Synthesis of Polysubstituted Tetrahydropyrans by Stereoselective Hydroalkoxylation of Silyl Alkenols: En Route to Tetrahydropyranyl Marine Analogues



Abstract
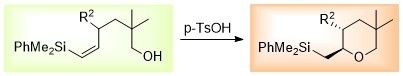
:
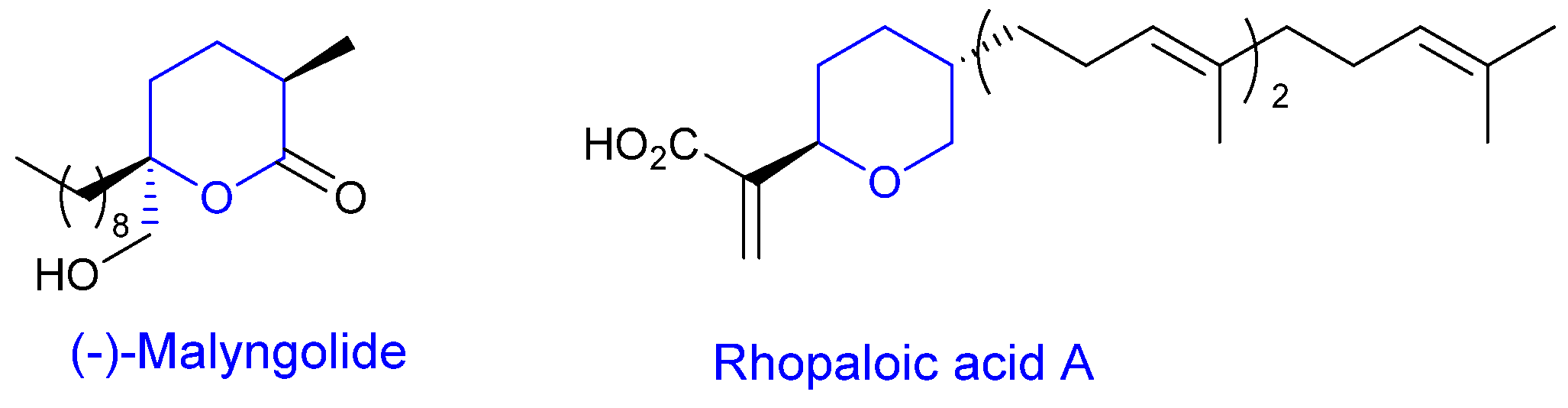
1. Introduction
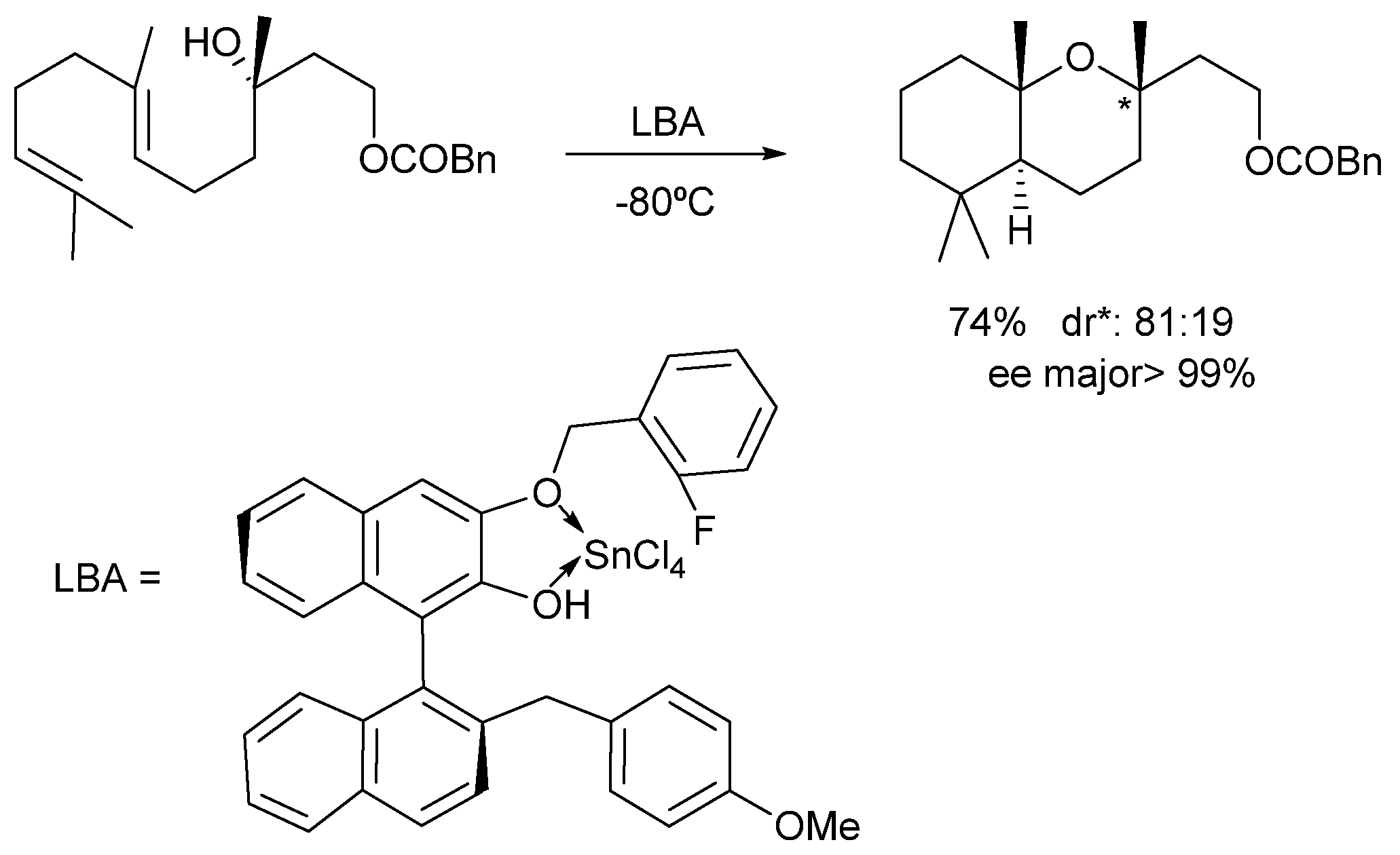
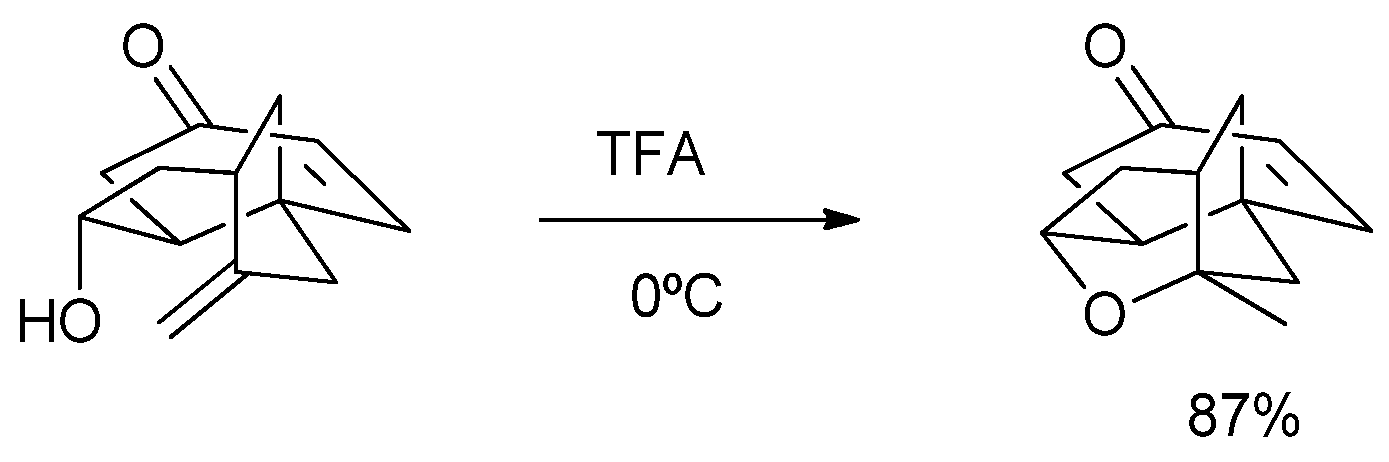
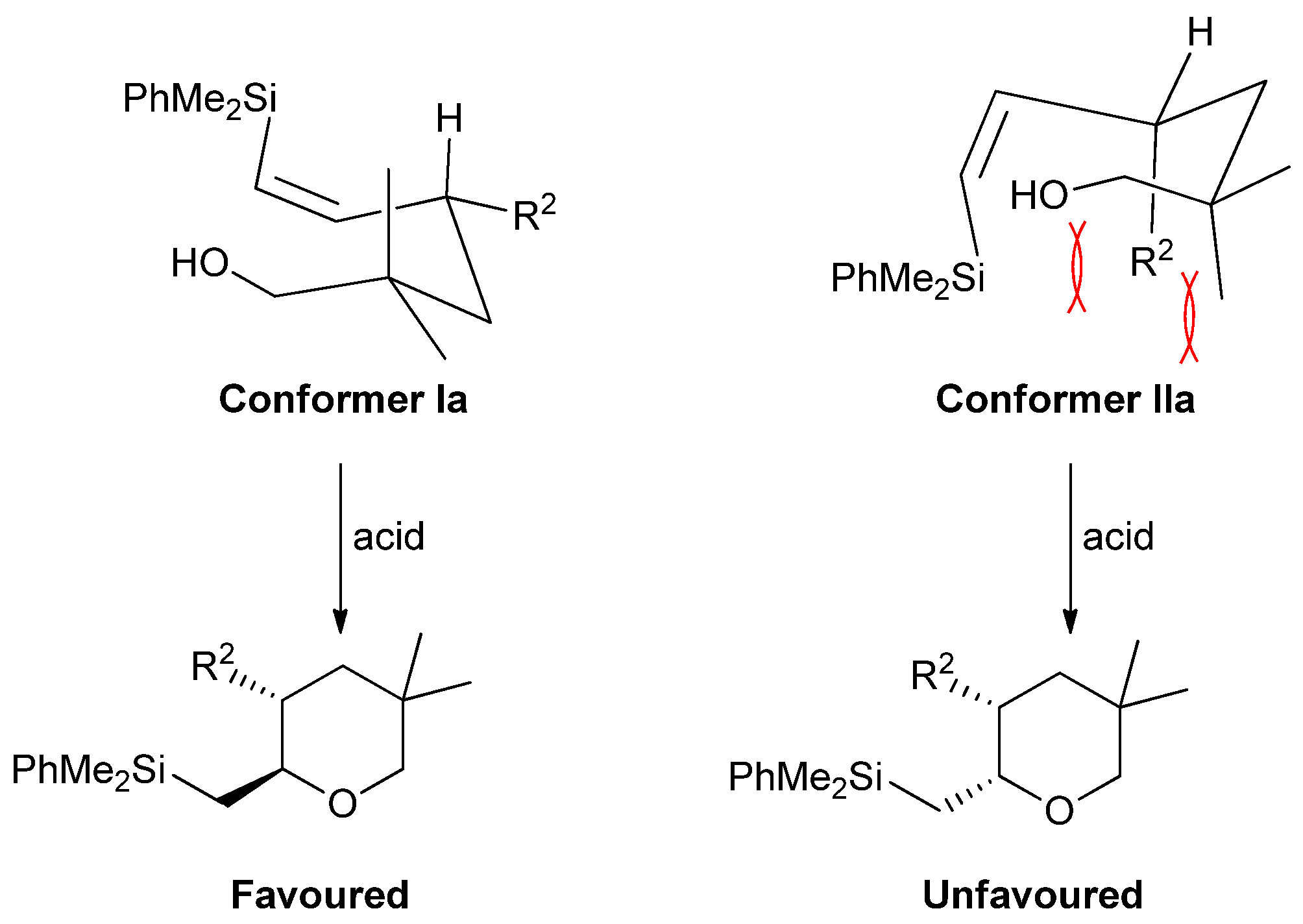
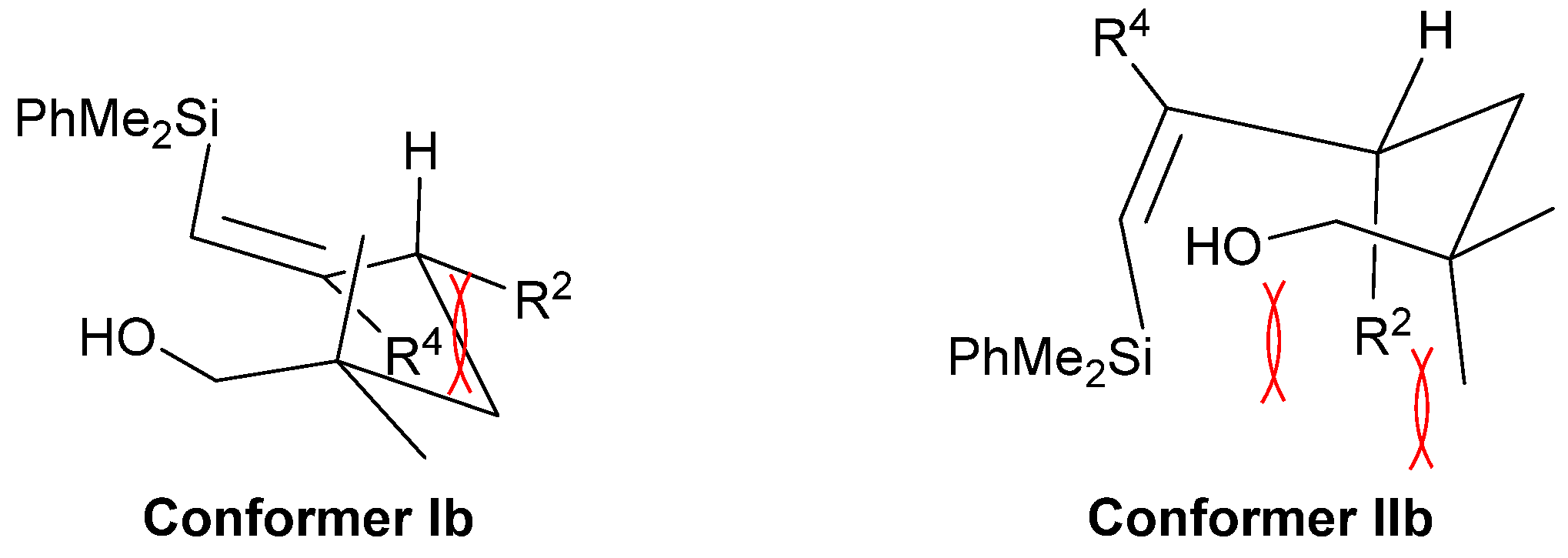
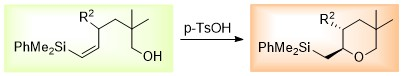
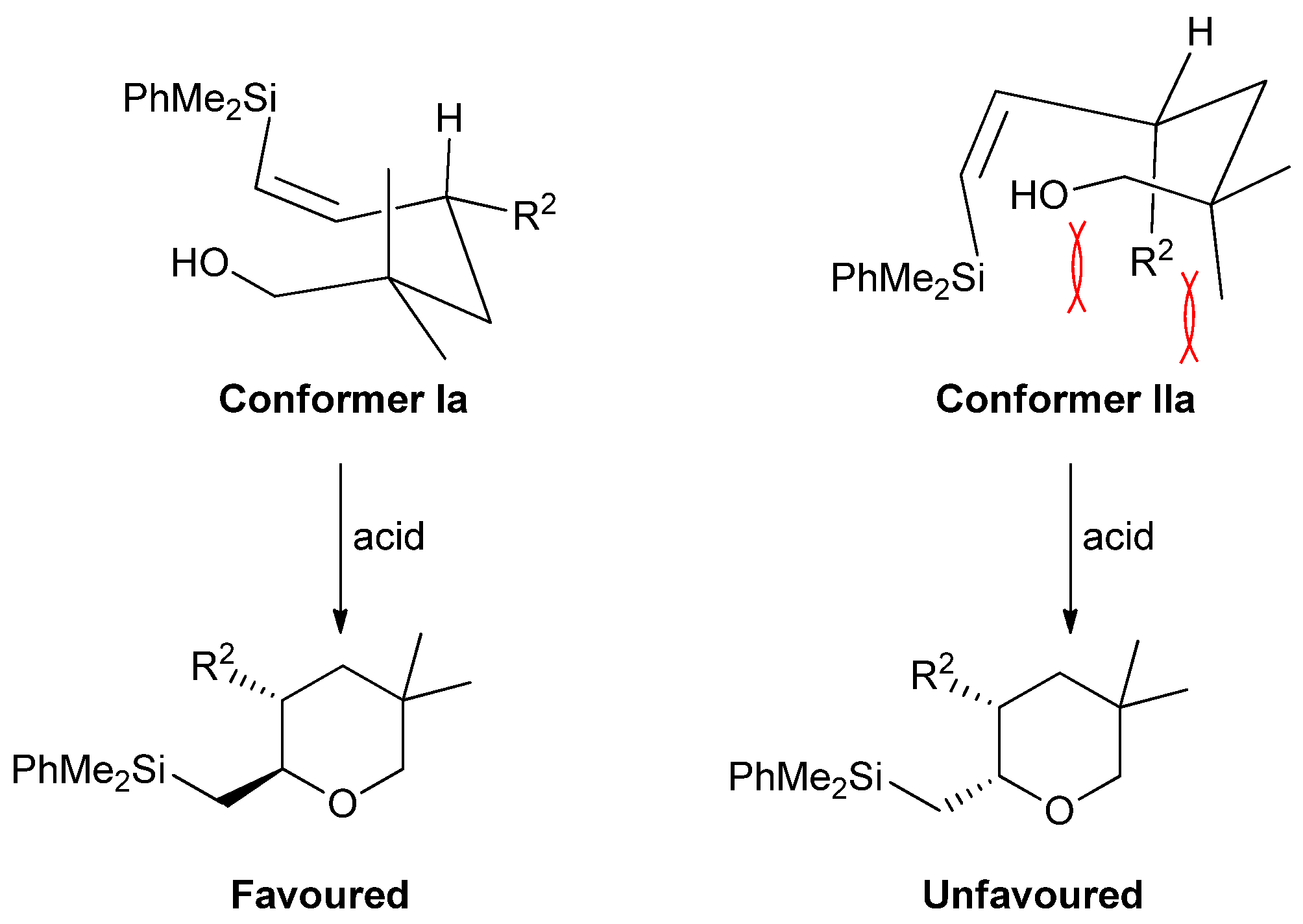
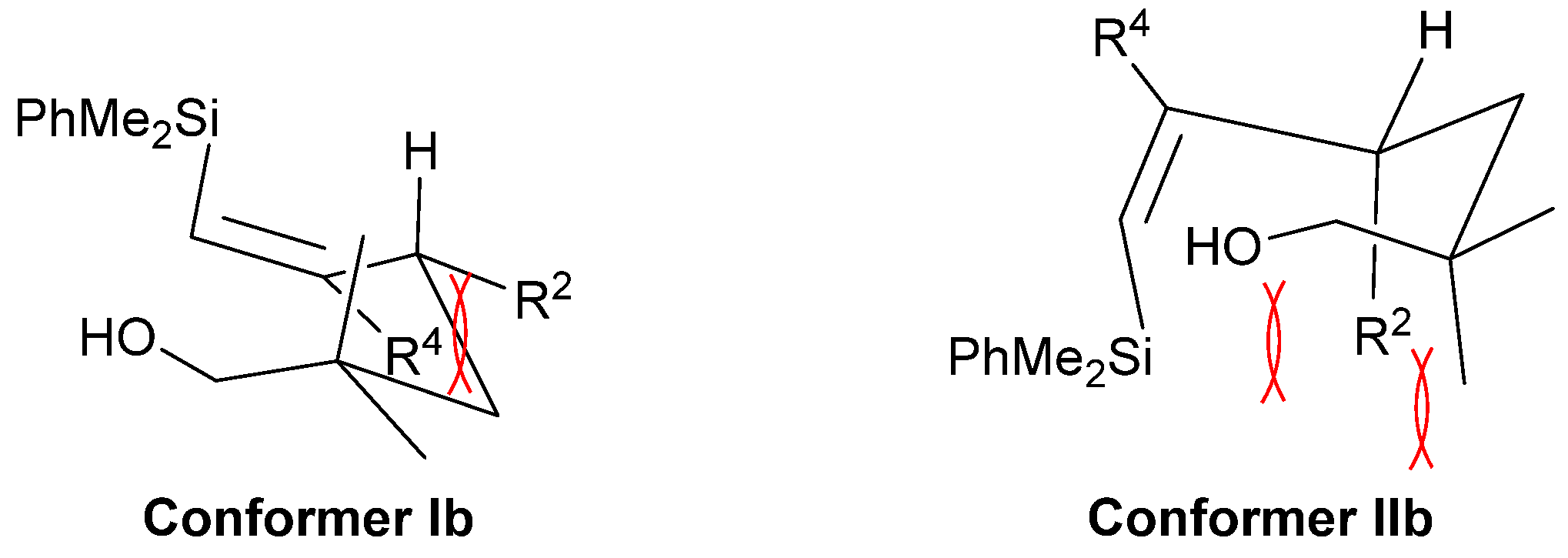
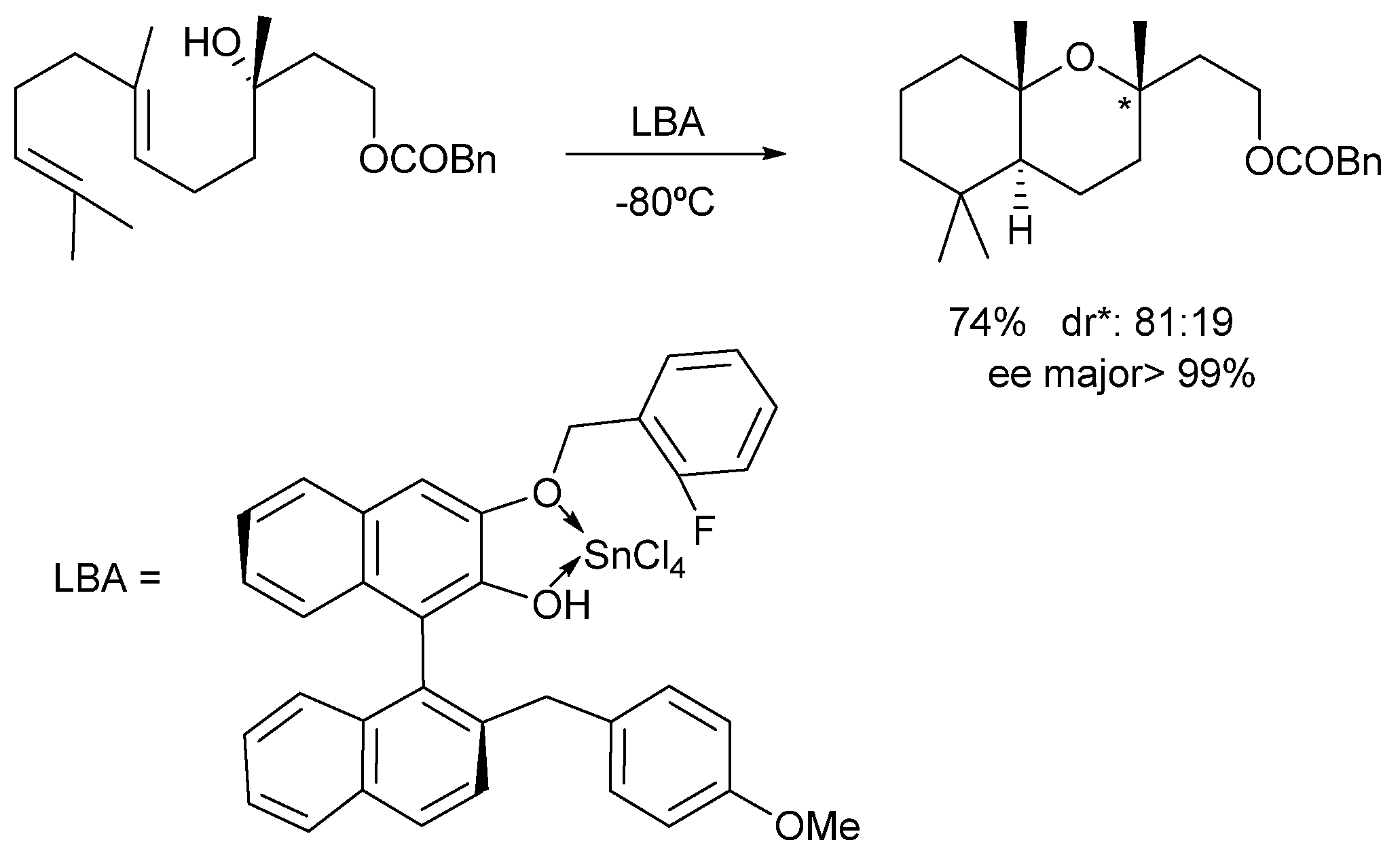
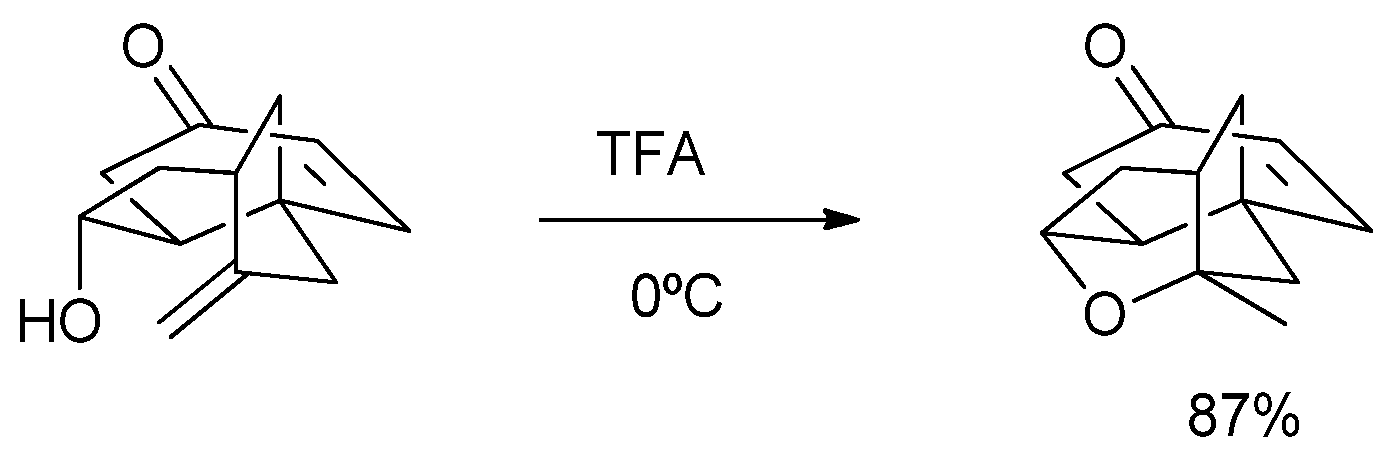
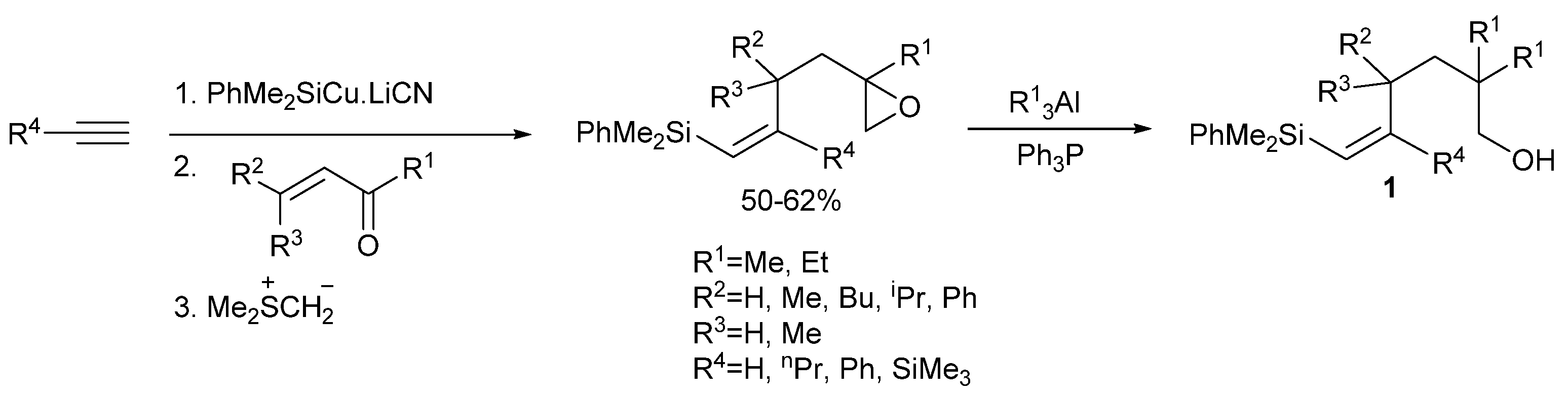
2. Results
3. Materials and Methods
3.1. General Procedures for the Acid-catalysed Cyclization of Vinylsilyl Alcohols
3.2. Trans-5,5-Dimethyl-2-Dimethylphenylsilylmethyl-3-Phenyl-Tetrahydropyran (2a)
3.3. 5,5-Dimethyl-2-Dimethylphenylsilylmethyl-Tetrahydropyran (2b)
3.4. 3,3,5,5-Tetramethyl-2-Dimethylphenylsilylmethyl-Tetrahydropyran (2c)
3.5. Trans-3-Butyl-5,5-Dimethyl-2-Dimethylphenylsilylmethyl-Tetrahydropyran (2d)
3.6. Trans-3-Isopropy-5,5-Dimethyl-2-Dimethylphenylsilylmethyl-Tetrahydropyran (2e)
3.7. 5,5-Dimethyl-2-Dimethylphenylsilylmethyl-3-Phenyl-2-Propyl-Tetrahydropyran (2f)
3.8. 5,5-Diethyl-3-Methyl-2-Dimethylphenylsilylmethyl-2-Propyl-Tetrahydropyran (2g)
3.9. 3,5,5-Trimethyl-2-Dimethylphenylsilylmethyl-2-Phenyl-Tetrahydropyrans 2i and 3i
3.10. 5,5-Dimethyl-2-Dimethylphenylsilylmethyl-2,3-Diphenyl-Tetrahydropyrans 2j and 3j
