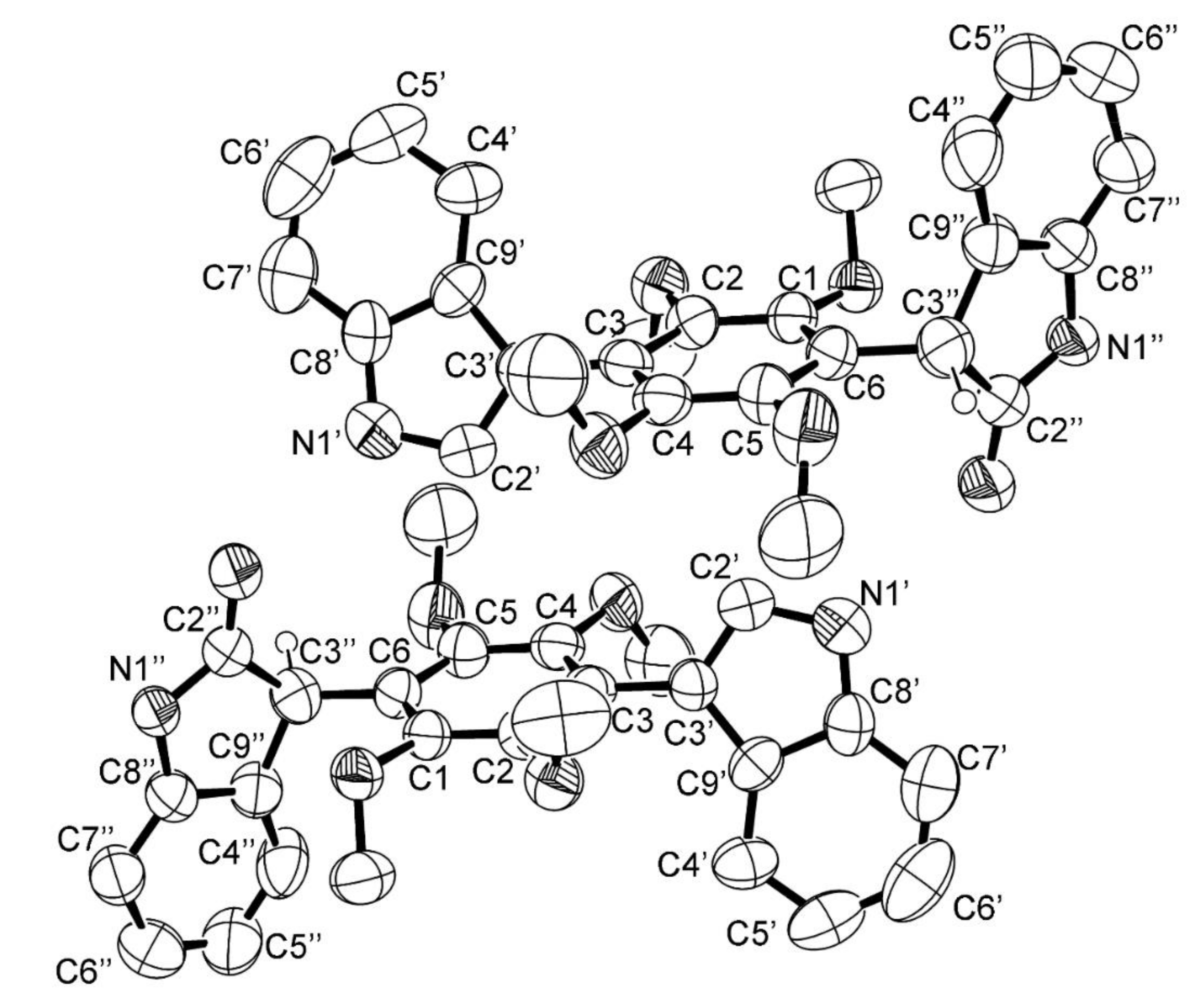
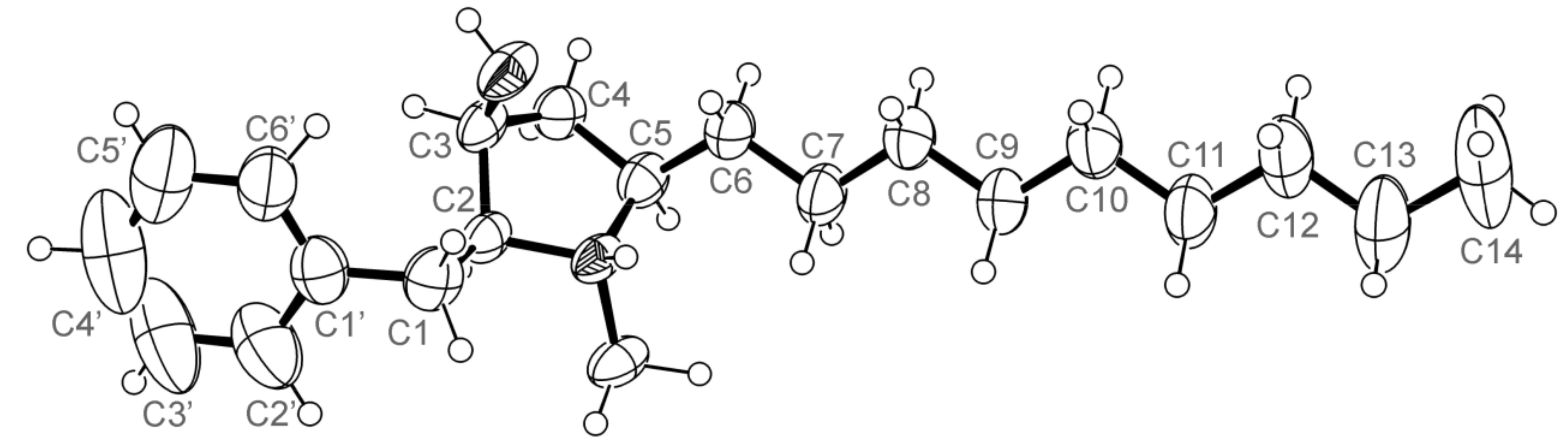
3.5. X-ray Crystal Structure of 3 and 5a
3.6. Antibacterial Activity Bioassays
3.6.1. Bacterial Strains and Growth Conditions
3.6.2. Antimicrobial Susceptibility Testing
3.6.3. Biofilm Formation Inhibition Assay
3.6.4. Antibiotic Synergy Testing
3.7. In Vitro Anticancer Activity Assays
3.7.1. Cell Lines
3.7.2. MTT Reduction Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Samson, R.A.; Visagie, C.M.; Houbraken, J.; Hong, S.B.; Hubka, V.; Klaassen, C.H.; Perrone, G.; Seifert, K.A.; Susca, A.; Tanney, J.B.; et al. Phylogeny, identification and nomenclature of the genus Aspergillus. Stud. Mycol. 2014, 78, 141–173. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Choi, S.Y.; Hwang, H.J.; Kim, Y.B. Fungal mycoflora and mycotoxins in Korean polished rice destined for humans. Int. J. Food Microbiol. 2005, 103, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Ismail, M.A.; Taligoola, H.K.; Chebon, S.K. Mycobiota associated with rice grains marketed in Uganda. J. Biol. Sci. 2004, 4, 271–278. [Google Scholar]
- Kozakiewicz, Z. Aspergillus species on stored products. Mycol. Pap. 1989, 161, 1–188. [Google Scholar]
- Richards, M.; Bird, A.E.; Munden, J.E. Chloflavonin a new fungal antibiotic. J. Antibiot. 1969, 22, 388–389. [Google Scholar] [CrossRef]
- Marchelli, R.; Vining, L.C. Terphenyllin, a novel p-terphenyl metabolite from Aspergillus candidus. J. Antibiot. 1975, 23, 328–331. [Google Scholar] [CrossRef]
- Takahashi, C.; Yoshihira, K.; Natori, S.; Umeda, M. The structure of toxic metabolites of Aspergillus candidus I. The compounds A and E, cytotoxic p-terphenyls. Chem. Pharm. Bull. 1976, 24, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Kurobane, I.; Vining, L.C.; McInnes, A.G.; Smith, D.G. 3-Hydroxyterphenyllin, a new metabolite of Aspergillus candidus. J. Antibiot. 1979, 32, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Takemura, A.; Nagano, H.; Kawazu, K. Candidusins A and B: New p-terphenyls with cytotoxic effects on sea urchin embryos. Agric. Biol. Chem. 1982, 6, 585–589. [Google Scholar] [CrossRef]
