Meroterpenoids and Isocoumarinoids from a Myrothecium Fungus Associated with Apocynum venetum

Abstract
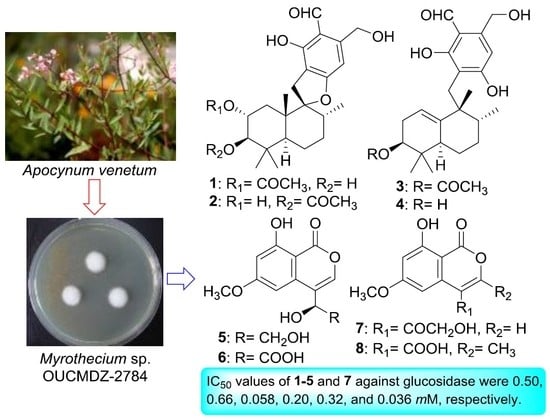
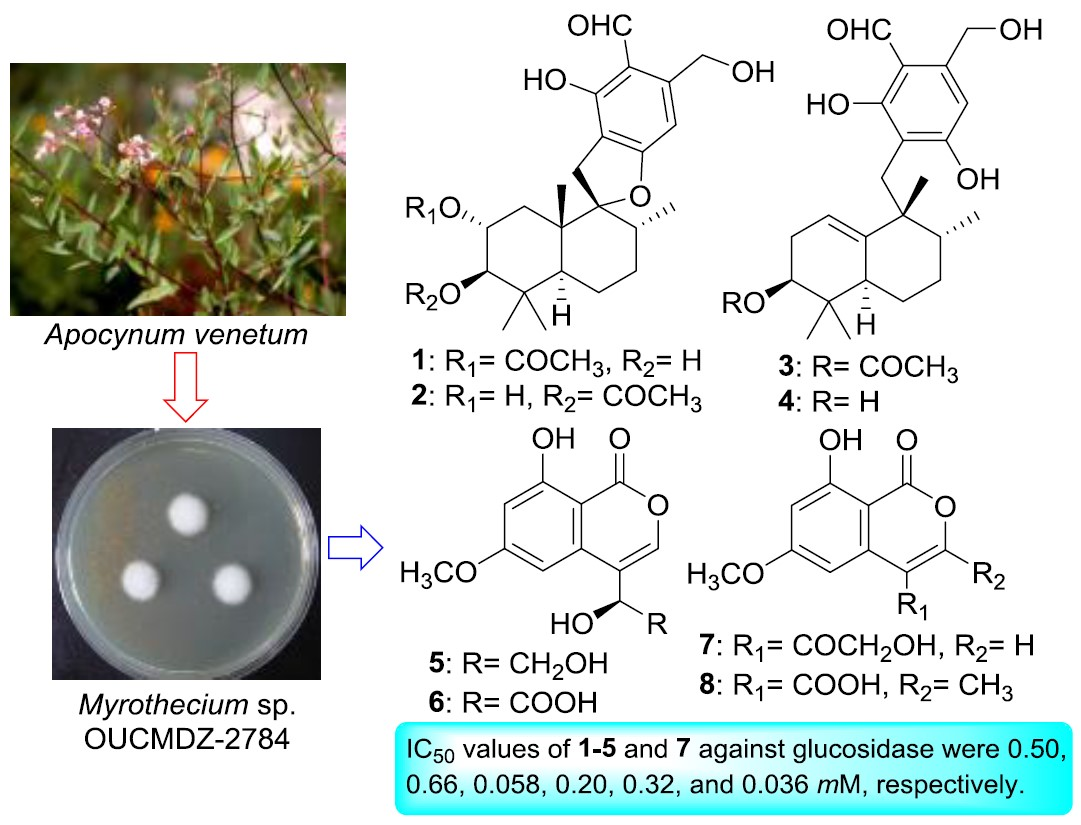
1. Introduction