3.11. 3-Isopropyl-5,5-Dimethyl-2-Dimethylphenylsilylmethyl-2-Phenyl-Tetrahydropyrans 2k and 3k
3.12. 5,5-Diethyl-3-Methyl-2-Dimethylphenylsilylmethyl-2-Phenyl-Tetrahydropyrans 2l and 3l
3.13. Procedure for the Fleming-Tamao Oxidation of Silyl Tetrahydropyran 2d
3.14. Trans-2-Hydroxymethyl-3-Butyl-5,5-Dimethyl-Tetrahydropyran (3d)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cardllina, J.H.; Moore, R.E. Structure and absolute configuration of malyngolide, an antibiotic from the marine blue-green alga Lyngbya majuscula Gomont. J. Org. Chem. 1979, 44, 4039–4042. [Google Scholar] [CrossRef]
- Yanai, M.; Ohta, S.; Ohta, E.; Ikegami, S. Novel norsesterterpenes, which inhibit gastrulation of the starfish embryo, from the marine sponge Rhopaloeides sp. Tetrahedron 1998, 54, 15607–15612. [Google Scholar] [CrossRef]
- Larrosa, I.; Romea, P.; Urpi, F. Synthesis of six-membered oxygenated heterocycles through carbon–oxygen bond-forming reactions. Tetrahedron 2008, 64, 2683–2723. [Google Scholar] [CrossRef]
- Cossy, J. Synthesis of Saturated Oxygenated Heterocycles. 5- and 6-Membered Rings I; Springer: Berlin, Germany, 2014; pp. 43–97. ISBN 13978-3-642-41472-5. [Google Scholar]
- Coulombel, L.; Duñach, E. Triflic acid-catalysed cyclisation of unsaturated alcohols. Green Chem. 2004, 6, 499–501. [Google Scholar] [CrossRef]
- Jeong, Y.; Kim, D.Y.; Choi, Y.; Ryu, J.S. Intramolecular hydroalkoxylation in Brønsted acidic ionic liquids and its application to the synthesis of (±)-centrolobine. Org. Biomol. Chem. 2011, 9, 374–378. [Google Scholar] [CrossRef] [PubMed]