- Kamigauchi, T.; Sakazaki, R.; Nagashima, K.; Kawamura, Y.; Yasuda, Y.; Matsushima, K.; Tani, H.; Takahashi, Y.; Suzuki, R.; Koizumi, K.; et al. Terprenins novel immunosuppressants produced by Aspergillus candidus. J. Antibiot. 1998, 51, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Inada, H.; Hayashi, A.; Higashimoto, K.; Pruksakorn, P.; Kamada, S.; Arai, A.; Ishida, S.; Kobayashi, M. Prenylterphenyllin and its dehydroxyl analogs, new cytotoxic substances from marine-derived fungus Aspergillus candidus IF10. J. Antibiot. 2007, 60, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Guo, Z.K.; Li, X.M.; Chen, F.X.; Zhan, X.F.; Shen, M.H. Spiculisporic acid analogues of the marine-derived fungus Aspergillus candidus strain HDf2, and their antibacterial activity. Antonie Leeuenhoek 2015, 108, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Semple, S.J.; Pyke, S.M.; Reynolds, G.D.; Flower, R.L. In vitro antiviral activity of the anthraquinone chrysophanic acid against poliovirus. Antivir. Res. 2001, 49, 169–178. [Google Scholar] [CrossRef]
- Noinart, J.; Buttachon, S.; Dethoup, T.; Gales, L.; Pereira, J.A.; Urbatzka, R.; Freitas, S.; Lee, M.; Silva, A.M.S.; Pinto, M.M.M.; et al. A new ergosterol analog, a new bis-anthraquinone and anti-obesity activity of anthraquinones from the marine sponge-associated fungus Talaromyces stipitatus KUFA 0207. Mar. Drugs 2017, 15, 139. [Google Scholar] [CrossRef] [PubMed]
- Lacey, H.J.; Vuong, D.; Pitt, J.; Lacey, E.; Piggott, A.M. Kumbicins A–D: Bis-indolyl benzenoids and benzoquinone from an Australian soil fungus, Aspergilus kumbius. Aust. J. Chem. 2016, 69, 152–160. [Google Scholar] [CrossRef]
- Ooike, M.; Nozawa, K.; Udagawa, S.; Kawai, K. Bisindolylbenzenoids from ascostromata of Petromyces muricatus. Can. J. Chem. 1997, 75, 625–628. [Google Scholar] [CrossRef]
- Whyte, A.C.; Joshi, B.K.; Gloer, J.B.; Wicklow, D.T.; Dowd, P.F. New cyclic peptide and bisindolyl benzenoid metabolites from the sclerotia of Aspergillus sclerotiorum. J. Nat. Prod. 2000, 63, 1006–1009. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, R.E.; Liesch, J.; Hensens, O.; Zitano, L.; Honeycutt, S.; Garrity, G.; Fronmtling, R.A.; Onishi, J.; Monaghan, R. L-657,398, a novel antifungal agent: Fermentation, isolation, structural elucidation and biological properties. J. Antibiot. 