A General Strategy for the Stereoselective Synthesis of the Furanosesquiterpenes Structurally Related to Pallescensins 1–2


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
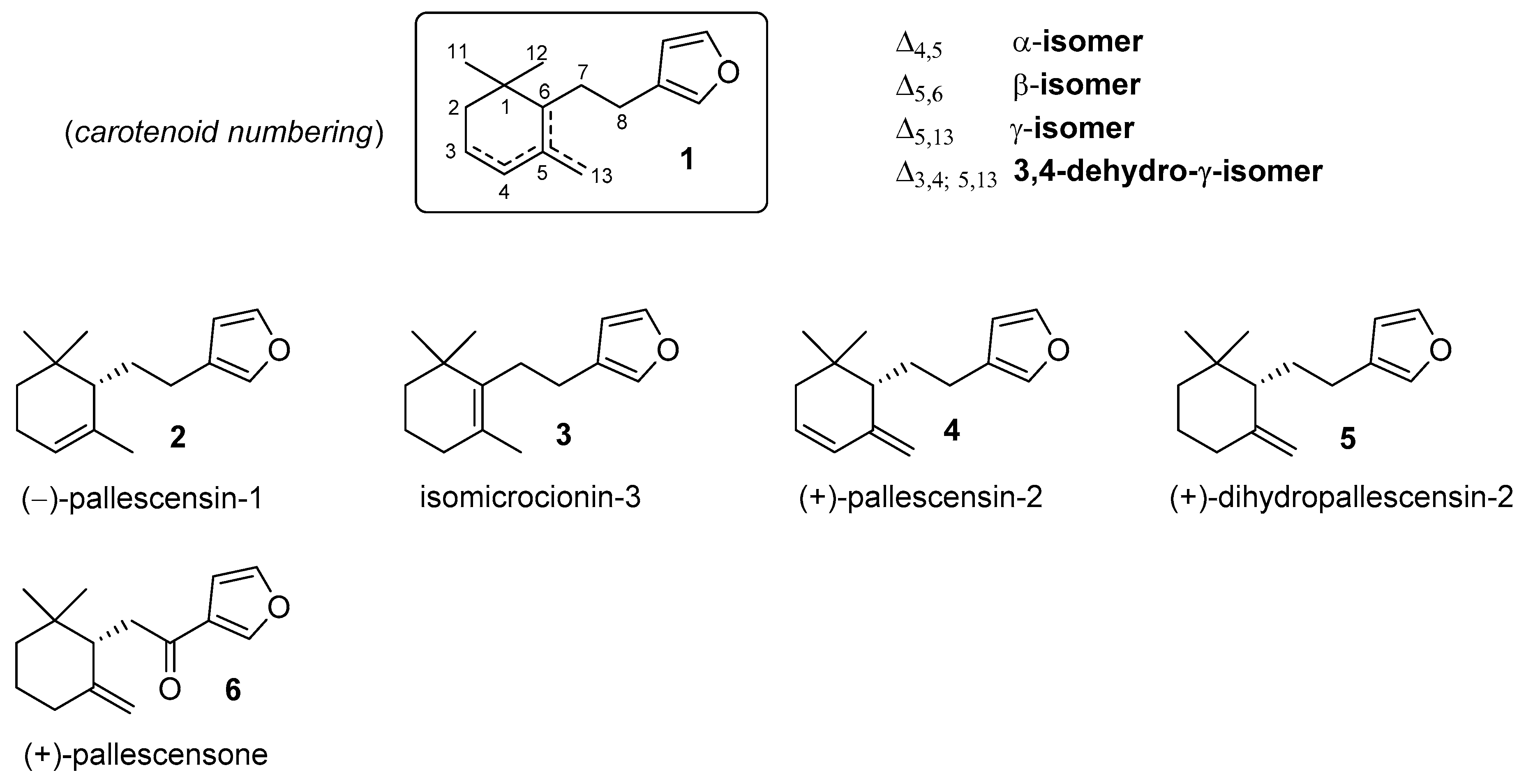
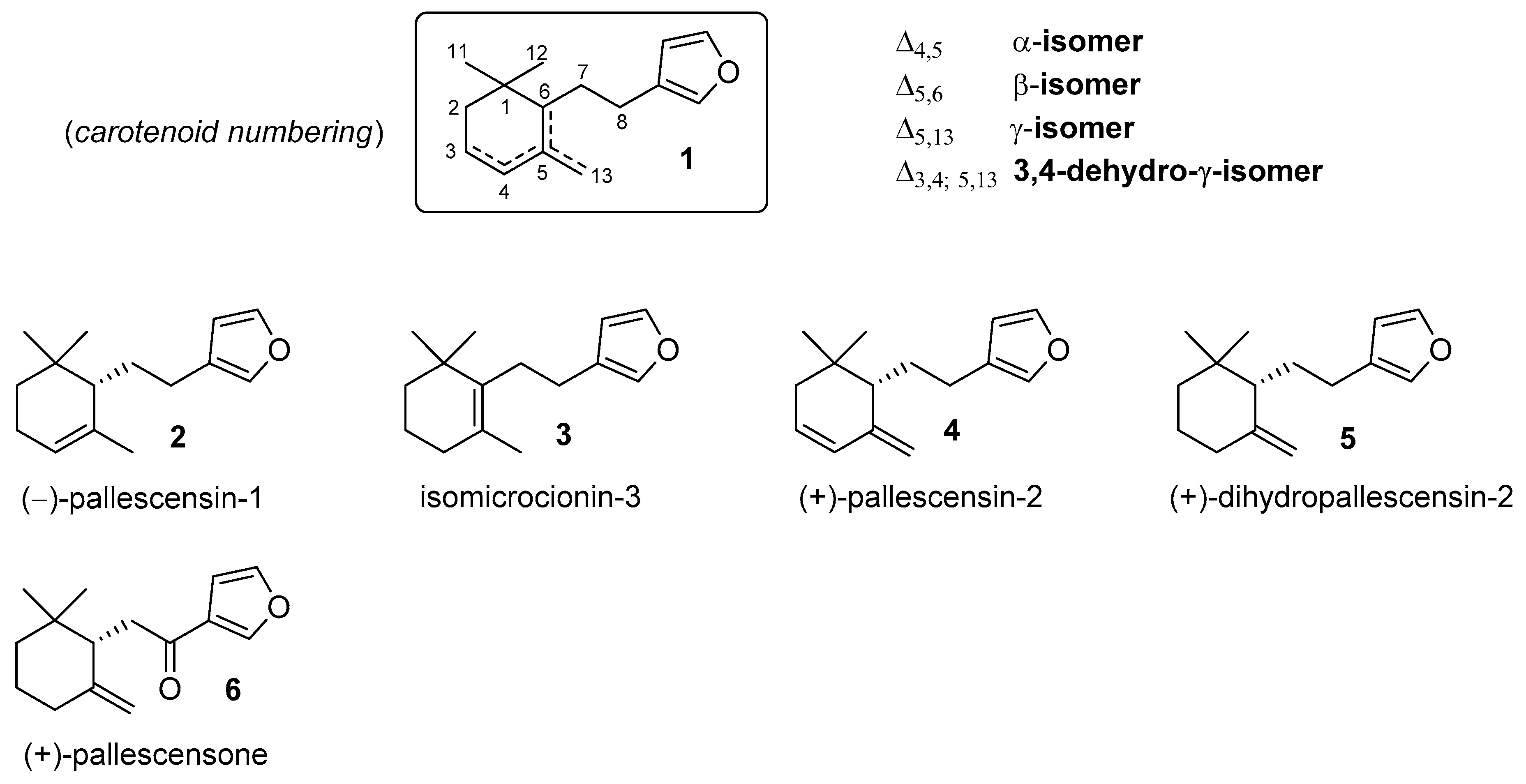
:1. Introduction
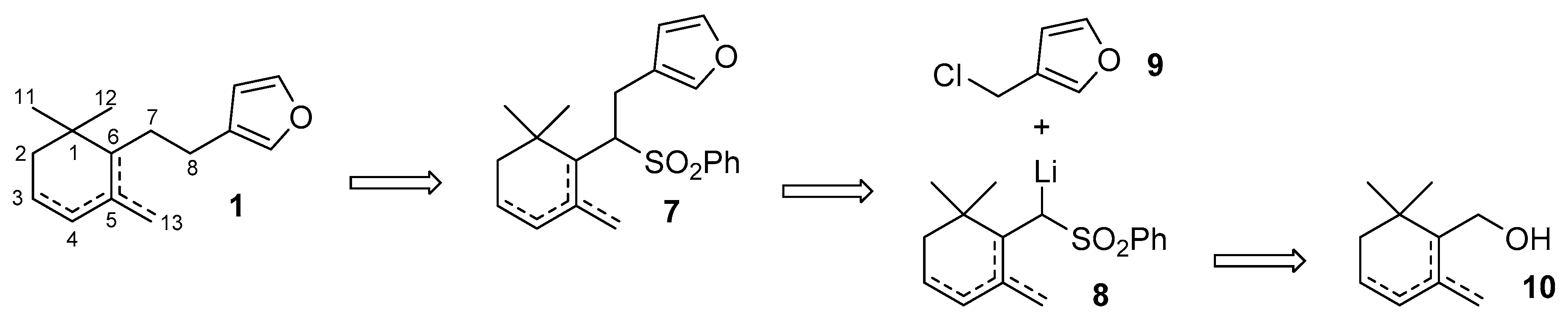
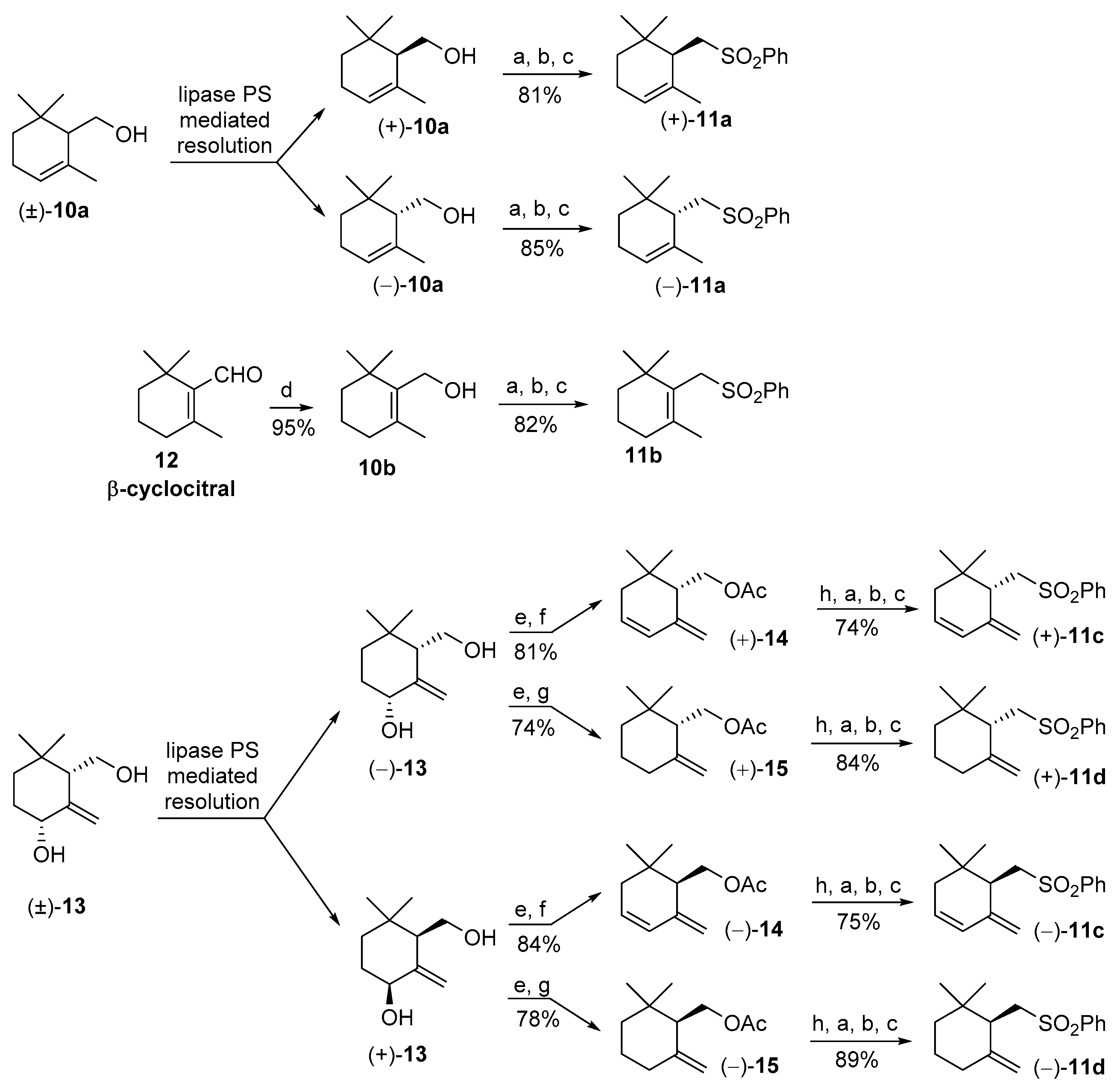
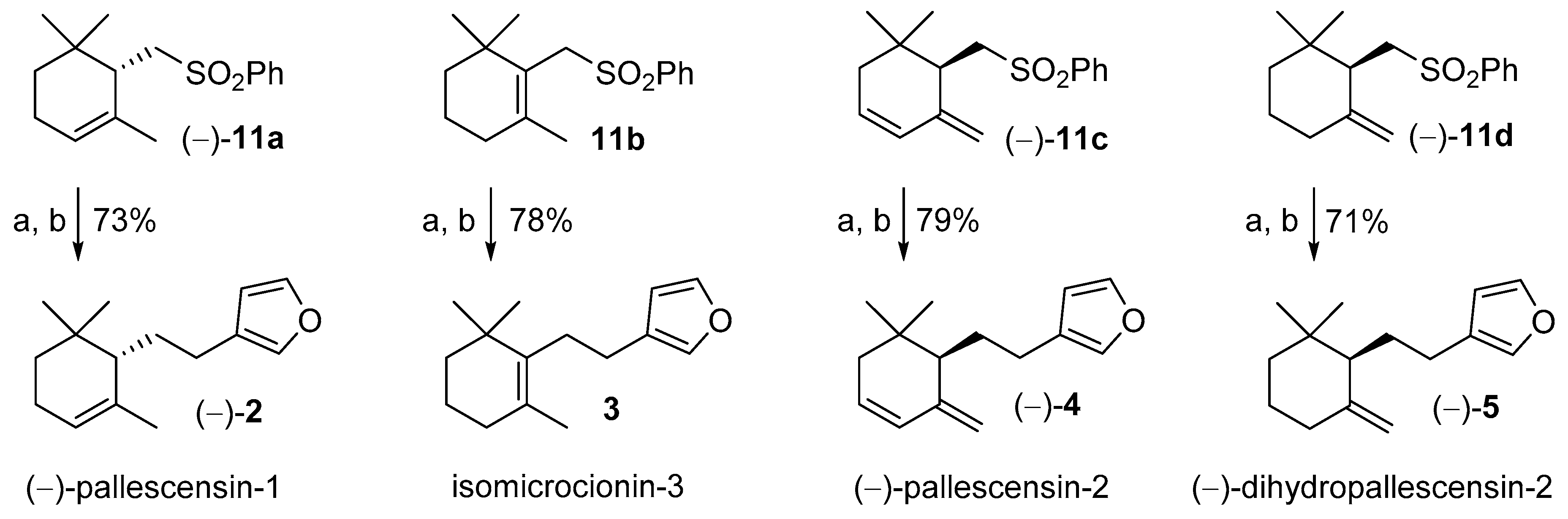
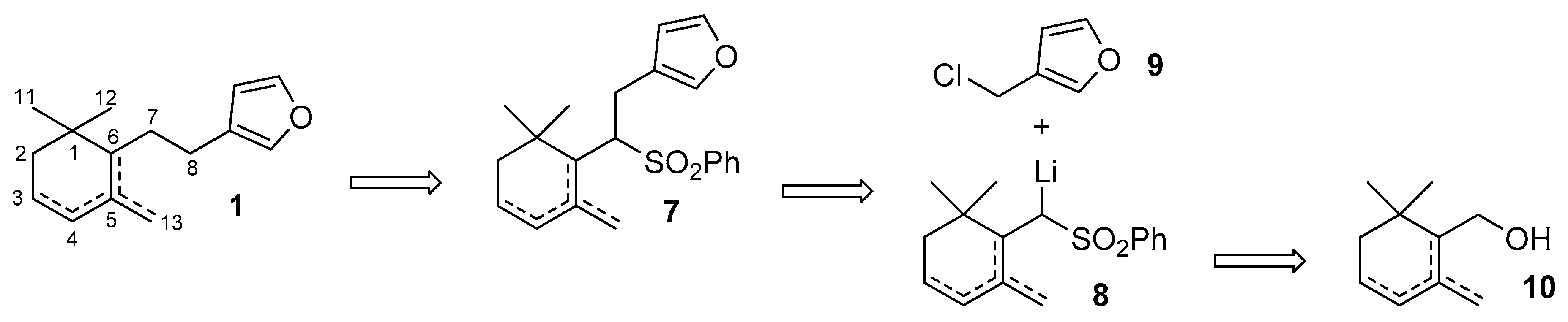
2. Results and Discussion
3. Materials and Methods
3.1. Materials and General Methods
3.2. Analytical Methods and Characterization of the Chemical Compounds
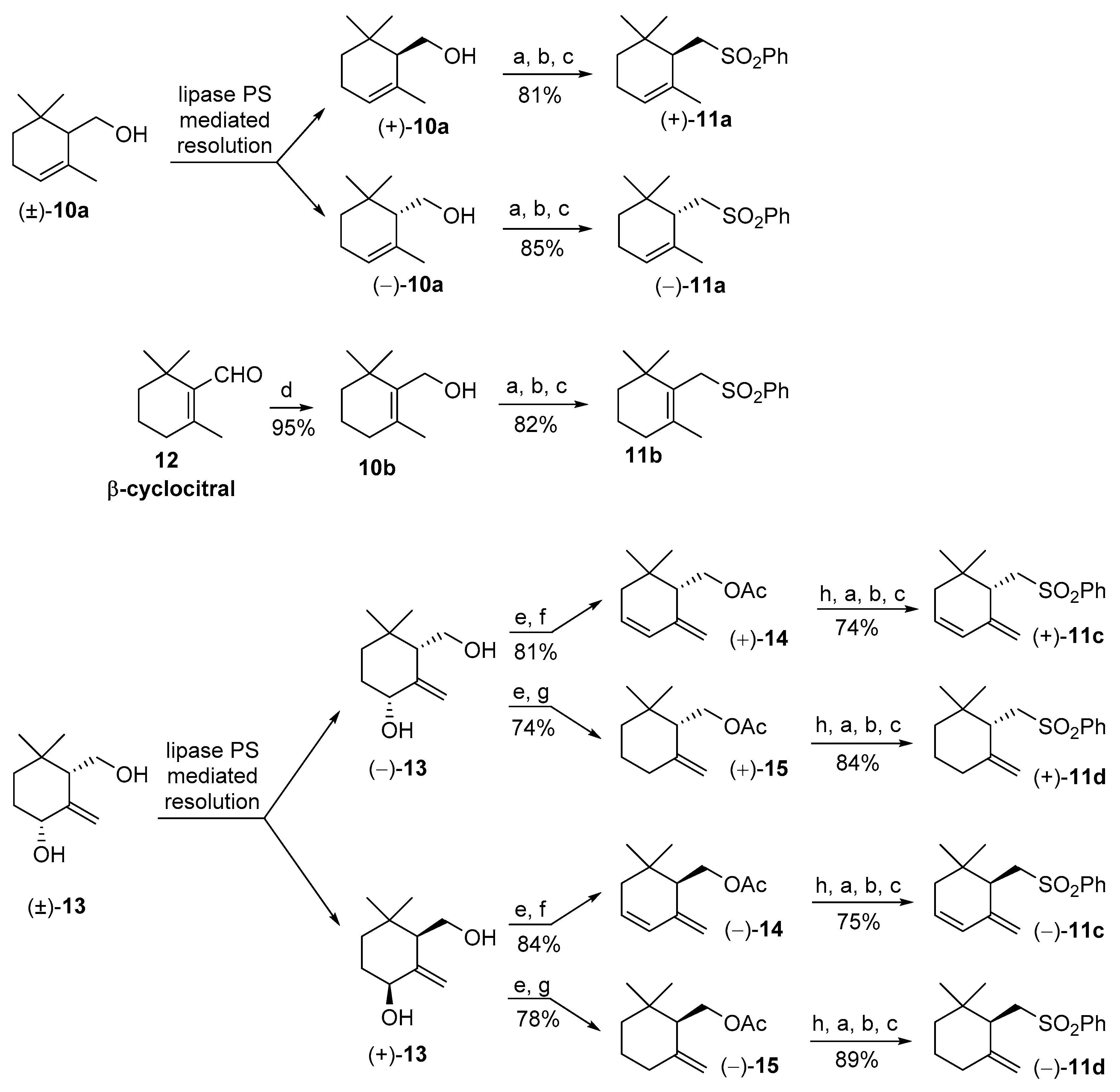
3.3. Stereoselective Preparation of (R) and (S) Enantiomers of 3,4-Dehydro-γ-cyclogeraniol Acetate 14 and γ-Cyclogeraniol Acetate 15
3.3.1. (R)-3,4-Dehydro-γ-cyclogeraniol Acetate (−)-14
3.3.2. (S)-3,4-Dehydro-γ-cyclogeraniol Acetate (+)-14
3.3.3. (R)-γ-Cyclogeraniol Acetate (−)-15
3.3.4. (S)-γ-Cyclogeraniol Acetate (+)-15
3.4. Synthesis of the Enantiomeric Forms of the Cyclogeranylsulfonylbenzene Derivatives 11a–d
3.4.1. General Procedure
3.4.2. (S)-((2,6,6-Trimethylcyclohex-2-enyl)methylsulfonyl)benzene (−)-11a
3.4.3. (R)-((2,6,6-Trimethylcyclohex-2-enyl)methylsulfonyl)benzene (+)-11a
3.4.4. 2,6,6-((Trimethylcyclohex-1-enyl)methylsulfonyl)benzene 11b
3.4.5. (R)-((2,2-Dimethyl-6-methylenecyclohexyl)methylsulfonyl)benzene (−)-11c
3.4.6. (S)-((2,2-Dimethyl-6-methylenecyclohexyl)methylsulfonyl)benzene (+)-11c