- Linares-Palomino, P.J.; Salido, S.; Altarejo, J.; Sánchez, A. Chlorosulfonic acid as a convenient electrophilic olefin cyclization agent. Tetrahedron Lett. 2003, 44, 6651–6655. [Google Scholar] [CrossRef]
- Coulombel, L.; Favier, I.; Duñach, E. Catalytic formation of C–O bonds by alkene activation: Lewis acid-cycloisomerisation of olefinic alcohols. Chem. Commun. 2005, 2286–2288. [Google Scholar] [CrossRef] [PubMed]
- Dzudza, A.; Marks, T.J. Efficient Intramolecular Hydroalkoxylation of Unactivated Alkenols Mediated by Recyclable Lanthanide Triflate Ionic Liquids: Scope and Mechanism. Chem. Eur. J. 2010, 16, 3403–3422. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Li, G.; Xu, F.; Zhang, Y.; Xue, M.; Shen, Q. Investigation and mechanistic study into intramolecular hydroalkoxylation of unactivated alkenols catalysed by cationic lanthanide complexes. Tetrahedron 2017, 73, 1451–1458. [Google Scholar] [CrossRef]
- Rosenfeld, D.C.; Shekharm, S.; Takemiya, A.; Utsunomiya, M.; Hartwig, J.F. Hydroamination and Hydroalkoxylation Catalyzed by Triflic Acid. Parallels to Reactions Initiated with Metal Triflates. Org. Lett. 2006, 8, 4179–4182. [Google Scholar] [CrossRef] [PubMed]
- Oe, Y.; Ohta, T.; Ito, Y. Ruthenium catalysed addition reaction of carboxylic acid across olefins without β-hydride elimination. Chem. Commun. 2004, 1620–1621. [Google Scholar] [CrossRef] [PubMed]
