Unstable Tetramic Acid Derivatives from the Deep-Sea-Derived Fungus Cladosporium sphaerospermum EIODSF 008

Abstract
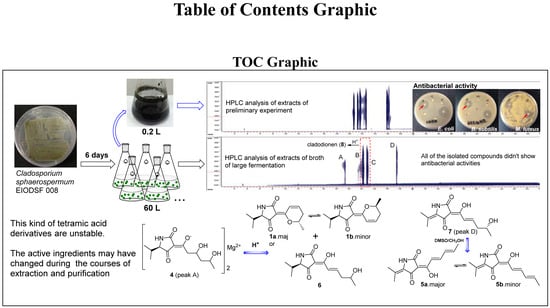
:
1. Introduction
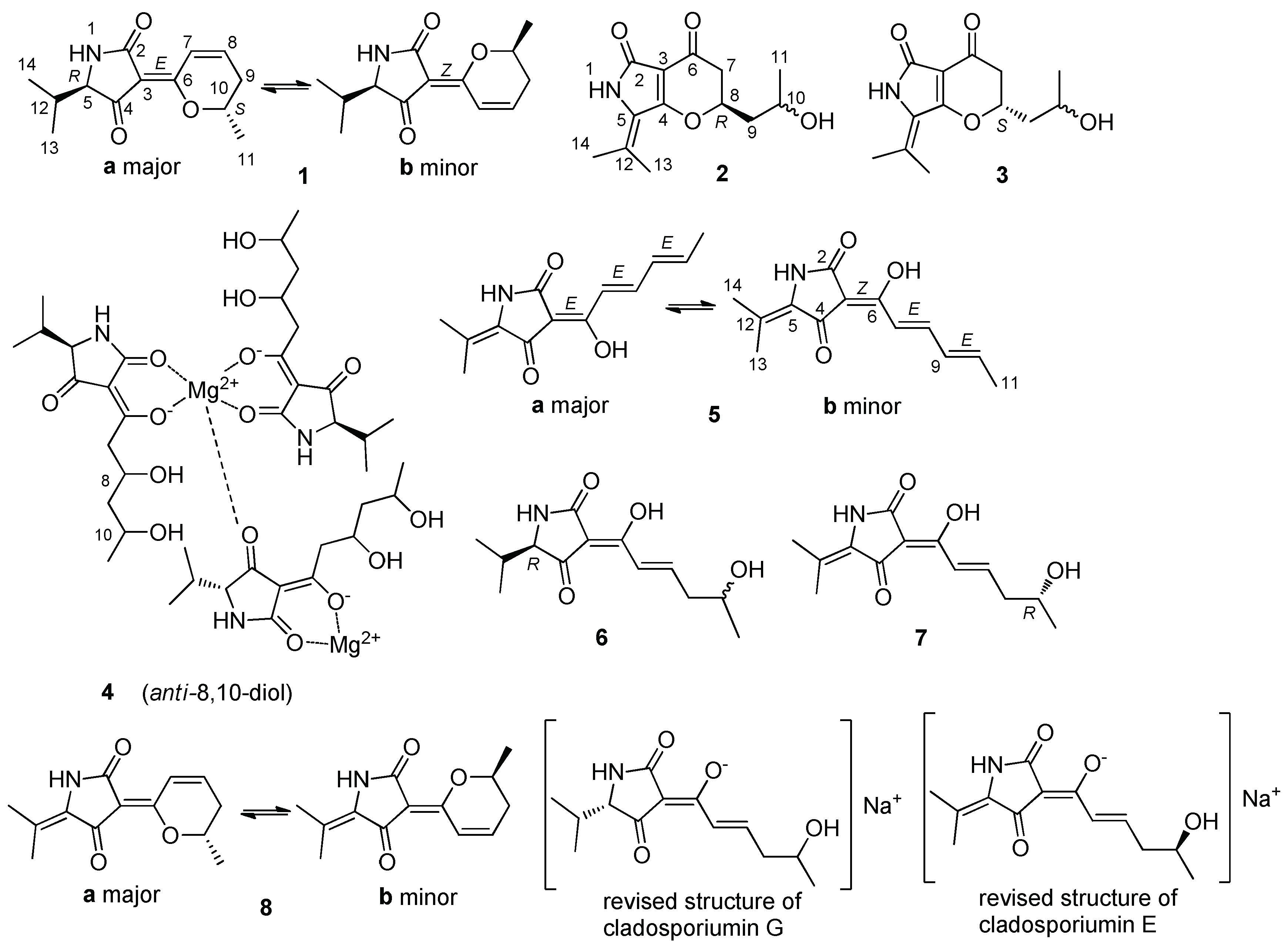
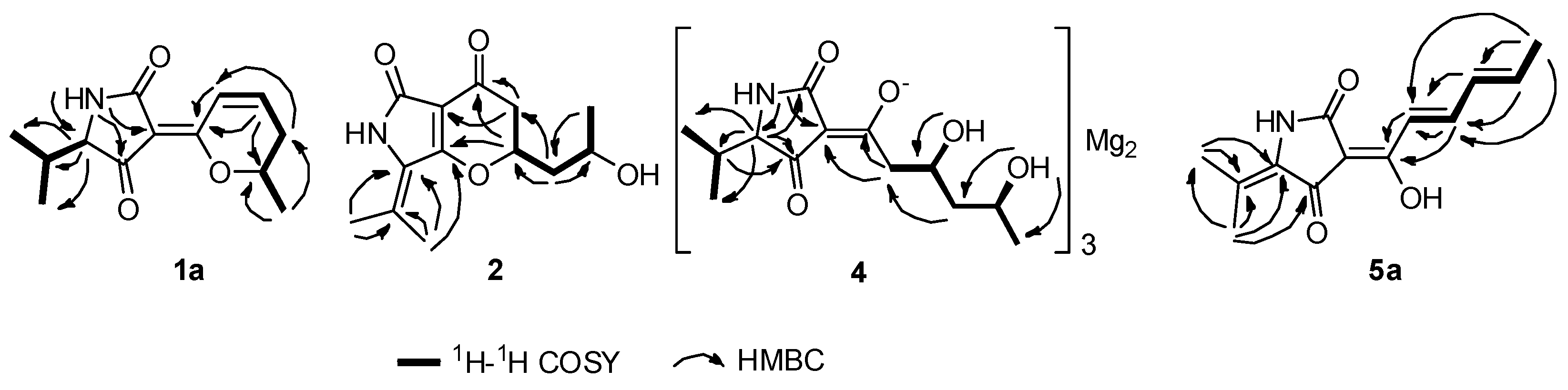
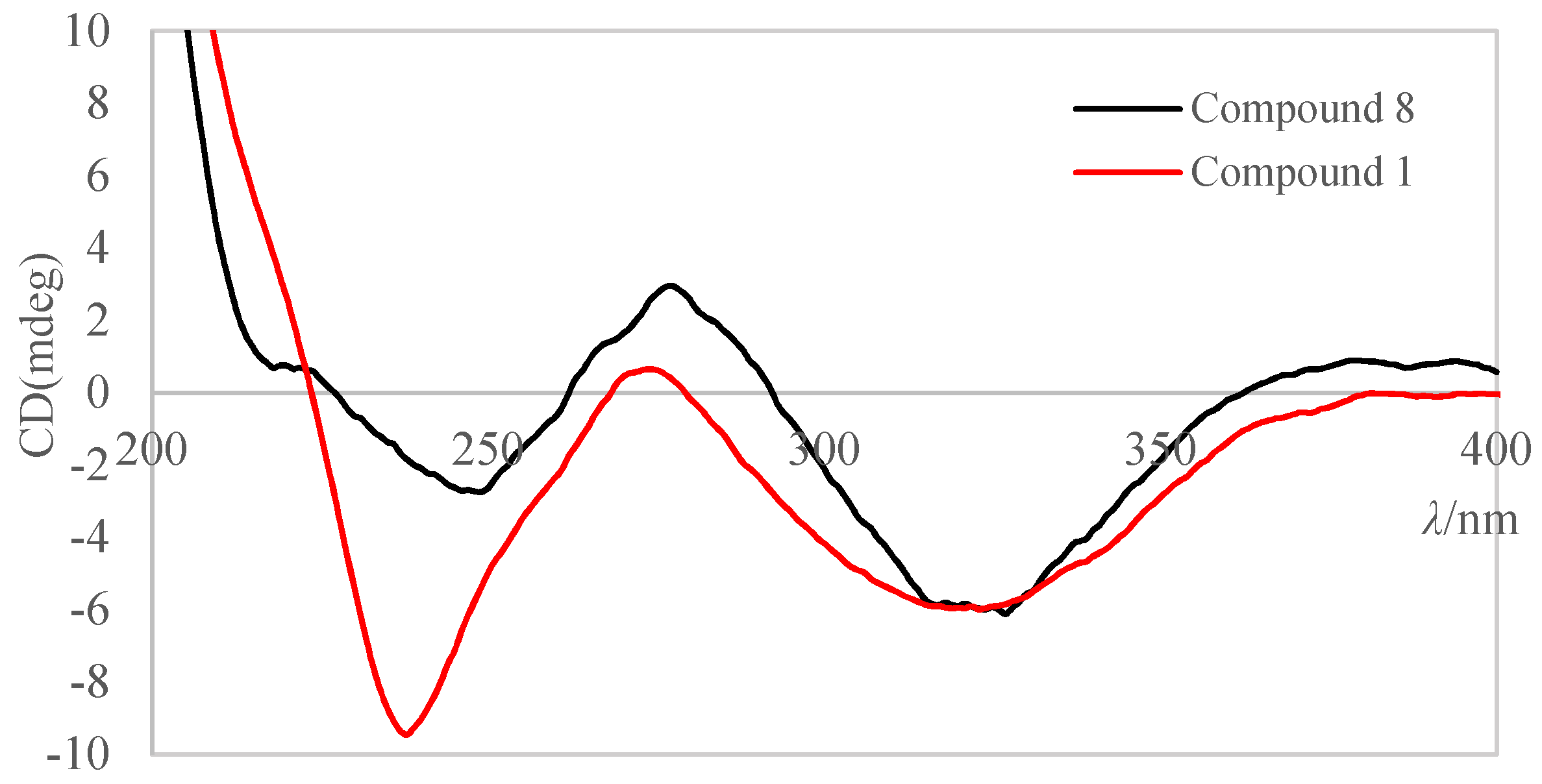
2. Result and Discussion
3. Experimental Section
3.1. General Experimental Procedures
3.2. Fungal Materials
3.3. Fermentation and Extraction
3.4. Isolation and Purification
3.5. Analysis of Magnesium Content in 4
3.6. Determination of Absolute Configuration of the Valine Unit in 4
3.7. Computational Methods to Calculate the ECD Spectra of 2 and 3 and the Equilibrium Populations of 1a and 1b
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Henning, H.G.; Gelbin, A. Advances in heterocyclic chemistry. Adv. Heterocycl. Chem. 1993, 57, 139–185. [Google Scholar]
- Ghisalberti, E.L. Bioactive tetramic acid metabolites. Stud. Nat. Prod. Chem. 2003, 28, 109–163. [Google Scholar]
- Mo, X.; Li, Q.; Ju, J. Naturally occurring tetramic acid products: Isolation, structure elucidation and biological activity. RSC Adv. 2014, 4, 50566–50593. [Google Scholar] [CrossRef]
- Saito, K.; Yamaguchi, T. NMR spectroscopic studies of the tautomerism in tetramic acid analogs and their anilides. III. Polar solvent effects on the tautomeric populations. Bull. Chem. Soc. Jpn. 1978, 51, 651–652. [Google Scholar] [CrossRef]
- Nolte, M.J.; Steyn, P.S.; Wessels, P.L. Structural investigations of 3-acylpyrrolidine-2,4-diones by nuclear magnetic-resonance spectroscopy and X-ray crystallography. J. Chem. Soc. Perkin. Trans. 1980, 1, 1057–1065. [Google Scholar] [CrossRef]