3.4.7. (R)-((2,2-Dimethyl-6-methylenecyclohexyl)methylsulfonyl)benzene (−)-11d
3.4.8. (S)-((2,2-Dimethyl-6-methylenecyclohexyl)methylsulfonyl)benzene (+)-11d
3.5. Synthesis of the Furanosesquiterpenes (−)-Pallescensin-1 (2), Isomicrocionin-3 (3), (−)-Pallescensin-2 (4), and (−)-Dihydropallescensis-2 (5)
3.5.1. General Procedure
3.5.2. Synthesis of (−)-Pallescensin-1 (2)
3.5.3. Synthesis of Isomicrocionin-3 (3)
3.5.4. Synthesis of (−)-Pallescensin-2 (4)
3.5.5. Synthesis of (−)-Dihydropallescensis-2 (5)
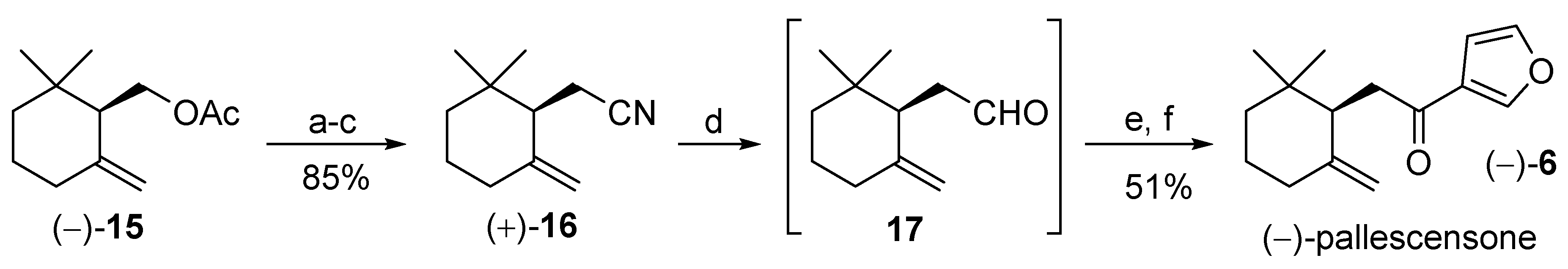
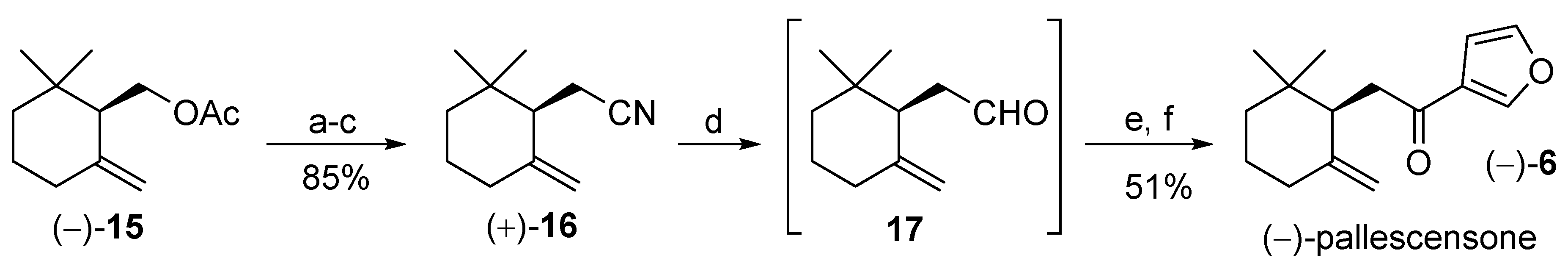
3.6. Synthesis of (−)-Pallescensone (6) Starting from (R)-γ-Cyclogeraniol Acetate
3.6.1. Synthesis of (R)-2-(2,2-Dimethyl-6-methylenecyclohexyl)acetonitrile (+)-16
3.6.2. Synthesis of (−)-Pallescensone (6)
Funding
Conflicts of Interest
References
- Cimino, G.; De Stefano, S.; Guerriero, A.; Minale, L. Furanosesquiterpenoids in sponges―I: Pallescensin-1, -2 and -3 from Disidea pallescens. Tetrahedron Lett. 1975, 16, 1417–1420. [Google Scholar] [CrossRef]
- Guella, G.; Guerriero, A.; Pietra, F. Sesquiterpenoids of the sponge Dysidea fragilis of the north-brittany sea. Helv. Chim. Acta 1985, 68, 39–48. [Google Scholar] [CrossRef]
- Gaspar, H.; Santos, S.; Carbone, M.; Rodrigues, A.S.; Rodrigues, A.I.; Uriz, M.J.; Savluchinske Feio, S.M.; Melck, D.; Humanes, M.; Gavagnin, M. Isomeric furanosesquiterpenes from the portuguese marine sponge Fasciospongia sp. J. Nat. Prod. 2008, 71, 2049–2052. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.E.; Walker, R.P.; Wratten, S.J.; Faulkner, D.J. A chemical defense mechanism for the nudibranch Cadlina luteomarginata. Tetrahedron 1982, 38, 1865–1873. [Google Scholar] [CrossRef]
- Butler, M.; Capon, R. Beyond polygodial: New drimane sesquiterpenes from a southern Australian marine sponge, Dysidea sp. Aust. J. Chem. 1993, 46, 1255–1267. [Google Scholar] [CrossRef]
- Fontana, A.; Muniaín, C.; Cimino, G. First chemical study of patagonian nudibranchs: A new seco-11,12-spongiane, tyrinnal, from the defensive organs of Tyrinna nobilis. J. Nat. Prod. 1998, 61, 1027–1029. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.-C.; Li, J.; Li, Z.-Y.; Shi, L.; Guo, Y.-W. Sesquiterpenes from the Hainan sponge Dysidea septosa. J. Nat. Prod. 2008, 71, 1399–1403. [Google Scholar] [CrossRef]
- Cambie, R.C.; Craw, P.A.; Bergquist, P.R.; Karuso, P. Chemistry of sponges, II. Pallescensone, a furanosesquiterpenoid from Dictyodendrilla cavernosa. J. Nat. Prod. 1987, 50, 948–949. [Google Scholar] [CrossRef]
- Mudianta, I.W.; Challinor, V.L.; Winters, A.E.; Cheney, K.L.; De Voss, J.J.; Garson, M.J. Synthesis and determination of the absolute configuration of (−)-(5R,6Z)-dendrolasin-5-acetate from the nudibranch Hypselodoris jacksoni. Beilstein J. Org. Chem. 2013, 9, 2925–2933. [Google Scholar] [CrossRef] [PubMed]