1988, 41, 1774–1779. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.H.; Phillipson, D.W.; Kahle, A.D. The relative and absolute stereochemistry of the antifungal preussin. J. Antibiot. 1989, 42, 1184–1185. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Sudoh, Y.; Tsuchirya, Y.; Okuda, T.; Igarashi, Y. Isolation and biosynthesis of preussin B, a pyrrolidine alkaloid from Simplicillium lanosoniveum. J. Nat. Prod. 2014, 77, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.M.; Ding, G.Z.; Fang, L.; Dai, J.G.; Yu, S.S.; Wang, Y.H.; Chen, X.G.; Ma, S.G.; Qu, J.; Xu, S.; et al. Thiodiketopiperazines produced by the endophytic fungus Epicoccum nigrum. J. Nat. Prod. 2010, 73, 1240–1249. [Google Scholar] [CrossRef] [PubMed]
- Parish, C.A.; Huber, J.; Baxter, J.; González, A.; Collado, J.; Platas, G.; Diez, M.T.; Vicente, F.; Dorso, K.; Abruzzo, G.; et al. A new ene-triyne antibiotic from the fungus Baeospora myosura. J. Nat. Prod. 2004, 67, 1900–1902. [Google Scholar] [CrossRef] [PubMed]
- Dzeha, T.; Jaspars, M.; Tabudravu, J. Clionasterol, a triterpenoid from the Kenyan marine green macroalga Halimeda macroloba. Western Indian Ocean J. Mar. Sci. 2003, 2, 157–161. [Google Scholar]
- Cantu, M.D.; Hillebrand, S.; Carrilho, E. Determination of the dissociation constants (pKa) of secondary and tertiary amines in organic media by capillary electrophoresis and their role in the electrophoretic mobility order inversion. J. Chromatogr. A 2005, 1068, 99–105. [Google Scholar] [CrossRef] [PubMed]
- EUCAST. Recommendations for MIC Determination of Colistin (Polymixin E) as Recommended by the Joint CLSI-EUCAST Polymixin Breakpoints Working Group. Available online: http://www.bioconnections.co.uk/files/merlin/Recommendations_for_MIC_determination_of_colistin_March_2016.pdf (accessed on 1 March 2018).
- Ravizza, R.; Gariboldi, M.B.; Passarelli, L.; Monti, E. Role of the p53/p21 system in the response of human colon carcinoma cells to doxorubicin. BMC Cancer 2004, 4, 92. [Google Scholar] [CrossRef] [PubMed]