- Steyn, P.S.; Wessels, P.L. Tautomerism in tetramic acids: 13C NMR determination of the structures and ratios of the tautomers in 3-acetyl-5-isopropylpyrrolidine-2,4-dione. Tetrahedron Lett. 1978, 19, 4707–4710. [Google Scholar] [CrossRef]
- Imamura, N.; Adachi, K.; Sano, H. Magnesidin A, a component of marine antibiotic magnesidin, produced by Vibrio gazogenes ATCC29988. J. Antibiot. 1994, 47, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Ohta, E.; Ohta, S.; Ikegami, S. Ancorinoside A Mg salt from the marine sponge, Ancorina sp., which specifically inhibits blastulation of starfish embryos. Tetrahedron 2001, 57, 4699–4703. [Google Scholar] [CrossRef]
- Capon, R.J.; Skene, C.; Lacey, E.; Gill, J.H.; Wadsworth, D.; Friedel, T. Geodin A magnesium salt: A novel nematocide from a southern Australian marine sponge, Geodia. J. Nat. Prod. 1999, 62, 1256–1259. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.H.; Wu, G.W.; Zhu, T.J.; Gu, Q.Q.; Li, D.H. Cladosins F and G, two new hybrid polyketides from the deep-sea-derived Cladosporium sphaerospermum 2005-01-E3. J. Asian Nat. Prod. Res. 2015, 17, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.W.; Sun, X.H.; Yu, G.H.; Wang, W.; Zhu, T.J.; Gu, Q.Q.; Li, D.H. Cladosins A-E, hybrid polyketides from a deep-sea-derived fungus, Cladosporium sphaerospermum. J. Nat. Prod. 2014, 77, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Kong, F.; Wang, Y.; Fu, P.; Zhu, W. Cladodionen, a cytotoxic hybrid polyketide from the marine-derived Cladosporium sp. OUCMDZ-1635. Mar. Drugs 2018, 16, 71. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.H.; Nong, X.H.; Liang, X.; Qi, S.H. New tetramic acid derivatives from the deep-sea-derived fungus Cladosporium sp SCSIO z0025. Tetrahedron 2018, 74, 2620–2626. [Google Scholar] [CrossRef]