- Winters, A.E.; White, A.M.; Dewi, A.S.; Mudianta, I.W.; Wilson, N.G.; Forster, L.C.; Garson, M.J.; Cheney, K.L. Distribution of defensive metabolites in nudibranch molluscs. J. Chem. Ecol. 2018, 44, 384–396. [Google Scholar] [CrossRef]
- Matsumoto, T.; Usui, S. Furanosesquiterpenoids absolute configuration of pallescensin-1, -2, and -A. Chem. Lett. 1978, 7, 105–108. [Google Scholar] [CrossRef]
- Tius, M.A.; Takaki, K.S. Biomimetic synthesis of (+/−)-pallescensin 1. J. Org. Chem. 1982, 47, 3166–3168. [Google Scholar] [CrossRef]
- Matsumoto, T.; Usui, S. Furanosesquiterpenoids: Total synthesis of pallescensins 2, F, and G. Bull. Chem. Soc. Jpn. 1983, 56, 491–493. [Google Scholar] [CrossRef]
- Baker, R.; Cottrell, I.F.; Ravenscroft, P.D.; Swain, C.J. Stereoselective synthesis of (+/−)-ancistrofuran and its stereoisomers. J. Chem. Soc. Perkin Trans. 1 1985, 2463–2468. [Google Scholar] [CrossRef]
- Kurth, M.J.; Soares, C.J. Asymmetric aza-Claisen rearrangement: Synthesis of (+)-dihydropallescensin-2 [(+)-penlanpallescensin]. Tetrahedron Lett. 1987, 28, 1031–1034. [Google Scholar] [CrossRef]
- Eicher, T.; Massonne, K.; Herrmann, M. Synthese von bryophyten-inhaltsstoffen 4. Synthesen des ricciocarpins A. Synthesis 1991, 1173–1176. [Google Scholar] [CrossRef]
- Cruz Almanza, R.; Hinojosa Reyes, A. A simple synthesis of γ-cyclohomocitral. Synthetic Commun. 1993, 23, 867–874. [Google Scholar] [CrossRef]
- Vidari, G.; Lanfranchi, G.; Masciaga, F.; Moriggi, J.-D. Enantioselective synthesis of γ-cyclohomocitral, pallescensone, and ancistrodial. Tetrahedron Asymmetry 1996, 7, 3009–3020. [Google Scholar] [CrossRef]