- Uyanik, M.; Ishihara, K.; Yamamoto, H. Biomimetic synthesis of acid-sensitive (−)- and (+)-caparrapi oxides, (−)- and (+)-8-epicaparrapi oxides and (+)-dysifragin induced by artificial cyclases. Bioorg. Med. Chem. 2005, 13, 5055–5065. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Li, A.; Edmonds, D.J. Total Synthesis of Platensimycin. Angew. Chem. Int. Ed. 2006, 45, 7086–7090. [Google Scholar] [CrossRef] [PubMed]
- Barbero, A.; Blanco, Y.; Pulido, F.J. Silylcuprates from Allene and Their Reaction with α,β-Unsaturatedd Nitriles and Imines. Synthesis of Silylated Oxo Compounds Leading to Cyclopentane and Cycloheptane Ring Formation. J. Org. Chem. 2005, 70, 6876–6883. [Google Scholar] [CrossRef] [PubMed]
- Barbero, A.; Castreño, P.; Pulido, F.J. Spiro-Cyclopropanation from Oxoallylsilanes. J. Am. Chem. Soc. 2005, 127, 8022–8023. [Google Scholar] [CrossRef] [PubMed]
- Barbero, A.; Castreño, P.; Pulido, F.J. Acid-Catalyzed Cyclization of Epoxyallylsilanes. An Unusual Rearrangement Cyclization Process. Org. Lett. 2003, 5, 4045–4048. [Google Scholar] [CrossRef] [PubMed]
- Diez-Varga, A.; Barbero, H.; Pulido, F.J.; González-Ortega, A.; Barbero, A. Competitive Silyl–Prins Cyclization versus Tandem Sakurai–Prins Cyclization: An Interesting Substitution Effect. Chem. Eur. J. 2014, 20, 14112–14119. [Google Scholar] [CrossRef] [PubMed]
- Barbero, A.; Diez-Varga, A.; Herrero, M.; Pulido, F.J. From Silylated Trishomoallylic Alcohols to Dioxaspiroundecanes or Oxocanes: Catalyst and Substitution Influence. J. Org. Chem. 2016, 81, 2704–2712. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Okajima, S.; Hondo, T.; Hosomi, A. Silicon-directed cyclization of vinylsilanes bearing hydroxy group catalysed by an acid. Tetrahedron Lett. 1995, 36, 1483–1486. [Google Scholar] [CrossRef]