- Shu, Y.Z.; Ye, Q.; Kolb, J.M.; Huang, S.; Veitch, J.A.; Lowe, S.E.; Manly, S.P. Bripiodionen, a new inhibitor of human cytomegalovirus protease from Streptomyces sp. WC76599. J. Nat. Prod. 1997, 60, 529–532. [Google Scholar] [CrossRef] [PubMed]
- Kasettrathat, C.; Ngamrojanavanich, N.; Wiyakrutta, S.; Mahidol, C.; Ruchirawat, S.; Kittakoop, P. Cytotoxic and antiplasmodial substances from marine-derived fungi, Nodulisporium sp. and CRI247-01. Phytochemistry 2008, 69, 2621–2626. [Google Scholar] [CrossRef] [PubMed]
- Isaka, M.; Chinthanom, P.; Rachtawee, P.; Somyong, W.; Luangsa-ard, J.J.; Hywel-Jones, N.L. Cordylactam, a new alkaloid from the spider pathogenic fungus Cordyceps sp. BCC 12671. Phytochem. Lett. 2013, 6, 162–164. [Google Scholar] [CrossRef]
- Riko, R.; Nakamura, H.; Shindo, K. Studies on pyranonigrins-isolation of pyranonigrin E and biosynthetic studies on pyranonigrin A. J. Antibiot. 2014, 67, 179–181. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Tsunematsu, Y.; Noguchi, H.; Hotta, K.; Watanabe, K. Elucidation of pyranonigrin biosynthetic pathway reveals a mode of tetramic acid, fused γ-pyrone, and exo-methylene formation. Org. Lett. 2015, 17, 4992–4995. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Kobayashi, Y.; Nakamura, M.; Tamura, O.; Kogen, H. Establishment of relative and absolute configurations of phaeosphaeride A: Total synthesis of ent-phaeosphaeride A. J. Org. Chem. 2015, 80, 1243–1248. [Google Scholar] [CrossRef] [PubMed]
- Maloney, K.N.; Hao, W.; Xu, J.; Gibbons, J.; Hucul, J.; Roll, D.; Brady, S.F.; Schroeder, F.C.; Clardy, J. Phaeosphaeride A, an inhibitor of STAT3-dependent signaling isolated from an endophytic fungus. Org. Lett. 2006, 8, 4067–4070. [Google Scholar] [CrossRef] [PubMed]