- Palombo, E.; Audran, G.; Monti, H. First enantioselective synthesis and determination of the absolute configuration of natural (+)-dehydro-β-monocyclonerolidol. Tetrahedron Lett. 2003, 44, 6463–6464. [Google Scholar] [CrossRef]
- Serra, S. A practical, enantiospecific synthesis of (S)-trans-γ-monocyclofarnesol. Nat. Prod. Commun. 2012, 7, 1569–1572. [Google Scholar] [CrossRef]
- Serra, S.; Cominetti, A.A.; Lissoni, V. A general synthetic approach to hydroquinone meroterpenoids: Stereoselective synthesis of (+)-(S)-metachromin V and alliodorol. Nat. Prod. Commun. 2014, 9, 303–308. [Google Scholar] [CrossRef]
- Serra, S.; Cominetti, A.A.; Lissoni, V. Use of (S)-trans-γ-monocyclofarnesol as a useful chiral building block for the stereoselective synthesis of diterpenic natural products. Nat. Prod. Commun. 2014, 9, 329–335. [Google Scholar] [CrossRef]
- Serra, S.; Lissoni, V. First enantioselective synthesis of marine diterpene ambliol-A. Eur. J. Org. Chem. 2015, 2226–2234. [Google Scholar] [CrossRef]
- Serra, S.; Gatti, F.G.; Fuganti, C. Lipase-mediated resolution of the hydroxy-cyclogeraniol isomers: Application to the synthesis of the enantiomers of karahana lactone, karahana ether, crocusatin C and γ-cyclogeraniol. Tetrahedron Asymmetry 2009, 20, 1319–1329. [Google Scholar] [CrossRef]
- Bovolenta, M.; Castronovo, F.; Vadalà, A.; Zanoni, G.; Vidari, G. A simple and efficient highly enantioselective synthesis of α-ionone and α-damascone. J. Org. Chem. 2004, 69, 8959–8962. [Google Scholar] [CrossRef]