- Miura, K.; Okajima, S.; Hondo, T.; Nakagawa, T.; Takahashi, T.; Hosomi, A. Acid-Catalyzed Cyclization of Vinylsilanes Bearing a Hydroxy Group: A New Method for Stereoselective Synthesis of Disubstituted Tetrahydrofurans. J. Am. Chem. Soc. 2000, 122, 11348–11357. [Google Scholar] [CrossRef]
- Pulido, F.J.; Barbero, A.; Val, P.; Diez, A.; González-Ortega, A. Efficiency of Acid- and Mercury-Catalyzed Cyclization Reactions in the Synthesis of Tetrahydrofurans from Allylsilyl Alcohols. Eur. J. Org. Chem. 2012, 5350–5356. [Google Scholar] [CrossRef]
- Barbero, A.; Barbero, H.; González-Ortega, A.; Pulido, F.J.; Val, P.; Diez-Varga, A.; Morán, J.R. Efficient access to polysubstituted tetrahydrofurans by electrophilic cyclization of vinylsilyl alcohols. RSC Adv. 2015, 5, 49541–49551. [Google Scholar] [CrossRef]
- Schneider, C.; Brauner, J. Lewis Base-Catalyzed Addition of Trialkylaluminum Compounds to Epoxides. Eur. J. Org. Chem. 2001, 4445–4450. [Google Scholar] [CrossRef]
- Hatakeyama, S.; Sugawara, K.; Takano, S. Stereocontrolled construction of substituted pyrrolidines based on intramolecular protodesilylation reaction. Enantiospecific synthesis of (–)-kainic acid and (+)-allokainic acid from L-serine. J. Chem. Soc. Chem. Commun. 1993, 2, 125–127. [Google Scholar] [CrossRef]