- Li, C.S.; Ding, Y.; Yang, B.J.; Miklossy, G.; Yin, H.Q.; Walker, L.A.; Turkson, J.; Cao, S. A new metabolite with a unique 4-pyranone-γ-lactam-1,4-thiazine moiety from a Hawaiian-plant associated fungus. Org. Lett. 2015, 17, 3556–3559. [Google Scholar] [CrossRef] [PubMed]
- Sodeoka, M.; Sampe, R.; Kojima, S.; Baba, Y.; Morisaki, N.; Hashimoto, Y. Asymmetric synthesis of a 3-acyltetronic acid derivative, RK-682, and formation of its calcium salt during silica gel column chromatography. Chem. Pharm. Bull. 2001, 49, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Biersack, B.; Diestel, R.; Jagusch, C.; Sasse, F.; Schobert, R. Metal complexes of natural melophlins and their cytotoxic and antibiotic activities. J. Inorg. Biochem. 2009, 103, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Romano, A.A.; Hahn, T.; Davis, N.; Lowery, C.A.; Struss, A.K.; Janda, K.D.; Bottger, L.H.; Matzanke, B.F.; Carrano, C.J. The Fe(III) and Ga(III) coordination chemistry of 3-(1-hydroxymethylidene) and 3-(1-hydroxydecylidene)-5-(2-hydroxyethyl)pyrrolidine-2,4-dione: novel tetramic acid degradation products of homoserine lactone bacterial quorum sensing molecules. J. Inorg. Biochem. 2012, 107, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Lebrun, M.H.; Duvert, P.; Gaudemer, F.; Gaudemer, A.; Deballon, C.; Boucly, P. Complexation of the fungal metabolite tenuazonic acid with copper (II), iron (III), nickel (II), and magnesium (II) ions. J. Inorg. Biochem. 1985, 24, 167–181. [Google Scholar] [CrossRef]
- Aoki, S.; Higuchi, K.; Ye, Y.; Satari, R.; Kobayashi, M. Melophlins A and B, novel tetramic acids reversing the phenotype of ras-transformed cells, from the marine sponge Melophlus sarassinorum. Tetrahedron 2000, 56, 1833–1836. [Google Scholar] [CrossRef]