- Luparia, M.; Boschetti, P.; Piccinini, F.; Porta, A.; Zanoni, G.; Vidari, G. Enantioselective synthesis and olfactory evaluation of 13-alkyl-substituted α-ionones. Chem. Biodivers. 2008, 5, 1045–1057. [Google Scholar] [CrossRef]
- Serra, S. Preparation and use of enantioenriched 2-aryl-propylsulfonylbenzene derivatives as valuable building blocks for the enantioselective synthesis of bisabolane sesquiterpenes. Tetrahedron Asymmetry 2014, 25, 1561–1572. [Google Scholar] [CrossRef]
- Nakagawa, I.; Aki, K.; Hata, T. Synthesis of 5′-alkylthio-5′-deoxynucleosides from nucleosides in a one-pot reaction. J. Chem. Soc. Perkin Trans. 1 1983, 1315–1318. [Google Scholar] [CrossRef]
- Surburg, H.; Panten, J. Common Fragrance and Flavor Materials: Preparation, Properties and Uses, 6th ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2016; ISBN 9783527331604. [Google Scholar]
- Serra, S. Recent advances in the synthesis of carotenoid-derived flavours and fragrances. Molecules 2015, 20, 12817–12840. [Google Scholar] [CrossRef]
- Serra, S.; Fuganti, C.; Brenna, E. Synthesis, olfactory evaluation, and determination of the absolute configuration of the 3,4-didehydroionone stereoisomers. Helv. Chim. Acta 2006, 89, 1110–1122. [Google Scholar] [CrossRef]
- Smith, P.M.; Thomas, E.J. Approaches to a synthesis of galbonolide B. J. Chem. Soc. Perkin Trans. 1 1998, 3541–3556. [Google Scholar] [CrossRef]