- Varga, J.; Frisvad, J.C.; Samson, R.A. Polyphasic taxonomy of Aspergillus section Candidi based on molecular, morphological and physiological data. Stud. Mycol. 2007, 59, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4325. [Google Scholar] [CrossRef] [PubMed]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: New York, NY, USA, 1990; pp. 315–322. [Google Scholar]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 72, 5463–5467. [Google Scholar] [CrossRef]
- Austin, A.; Petersson, G.A.; Frisch, M.J.; Dobek, F.J.; Scalmani, G.; Throssel, K. A density functional with spherical atom dispersion terms. J. Chem. Theory Comput. 2012, 8, 4989–5007. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Harada, N. ECD Cotton effect approximated by the Gaussian curve and other methods. Chirality 2010, 22, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Inoue, Y.; Grimme, S. Time dependent density functional theory calculations for electronic circular dichroism spectra and optical rotations of conformationally flexible chiral donor-acceptor dyad. J. Org. Chem. 2006, 71, 9797–9806. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short story of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Simões, R.R.; Aires-de-Sousa, M.; Conceicao, T.; Antunes, F.; da Costa, P.M.; de Lencastre, H. High prevalence of EMRSA-15 in Portuguese public buses: A worrisome finding. PLoS ONE 2011, 6, e17630. [Google Scholar] [CrossRef] [PubMed]
- Bessa, L.J.; Barbosa-Vasconcelos, A.; Mendes, A.; Vaz-Pires, P.; Martins da Costa, P. High prevalence of multidrug-resistant Escherichia coli and Enterococcus spp. in river water, upstream and downstream of a wastewater treatment plant. J. Water Health 2014, 12, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Clinical and Laboratory Standards Institute. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically. In Approved Standard, 10th ed.; CLSI: Wayne, PA, USA, 2015; Document M07-A10. [Google Scholar]