- Lin, Z.J.; Lu, Z.Y.; Zhu, T.J.; Fang, Y.C.; Gu, Q.Q.; Zhu, W.M. Penicillenols from Penicillium sp. GQ-7, an endophytic fungus associated with Aegiceras corniculatum. Chem. Pharm. Bull. 2008, 56, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Kikuzaki, H.; Tsai, S.-M.; Nakatani, N. Gingerdiol related compounds from the rhizomes of Zingiber officinale. Phytochemistry 1992, 31, 1783–1786. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Tan, C.H.; Kishi, Y. Toward creation of a universal NMR database for stereochemical assignment: The case of 1,3,5-trisubstituted acyclic systems. Helv. Chim. Acta 2000, 83, 2562–2571. [Google Scholar] [CrossRef]
- Dippenaar, A.; Holzapfed, C.W.; Boeyens, J.C.A. Crystal structure of copper bis(tenuazonate) monohydrate. J. Cryst. Mol. Struct. 1977, 7, 189–197. [Google Scholar] [CrossRef]
- Kohl, H.; Bhat, S.V.; Patell, J.R.; Gandhi, N.M.; Nazareth, J.; Divekar, P.V.; de Souza, N.J.; Berscheid, H.G.; Fehlhaber, H.W. Structure of magnisidin, a new magnesium-containing antibiotic from Pseudomonas magnesiorubra. Tetrahedron Lett. 1974, 15, 983–986. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Tang, G.L.; Xu, X.Y.; Nong, X.H.; Qi, S.H. Insights into deep-sea sediment fungal communities from the East Indian Ocean using targeted environmental sequencing combined with traditional cultivation. PLoS ONE 2014, 9, e109118. [Google Scholar] [CrossRef] [PubMed]
- Fujii, K.; Ikai, Y.; Mayumi, T.; Oka, H.; Suzuki, M.; Harada, K. A nonempirical method using LC/MS for determination of the absolute configuration of constituent amino acids in a peptide: Elucidation of limitations of Marfey’s method and of its separation mechanism. Anal. Chem. 1997, 69, 3346–3352. [Google Scholar] [CrossRef]

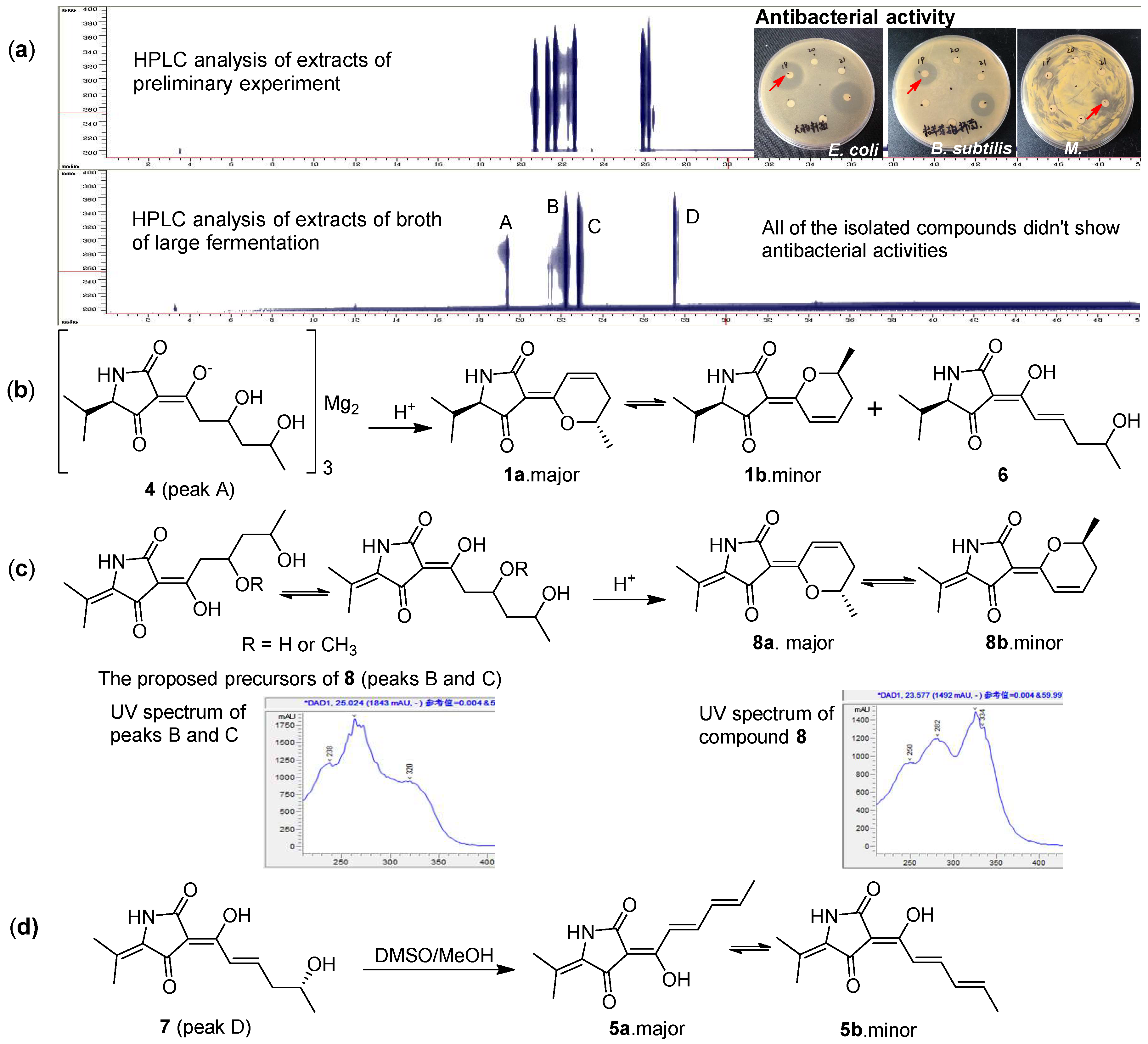

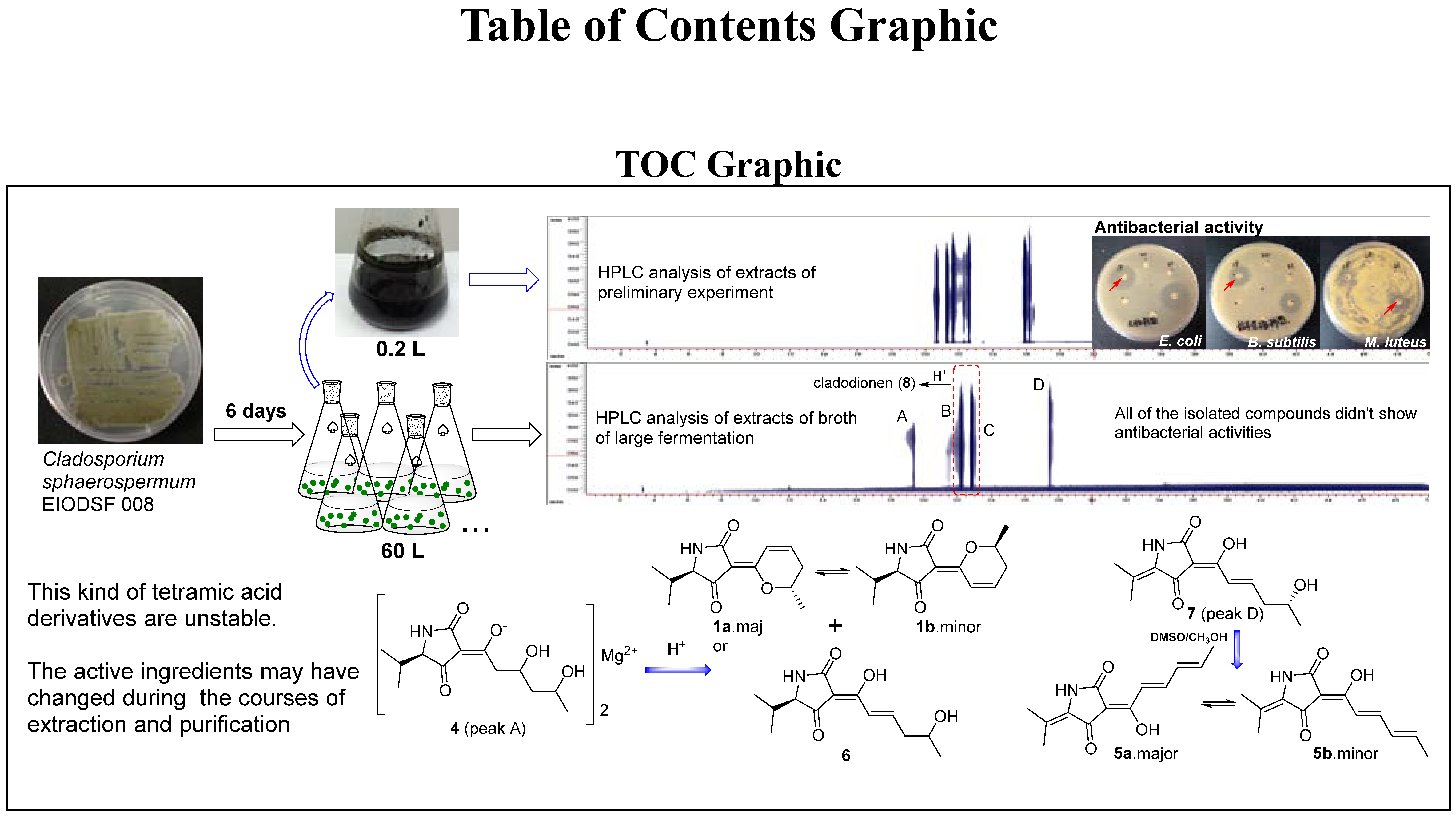{kind=link}
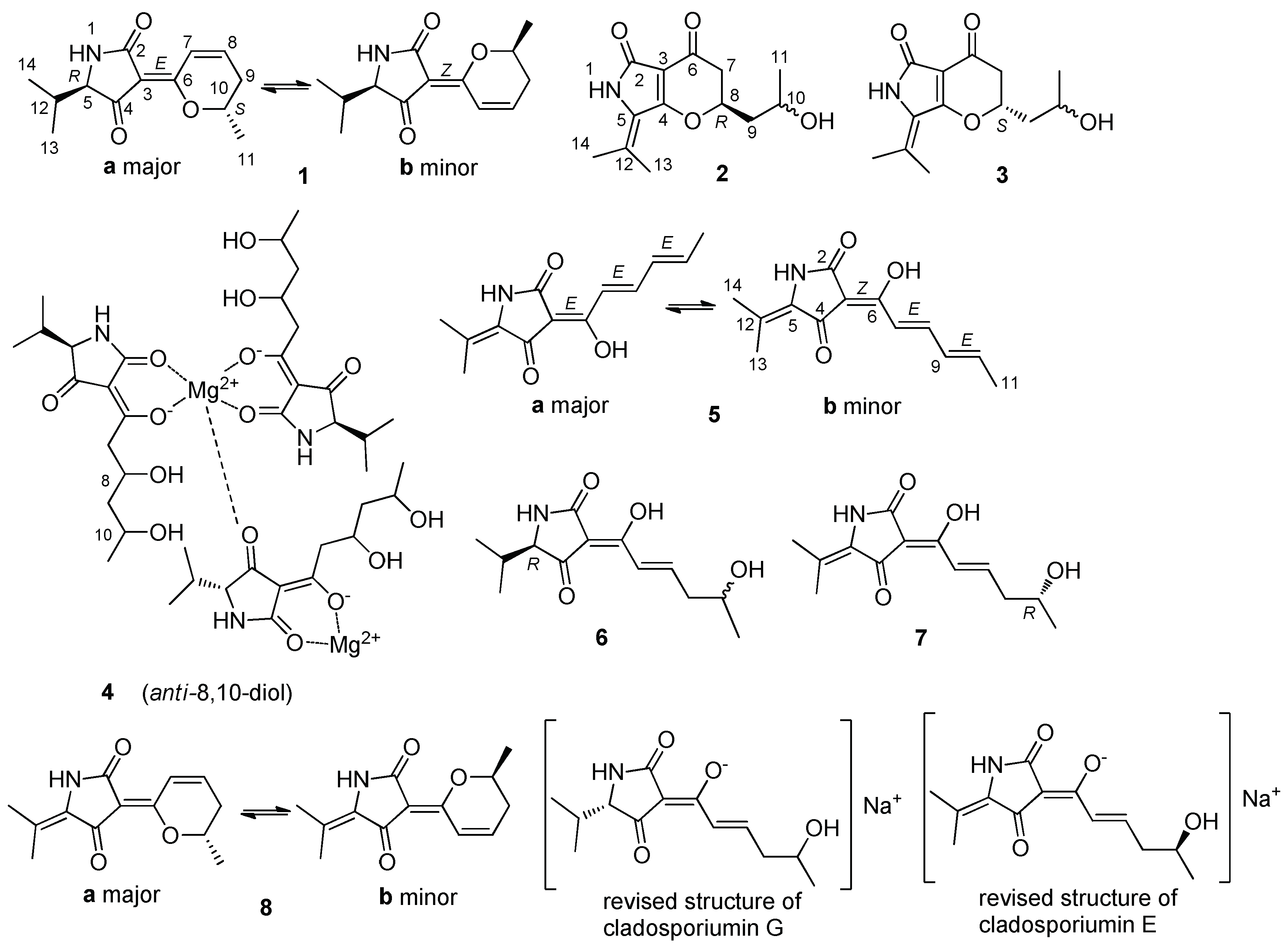{kind=link}