- Frigerio, M.; Santagostino, M. A mild oxidizing reagent for alcohols and 1,2-diols: O-iodoxybenzoic acid (IBX) in DMSO. Tetrahedron Lett. 1994, 35, 8019–8022. [Google Scholar] [CrossRef]
- Bourdron, J.; Commeiras, L.; Audran, G.; Vanthuyne, N.; Hubaud, J.C.; Parrain, J.-L. First total synthesis and assignment of the stereochemistry of crispatenine. J. Org. Chem. 2007, 72, 3770–3775. [Google Scholar] [CrossRef]
- Vidari, G.; Lanfranchi, G.; Sartori, P.; Serra, S. Saponaceolides: Differential cytotoxicity and enantioselective synthesis of the right-hand lactone moiety. Tetrahedron Asymmetry 1995, 6, 2977–2990. [Google Scholar] [CrossRef]
- Tanis, S.P. A simple synthesis of 3-substituted furans. The preparations of dendrolasin, perillene and congeners. Tetrahedron Lett. 1982, 23, 3115–3118. [Google Scholar] [CrossRef]
- Meyers, A.I.; Collington, E.W. Facile and specific conversion of allylic alcohols to allylic chlorides without rearrangement. J. Org. Chem. 1971, 36, 3044–3045. [Google Scholar] [CrossRef]
- Fujii, M.; Morimoto, Y.; Ono, M.; Akita, H. Preparation of (S)-γ-cyclogeraniol by lipase-catalyzed transesterification and synthesis of (+)-trixagol and (+)-luffarin-P. J. Mol. Catal. B Enzym. 2016, 123, 160–166. [Google Scholar] [CrossRef]
- Tsangarakis, C.; Stratakis, M. Biomimetic cyclization of small terpenoids promoted by zeolite NaY: Tandem formation of α-ambrinol from geranyl acetone. Adv. Synth. Catal. 2005, 347, 1280–1284. [Google Scholar] [CrossRef]