- May Zin, W.W.; Buttachon, S.; Dethoup, T.; Pereira, J.A.; Gales, L.; Inácio, A.; Costa, P.M.; Lee, M.; Sekeroglu, N.; Silva, A.M.S.; et al. Antibacterial and antibiofilm activities of the metabolites isolated from the culture of the mangrove-derived endophytic fungus Eurotium chevalieri KUFA 0006. Phytochemistry 2017, 141, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Bessa, L.J.; Palmeira, A.; Gomes, A.S.; Vasconcelos, V.; Sousa, E.; Pinto, M.; Martins da Costa, P. Synergistic effects between thioxanthones and oxacillin against methicillin-resistant Staphylococcus aureus. Microb. Drug Resist. 2015, 21, 404–415. [Google Scholar] [CrossRef] [PubMed]
- Odds, F.C. Synergy, antagonism, and what the chequer board puts between them. J. Antimicrob. Chemother. 2003, 52. [Google Scholar] [CrossRef] [PubMed]
- Ramos, A.A.; Prata-Sena, M.; Castro-Carvalho, B.; Dethoup, T.; Buttachon, B.; Kijjoa, A.; Rocha, E. Testing the potential of four marine-derived fungi extracts as anti-proliferative and cell death-inducing agents in seven cancer cell lines. Asian Pac. J. Trop. Med. 2015, 8, 798–806. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δC, Type | δH, (J in Hz) |
|---|---|---|
| 1, 2, 4, 5 | 147.6, C | - |
| 3, 6 | 120.0, C | - |
| NH-1′, 1″ | - | 11.15, d (2.2) |
| 2′, 2″ | 126.0, CH | 7.43, d (2.5) |
| 3′, 3″ | 106.7, C | - |
| 4′, 4″ | 102.1, CH | 6.88, d (2.5) |
| 5′, 5″ | 153.1, C | - |
| 6′, 6″ | 110.9, CH | 6.78, dd (8.8, 2.5) |
| 7′, 7″ | 111.9, CH | 7.33, d (8.8) |
| 8′, 8″ | 131.0, C | - |
| 9′, 9″ | 127.7 | - |
| OCH3-1, 2, 4, 5 | 60.3, CH3 | 3.46, s |
| OCH3-5, 5′ | 55.3, CH2 | 3.72, s |
| 5a (DMSO, 300.13 and 75.4 MHz) | 5b (DMSO, 500.13 and 125.4 MHz) | ||||||
|---|---|---|---|---|---|---|---|
| Position | δC, Type | δH, (J in Hz) | δC, Type | δH, (J in Hz) | COSY | HMBC | NOESY |
| 1a | 30.6, CH2 | 3.69, dd (13.0, 9.4) | 31.3, CH2 | 3.11, dd (13.8, 8.0) | H-1b, 2 | C-2, 3 | H-1b, 2′, 6′ |
| b | 3.13, dd (12.7, 4.8) | 2.94, dd (13.8, 6.6) | H-1a, 2 | C-2, 3 | H-1a | ||
| 2 | 76.0, CH | 3.09, q (4.7) | 65.5, CH | 3.46, m | H-1b, 1b, 3 | C-1 | H-3 |
| 3 | 68.6, CH | 4.22, m | 68.7, CH | 4.09, dd (7.9, 4.0) | H-2, 4β, OH-3 | C-2, 5 | H-2, H-4β, OH-3 |
| 4α | 38.8, CH2 | 2.06, ddd (14.8, 9.3, 2.1) | 38.5, CH2 | 1.58, dd (13.9, 5.8) | H-3, 5 | C-3, 5, 6 | H-3, 4β, 5 |
| β | 2.69, ddd (15.0, 8.2, 8.2) | 2.37, ddd (13.9, 9.9, 5.5) | H-5 | C-2, 3, 5, 6 | H-4α, H-3 | ||
| 5 | 69.3, CH | 2.92,m | 57.6, CH | 3.41, m | H-4β, 4α, 6 | C-4, 6, 7 | |