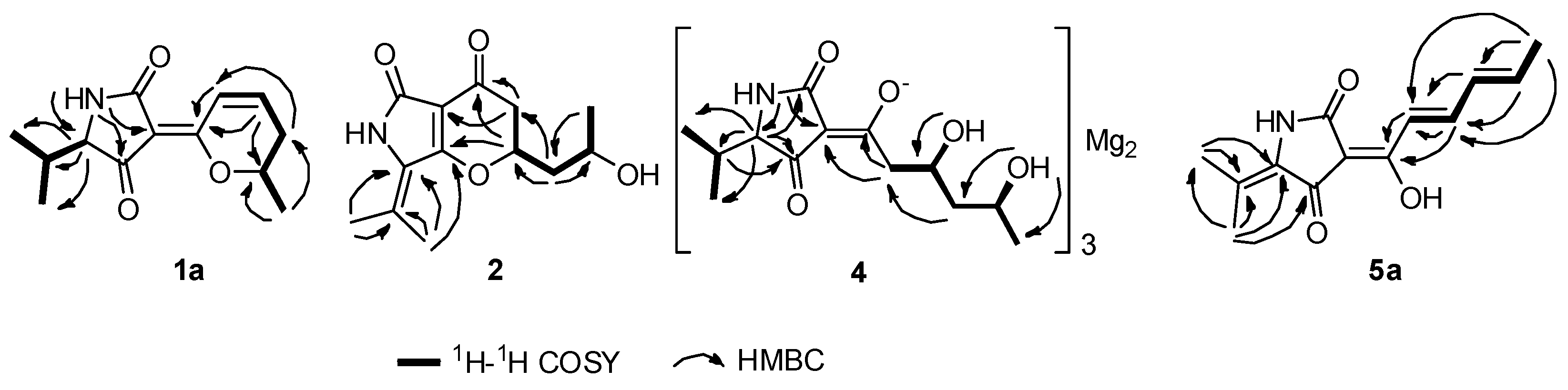{kind=link}
{kind=link}
{kind=link}
{kind=link}
| δH (J in Hz) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | 1 a | 2 a | 3 a | 4 a | 5 b | |||
| Position | a | b | a | b | a | b | ||
| 1-NH | 8.15 (s) | 7.97 (s) | 9.54 (s) | 9.54 (s) | 7.73 (s) | 7.80 (s) | 8.12 (s) | 8.64 (s) |
| 5 | 3.52 (d, 3.0) | 3.58 (d, 3.0) | 3.37 (s) | 3.42 (s) | ||||
| 7 | 7.75 (dd, 10.0, 2.0) | 7.56 (dd, 10.0, 2.0) | 2.44 (dd, 16.5, 3.5) 2.59 (dd, 16.5, 12.5) | 2.47 (dd, 17.0, 3.5) 2.61 (dd, 16.5, 12.5) | 2.81 (d, 6.5) | 2.85 (d, 6.5) | 7.15 (d, 15.5) | 7.23 (d, 15.5) |
| 8 | 6.92 (m) | 6.92 (m) | 4.95 (m) | 4.88 (m) | 4.03 (m) | 4.06 (m) | 7.50 (dd, 15.0, 10.5) | 7.45 (dd, 15.0, 10.0) |
| 9 | 2.34 (m) 2.54 (m) | 2.34 (m) 2.57 (m) | 1.63 (m) 1.91 (m) | 1.76 (m) 2.01 (m) | 1.36 (m) | 1.41 (m) | 6.36 (dd, 15.0, 10.0) | 6.36 (dd, 15.0, 10.0) |
| 10 | 4.39 (m) | 4.39 (m) | 3.88 (m) | 3.82 (m) | 3.80 (br s) | 3.80 (br s) | 6.29 (dq, 14.5, 7.5) | 6.29 (dq, 14.5, 7.5) |
| 11 | 1.37 (d, 6.5) | 1.37 (d, 6.5) | 1.12 (d, 6.5) | 1.13 (d, 6.5) | 1.03 (m) | 1.03 (m) | 1.91 (d, 6.5) | 1.92 (d, 6.0) |
| 12 | 1.98 (m) | 1.98 (m) | 1.99 (m) | 1.99 (m) | ||||
| 13 | 0.91 (d, 7.0) | 0.91 (d, 7.0) | 2.11 (s) | 2.11 (s) | 0.92 (m) | 0.92 (m) | 2.28 (s) | 2.28 (s) |
| 14 | 0.69 (d, 7.0) | 0.69 (d, 7.0) | 1.91 (s) | 1.91 (s) | 0.68 (m) | 0.68 (m) | 1.90 (s) | 1.88 (s) |
| 10-OH | 4.73 (d, 5.0) | 4.69 (br s) | 4.30 (br s) | 4.34 (br s) | ||||
| 8-OH | 4.30 (br s) | 4.30 (br s) | ||||||
| 6-OH | N.D. c | N.D. c | ||||||
| δC, Type | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | 1 a | 2 a | 3 a | 4 a | 5 b | |||
| Position | a | b | a | b | a | b | ||
| 2 | 169.6, C | 167.9, C | 163.5, C | 163.4, C | 177.6, C | 177.7, C | 166.0, C | 172.1, C |