- Proszenyák, Á.; Charnock, C.; Hedner, E.; Larsson, R.; Bohlin, L.; Gundersen, L.-L. Synthesis, antimicrobial and antineoplastic activities for agelasine and agelasimine analogs with a β-cyclocitral derived substituent. Arch. Pharm. 2007, 340, 625–634. [Google Scholar] [CrossRef]
- Trost, B.M.; Shen, H.C.; Surivet, J.-P. Biomimetic enantioselective total synthesis of (−)-siccanin via the Pd-catalyzed asymmetric allylic alkylation (AAA) and sequential radical cyclizations. J. Am. Chem. Soc. 2004, 126, 12565–12579. [Google Scholar] [CrossRef]

© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serra, S. A General Strategy for the Stereoselective Synthesis of the Furanosesquiterpenes Structurally Related to Pallescensins 1–2. Mar. Drugs 2019, 17, 245. https://doi.org/10.3390/md17040245
Serra S. A General Strategy for the Stereoselective Synthesis of the Furanosesquiterpenes Structurally Related to Pallescensins 1–2. Marine Drugs. 2019; 17(4):245. https://doi.org/10.3390/md17040245
Chicago/Turabian StyleSerra, Stefano. 2019. "A General Strategy for the Stereoselective Synthesis of the Furanosesquiterpenes Structurally Related to Pallescensins 1–2" Marine Drugs 17, no. 4: 245. https://doi.org/10.3390/md17040245
APA StyleSerra, S. (2019). A General Strategy for the Stereoselective Synthesis of the Furanosesquiterpenes Structurally Related to Pallescensins 1–2. Marine Drugs, 17(4), 245. https://doi.org/10.3390/md17040245