| 6 | 30.1, CH2 | 2.25, m | 33.6, CH2 | 1.73, m | H-5 | C-4, 5, 7 | H-4α |
| 1.95, m | 1.64, m | H-5 | C-4, 5, 7 | ||||
| 7 | 26.5, CH2 | 1.40, m | 26.1, CH2 | 1.26, brs | |||
| 8 | 26.5, CH2 | 1.22, brs | 28.6, CH2 | 1.26, brs | |||
| 9 | 29.4, CH2 | 1.22, brs | 28.9, CH2 | 1.26, brs | |||
| 10 | 29.2, CH2 | 1.22, brs | 28.8, CH2 | 1.26, brs | |||
| 11 | 29.2, CH2 | 1.22, brs | 28.7, CH2 | 1.26, brs | |||
| 12 | 29.4, CH2 | 1.22, brs | 31.6, CH2 | 1.26, brs | |||
| 13 | 22.7, CH2 | 1.22, brs | 22.1, CH2 | 1.26, brs | |||
| 14 | 14.1, CH3 | 0.88, t (6.5) | 14.0, CH3 | 0.86, t (6.8) | H-13 | C-12, 13 | |
| 1′ | 136.1, C | - | 137.4, C | - | |||
| 2′ | 129.4, CH | 7.31, m | 129.1, CH | 7.31, m | C-1 | H-1a, 1b, H2 | |
| 3′ | 128.9, CH | 7.35, m | 128.5, CH | 7.34, m | C-1 | ||
| 4′ | 127.2, CH | 7.25, m | 126.6, CH | 7.26, m | C-2′, 6′ | ||
| 5′ | 128.9, CH | 7.31, m | 128.5, CH | 7.34, m | C-1 | ||
| 6′ | 129.4, CH | 7.35, m | 129.1, CH | 7.31, m | C-1 | H-1a, 1b, H2 | |
| N-CH3 | 36.7, CH3 | 2.81, s | - | - | |||
| OH-3 | - | 5.12, d (11.7) | - | 5.62, d (4.0) | H-3 | C-2, 3, 4 | H-1a, 3, 4β |
| Bacterial Strain | 5a-Van | 5a-Ox | ||
|---|---|---|---|---|
| ΣFIC | Activity | ΣFIC | Activity | |
| E. faecalis B3/101 (VRE) | 0.4 | S | - | - |
| S. aureus 66/1 (MRSA) | - | - | 0.2 | S |
| Compound | µg/mL of 5a + Colistin | |||||||
|---|---|---|---|---|---|---|---|---|
| 5a | 0 | 1 | 2 | 4 | 8 | 16 | 32 | 64 |
| Colistin (MIC) | 8 R | 8 R | 8 R | 4 R | 1 S | 0.016 S | <0.008 S | <0.008 S |

| IC50 (95% CI) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compounds (µM) | HepG2 | HT29 | HCT116 | A549 | A375 | MCF7 | U251 | T98G |
| 2b | 56.3 | 34.8 | 60.8 | 84.1 | 82.8 | 94.8 | n.d. | n.d. |
| (37.9–83.7) | (22.0–55.0) | (41.6–88.8) | (60.3–117.1) | (50.5–123.8) | (63.2–142.3) | |||
| 2c | 123.8 | 113.7 | 72.9 | 109.0 | 146.4 | n.d. | n.d. | n.d. |
| (86.4–177.5) | (78.9–163.8) | (56.0–94.9) | (66.2–179.5) | (102.3–209.4) | ||||
| 2e | 118.9 | 111.6 | 73.2 | 105.8 | 123.1 | 186.8 | 212.5 | n.d. |
| (88.9–159.0) | (85.9–145.0) | (59.3–90.4) | (72.8–153.7) | (91.9–164.9) | (155.1–224.8) | (156.2–289.2) | ||
| 3 | n.d. | 43.2 | 64.1 | 85.2 | 109.8 | 99.7 | 120.2 | n.d. |