| 3 | 102.9, C | 103.0, C | 105.6, C | 105.6, C | 102.1, C | 102.1, C | 101.6, C | 103.5, C |
| 4 | 196.4, C | 199.6, C | 173.0, C | 173.1, C | 195.5, C | 195.5, C | 188.6, C | 182.4, C |
| 5 | 64.4, CH | 65.0, CH | 127.7, C | 127.8, C | 65.3, CH | 65.4, CH | 129.7, C | 128.4, C |
| 6 | 165.1, C | 166.7, C | 185.9, C | 185.8, C | 192.7, C | 192.7, C | 177.5, C | 174.2, C |
| 7 | 119.5, CH | 119.8, CH | 41.6, CH2 | 41.1, CH2 | 46.1, CH2 | 46.1, CH2 | 119.7, CH | 119.4, CH |
| 8 | 143.2, CH | 143.7, CH | 80.5, CH | 81.1, CH | 66.2, CH | 66.0, CH | 145.6, CH | 145.3, CH |
| 9 | 30.4, CH2 | 30.3, CH2 | 43.1, CH2 | 42.9, CH2 | 46.6, CH2 | 46.7, CH2 | 131.2, CH | 131.2, CH |
| 10 | 73.3, CH | 73.2, CH | 61.8, CH | 62.4, CH | 63.4, CH | 63.3, CH | 142.7, CH | 142.4, CH |
| 11 | 20.0, CH3 | 19.0, CH3 | 24.2, CH3 | 23.3, CH3 | 24.9, CH3 | 25.0, CH3 | 19.1, CH3 | 19.1, CH3 |
| 12 | 29.8, CH | 30.0, CH | 125.7, C | 125.6, C | 29.9, CH | 29.9, CH | 125.7, C | 124.7, C |
| 13 | 19.0, CH3 | 20.0, CH3 | 19.7, CH3 | 19.6, CH3 | 20.0, CH3 | 20.0, CH3 | 19.0, CH3 | 18.4, CH3 |
| 14 | 15.8, CH3 | 15.6, CH3 | 21.6, CH3 | 21.6, CH3 | 15.9, CH3 | 15.9, CH3 | 21.1, CH3 | 21.1, CH3 |
| Compound | 6 | Cladosporiumin G | 7 | Cladosporiumin E | ||||
|---|---|---|---|---|---|---|---|---|
| Position | δC, Type | δH (J in Hz) | δC | δH (J in Hz) | δC, Type | δH (J in Hz) | δC | δH (J in Hz) |
| 1-NH | 8.89 (br s) | 6.68 (s) | 10.06 (br s) | 8.34 (s) | ||||
| 2 | 175.6, C | 177.2 | N.D. a | 172.5 | ||||
| 3 | 100.0, C | 102.2 | 102.1, C | 102.7 | ||||
| 4 | 195.4, C | 196.5 | N.D. a | 184.4 | ||||
| 5 | 66.9, CH | 3.77 (br s) | 64.4 | 3.24 (d, 1.7) | 129.9, C | 131.7 | ||
| 6 | 173.2, C | 181.7 | N.D. a | 181.8 | ||||