- Fleming, I. Stereocontrol in Organic Synthesis using silicon compounds. In Frontiers in Natural Product Chemistry; Atta-ur-Rahman, Choudhary, M.I., Kahn, K.M., Eds.; Bentham Scientific Publishers: Karachi, Pakistan, 2005; pp. 55–64. ISBN 978-1-60805-676-7. [Google Scholar]
- Fleming, I.; Barbero, A.; Walter, D. Stereochemical Control in Organic Synthesis Using Silicon-Containing Compounds. Chem. Rev. 1997, 97, 2063–2192. [Google Scholar] [CrossRef] [PubMed]
- Paddon-Row, M.N.; Rondan, N.G.; Houk, K.N. Staggered models for asymmetric induction: Attack trajectories and conformations of allylic bonds from ab initio transition structures of addition reactions. J. Am. Chem. Soc. 1982, 104, 7162–7166. [Google Scholar] [CrossRef]
- Fleming, I.; Henning, R.; Parker, D.C.; Plaut, H.E.; Sanderson, P.E.J. The phenyldimethylsilyl group as a masked hydroxy group. J. Chem. Soc. Perkin Trans. 1 1995, 4, 317–337. [Google Scholar] [CrossRef]
- Maezaki, N.; Matsumori, Y.; Shogaki, T.; Soejima, M.; Ohishi, H.; Tanaka, T.; Iwata, C. Stereoselective Synthesis of a 2,2,5-Trisubstituted Tetrahydropyran Chiron via 1,3- and 1,6-Asymmetric Induction: A Total Synthesis of (−)-Malyngolide. Tetrahedron 1998, 54, 13087–13104. [Google Scholar] [CrossRef]


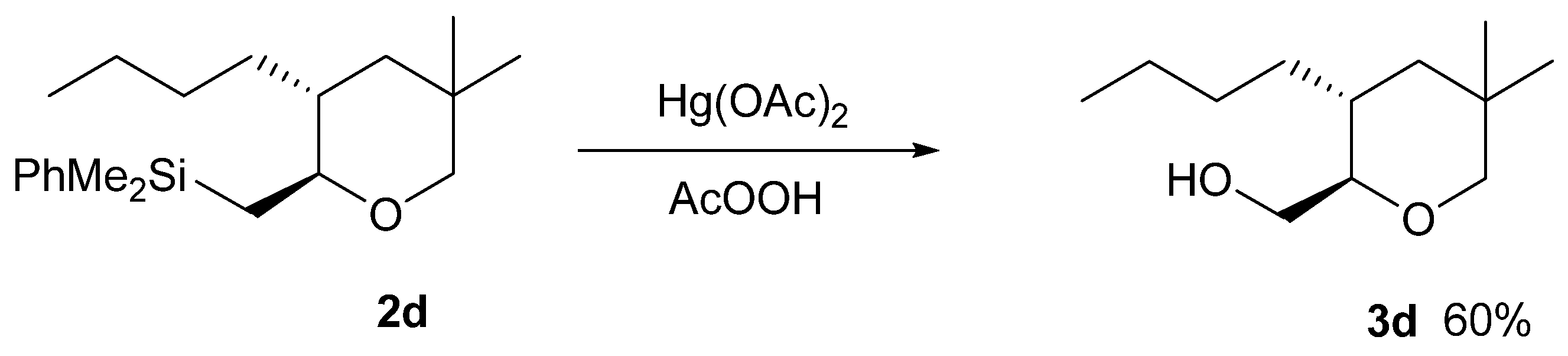
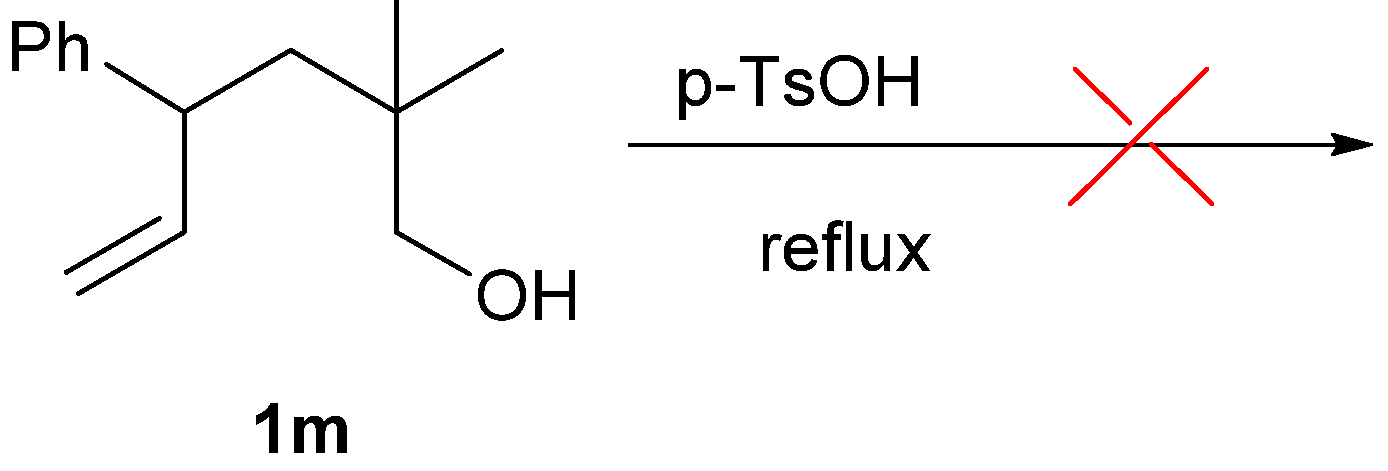

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
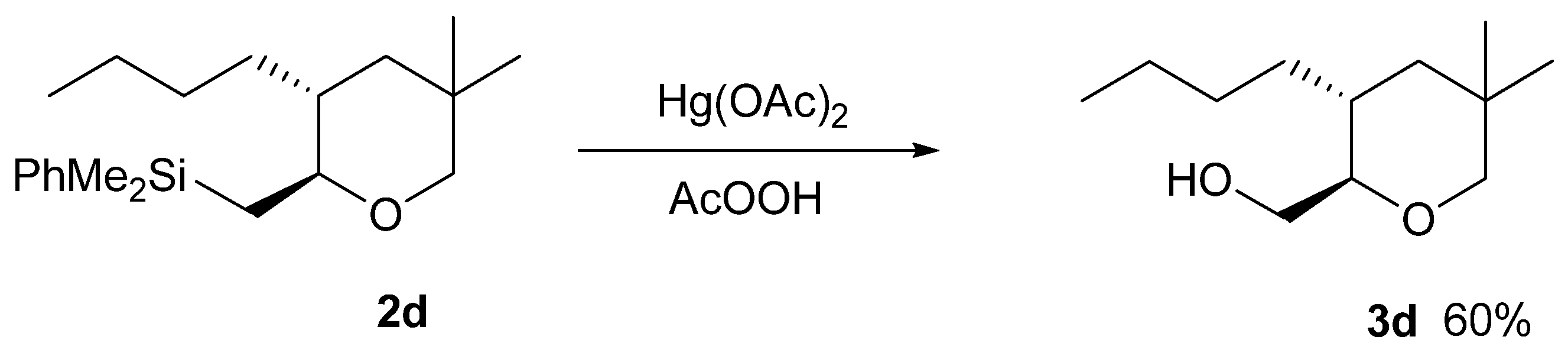{kind=link}
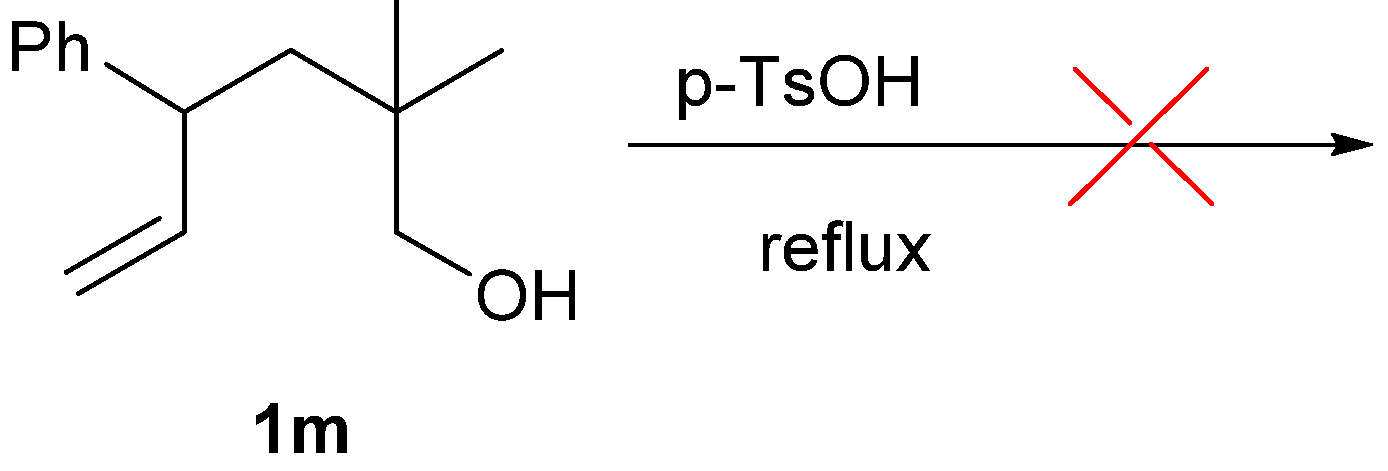{kind=link}

| Entry | Acid 1 | Temperature (°C) | Solvent | Ratio 2a/3a 2 | Product, Yield |