| (28.3–65.7) | (42.9–95.7) | (61.9–117.3) | (76.4–157.6) | (70.8–143.0) | (81.2–177.7) | |||
| 5a | 43.2 | 12.3 | 29.8 | 47.9 | 38.5 | 53.6 | 74.1 | 50.4 |
| (30.7–60.8) | (10.1–15.5) | (19.1–46.6) | (33.2–69.0) | (29.8–49.7) | (39.4–72.9) | (54–0–101.6) | (32.0–79.4) | |
| 5b | 153.4 | 57.2 | 124.8 | 215.7 | 148.5 | 128.8 | 128.6 | – |
| (96.2–244.7) | (41.7–78.4) | (94.9–164.1) | (161.4–288.1) | (103.7–212.4) | (94.5–175.5) | (86.7–190.8) | ||
| Dox | 0.12 | 0.63 | 0.29 | 0.24 | 0.05 | 0.36 | 1.11 | 15.4 |
| (0.07–0.22) | (0.26–1.12) | (0.16–0.54) | (0.13–0.08) | (0.03–0.08) | (0.16–0.84) | (0.43–2.85) | (10.4–22.9) | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buttachon, S.; Ramos, A.A.; Inácio, Â.; Dethoup, T.; Gales, L.; Lee, M.; Costa, P.M.; Silva, A.M.S.; Sekeroglu, N.; Rocha, E.; et al. Bis-Indolyl Benzenoids, Hydroxypyrrolidine Derivatives and Other Constituents from Cultures of the Marine Sponge-Associated Fungus Aspergillus candidus KUFA0062. Mar. Drugs 2018, 16, 119. https://doi.org/10.3390/md16040119
Buttachon S, Ramos AA, Inácio Â, Dethoup T, Gales L, Lee M, Costa PM, Silva AMS, Sekeroglu N, Rocha E, et al. Bis-Indolyl Benzenoids, Hydroxypyrrolidine Derivatives and Other Constituents from Cultures of the Marine Sponge-Associated Fungus Aspergillus candidus KUFA0062. Marine Drugs. 2018; 16(4):119. https://doi.org/10.3390/md16040119
Chicago/Turabian StyleButtachon, Suradet, Alice A. Ramos, Ângela Inácio, Tida Dethoup, Luís Gales, Michael Lee, Paulo M. Costa, Artur M. S. Silva, Nazim Sekeroglu, Eduardo Rocha, and et al. 2018. "Bis-Indolyl Benzenoids, Hydroxypyrrolidine Derivatives and Other Constituents from Cultures of the Marine Sponge-Associated Fungus Aspergillus candidus KUFA0062" Marine Drugs 16, no. 4: 119. https://doi.org/10.3390/md16040119
APA StyleButtachon, S., Ramos, A. A., Inácio, Â., Dethoup, T., Gales, L., Lee, M., Costa, P. M., Silva, A. M. S., Sekeroglu, N., Rocha, E., Pinto, M. M. M., Pereira, J. A., & Kijjoa, A. (2018). Bis-Indolyl Benzenoids, Hydroxypyrrolidine Derivatives and Other Constituents from Cultures of the Marine Sponge-Associated Fungus Aspergillus candidus KUFA0062. Marine Drugs, 16(4), 119. https://doi.org/10.3390/md16040119