| 7 | 122.8, CH | 7.04 (d, 16.0) | 132.4 | 7.57 (d, 15.5) | 123.6, CH | 7.19 (d, 15.5) | 132.5 | 7.59 (d, 15.5) |
| 8 | 147.6, CH | 7.12 (m) | 137.0 | 6.61 (m) | 147.3, CH | 7.12 (m) | 137.1 | 6.61 (m) |
| 9 | 42.9, CH2 | 2.36 (m) | 42.5 | 2.18 (m) 2.25 (m) | 43.0, CH2 | 2.36 (m) | 42.5 | 2.18 (m) 2.26 (m) |
| 10 | 65.7, CH | 3.81 (m) | 66.3 | 3.69 (m) | 65.7, CH | 3.88 (m) | 66.3 | 3.70 (m) |
| 11 | 23.9, CH3 | 1.09 (d, 6.0) | 23.7 | 1.05 (d, 6.1) | 23.9, CH3 | 1.09 (d, 6.0) | 23.7 | 1.05 (d, 6.1) |
| 12 | 30.2, CH | 2.06 (m) | 29.8 | 1.98 (m) | 124.4, C | 113.0 | ||
| 13 | 19.5, CH3 | 0.96 (d, 7.0) | 20.3 | 0.92 (d, 7.0) | 18.8, CH3 | 2.16 (s) | 18.2 | 2.14 (s) |
| 14 | 16.2, CH3 | 0.74 (d, 6.5) | 15.9 | 0.66 (d, 6.7) | 21.5, CH3 | 1.82 (s) | 21.3 | 1.68 (s) |
| 10-OH | N.D. a | 4.62 (s) | N.D. a | 4.59 (s) | ||||
| 6-OH | N.D. a | N.D. a | N.D. a | |||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, X.; Huang, Z.-H.; Ma, X.; Qi, S.-H. Unstable Tetramic Acid Derivatives from the Deep-Sea-Derived Fungus Cladosporium sphaerospermum EIODSF 008. Mar. Drugs 2018, 16, 448. https://doi.org/10.3390/md16110448
Liang X, Huang Z-H, Ma X, Qi S-H. Unstable Tetramic Acid Derivatives from the Deep-Sea-Derived Fungus Cladosporium sphaerospermum EIODSF 008. Marine Drugs. 2018; 16(11):448. https://doi.org/10.3390/md16110448
Chicago/Turabian StyleLiang, Xiao, Zhong-Hui Huang, Xuan Ma, and Shu-Hua Qi. 2018. "Unstable Tetramic Acid Derivatives from the Deep-Sea-Derived Fungus Cladosporium sphaerospermum EIODSF 008" Marine Drugs 16, no. 11: 448. https://doi.org/10.3390/md16110448
APA StyleLiang, X., Huang, Z.-H., Ma, X., & Qi, S.-H. (2018). Unstable Tetramic Acid Derivatives from the Deep-Sea-Derived Fungus Cladosporium sphaerospermum EIODSF 008. Marine Drugs, 16(11), 448. https://doi.org/10.3390/md16110448