|---|---|---|---|---|---|
| 1 | TMSOTf | −78 | CH2Cl2 | Complex mixture | |
| 2 | TMSOTf | −78 | Et2O | Complex mixture | |
| 3 | TiCl4 | −78 | CH2Cl2 | Complex mixture | |
| 4 | ZnCl2 | −78 | CH2Cl2 | n.r. 3 | |
| 5 | SiO2 | r. t. | AcOEt | n.r. 3 | |
| 6 | BF3·OEt2 | −78 | CH2Cl2 | n.r. 3 | |
| 7 | BF3·OEt2 | 0 | CH2Cl2 | 67:33 | 2a + 3a (69%) |
| 8 | SnCl4 | −78 | CH2Cl2 | 50:50 | 2a + 3a (63%) |
| 9 | CSA | reflux | CH2Cl2 | n.r. 3 | |
| 10 | p-TsOH | reflux | CH2Cl2 | >95:5 | 2a (77%) |

| Entry | R1 | R2 | R3 | R4 | Time (h) 1 | dr2 | Yield (%) 3 |
|---|---|---|---|---|---|---|---|
| 1 | Me | Ph | H | H | 1 | ˃95:5 | 2a (77) |
| 2 | Me | Ph | H | H | 4 | ˃95:5 | 2a (62) 4 |
| 3 | Me | Ph | H | H | 4 | ˃95:5 | 2a (67) 5 |
| 4 | Me | H | H | H | 1 | 2b (71) | |
| 5 | Me | Me | Me | H | 1 | 2c (70) | |
| 6 | Me | Bu | H | H | 1 | >95:5 | 2d (73) |
| 7 | Me | iPr | H | H | 1 | >95:5 | 2e (79) |
| 8 | Me | Ph | H | nPr | 1 | >95:5 | 2f (71) |
| 9 | Et | Me | H | nPr | 1 | >95:5 | 2g (70) |
| 10 | Me | Ph | H | SiMe3 | 3 | n.r. 6 |

| Entry | Acid 1 | Temperature (°C) | Solvent | Time (min) | Product (yield) |
|---|---|---|---|---|---|
| 1 | BF3·OEt2 | 0 | CH2Cl2 | 60 | Desilylated THP 2 (52%) |
| 2 | SnCl4 | −78 | CH2Cl2 | 90 | Desilylated THP (48%) |
| 3 | p-TsOH | reflux | CH2Cl2 | 60 | Desilylated THP (69%) |
| 4 | p-TsOH | r.t. | CH2Cl2 | 30 | 2i + 3i + Desilylated THP (55%) 3 |
| 5 | p-TsOH | 0 | CH2Cl2 | 10 | 2i + 3i (71%) 4 |
| 6 | CSA | r.t. | CH2Cl2 | 30 | 2i + 3i (70%) 4 |
| 7 | CSA | 0 | CH2Cl2 | 60 | 2i + 3i (69%) 4 |
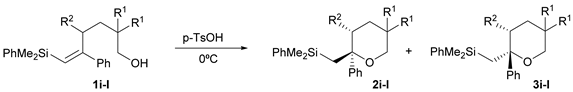
| Entry | R1 | R2 | Time (min) | Product (Ratio 2:3) | Yield (%) 1 |
|---|---|---|---|---|---|
| 1 | Me | Me | 10 | 78:22 | 2i + 3i (71) |
| 2 | Me | Ph | 10 | 79:21 | 2j + 3j (65) |
| 3 | Me | iPr | 10 | 91:9 | 2k + 3k (72) |
| 4 | Et | Me | 10 | 86:14 | 2l + 3l (73) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díez-Poza, C.; Val, P.; Pulido, F.J.; Barbero, A. Synthesis of Polysubstituted Tetrahydropyrans by Stereoselective Hydroalkoxylation of Silyl Alkenols: En Route to Tetrahydropyranyl Marine Analogues. Mar. Drugs 2018, 16, 421. https://doi.org/10.3390/md16110421
Díez-Poza C, Val P, Pulido FJ, Barbero A. Synthesis of Polysubstituted Tetrahydropyrans by Stereoselective Hydroalkoxylation of Silyl Alkenols: En Route to Tetrahydropyranyl Marine Analogues. Marine Drugs. 2018; 16(11):421. https://doi.org/10.3390/md16110421
Chicago/Turabian StyleDíez-Poza, Carlos, Patricia Val, Francisco J. Pulido, and Asunción Barbero. 2018. "Synthesis of Polysubstituted Tetrahydropyrans by Stereoselective Hydroalkoxylation of Silyl Alkenols: En Route to Tetrahydropyranyl Marine Analogues" Marine Drugs 16, no. 11: 421. https://doi.org/10.3390/md16110421
APA StyleDíez-Poza, C., Val, P., Pulido, F. J., & Barbero, A. (2018). Synthesis of Polysubstituted Tetrahydropyrans by Stereoselective Hydroalkoxylation of Silyl Alkenols: En Route to Tetrahydropyranyl Marine Analogues. Marine Drugs, 16(11), 421. https://doi.org/10.3390/md16110421