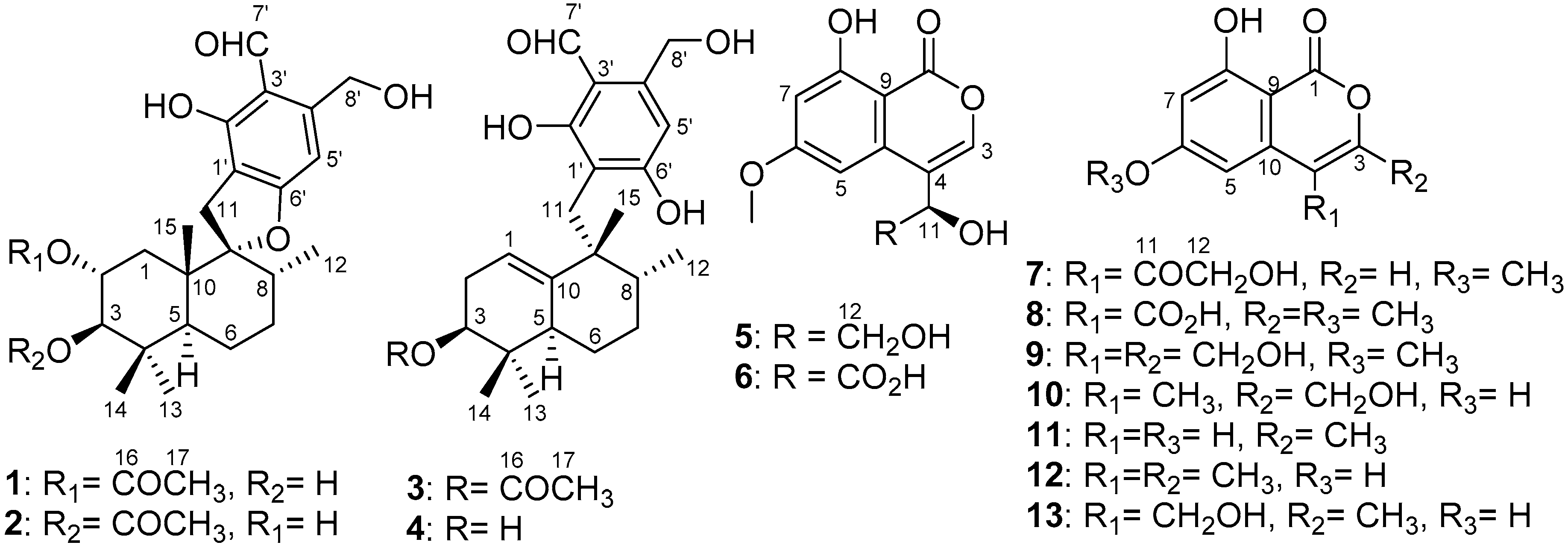
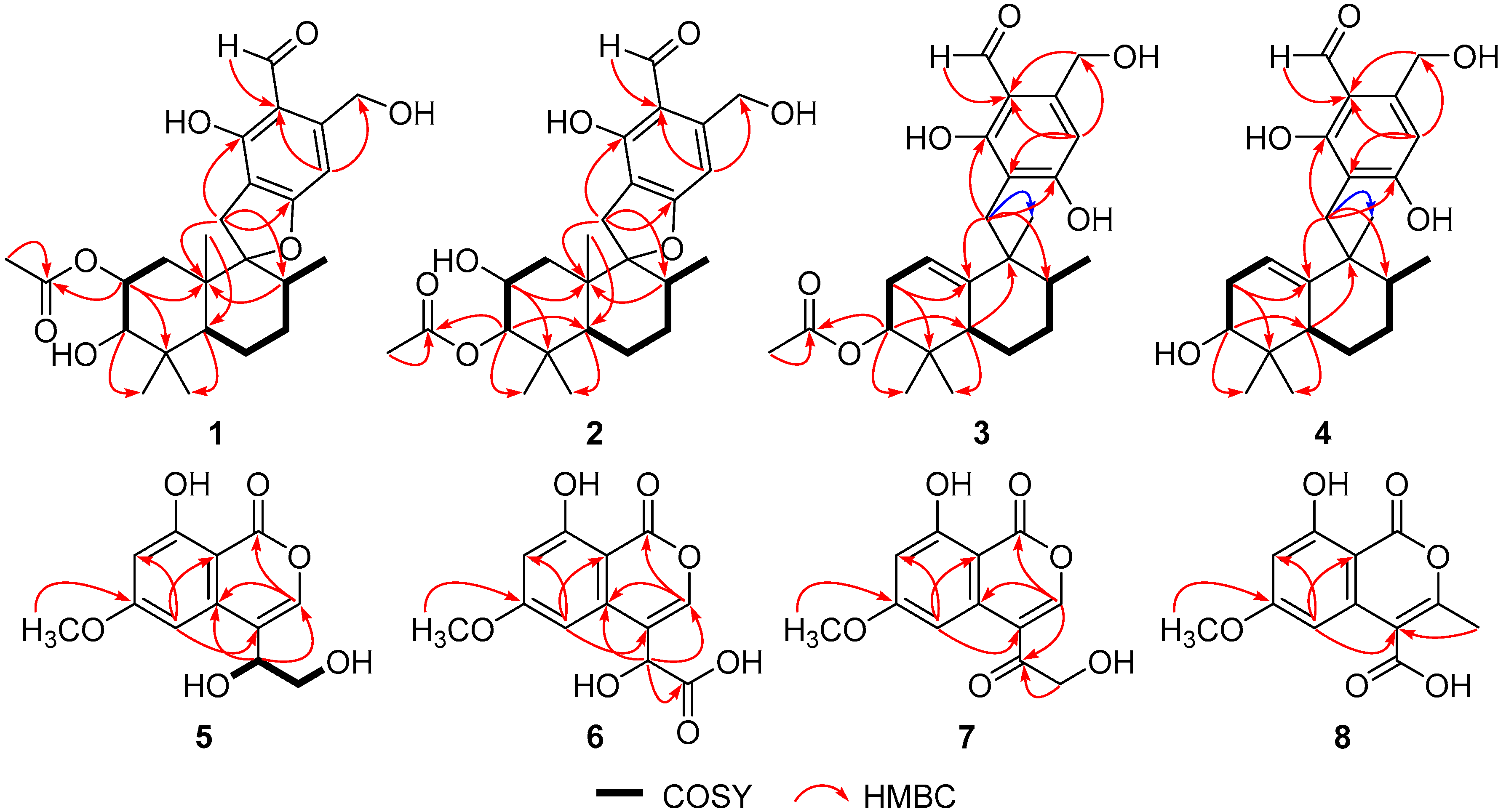
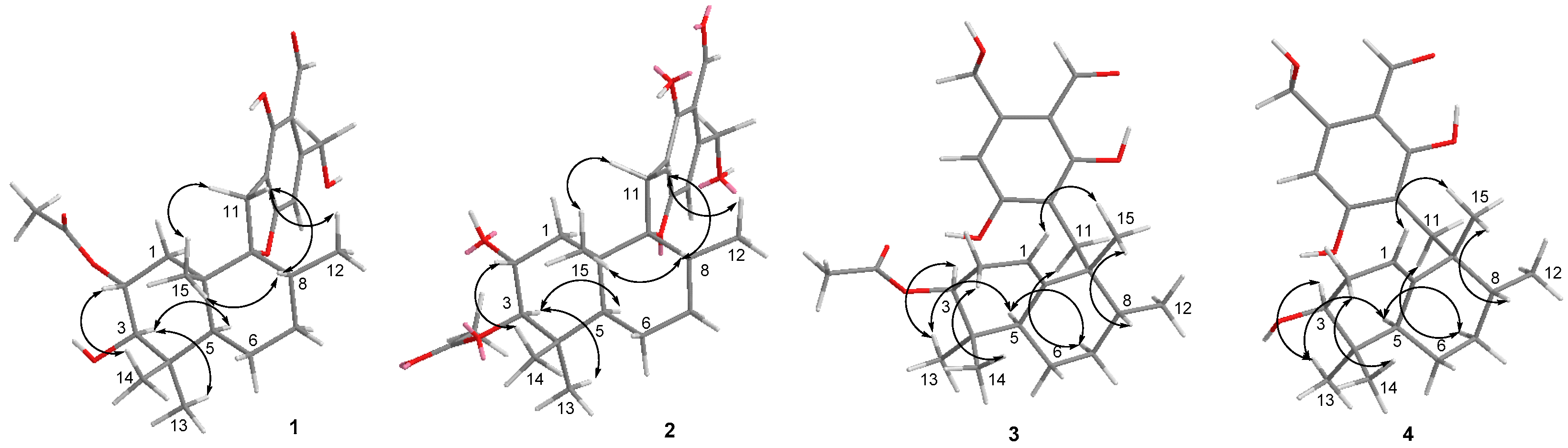
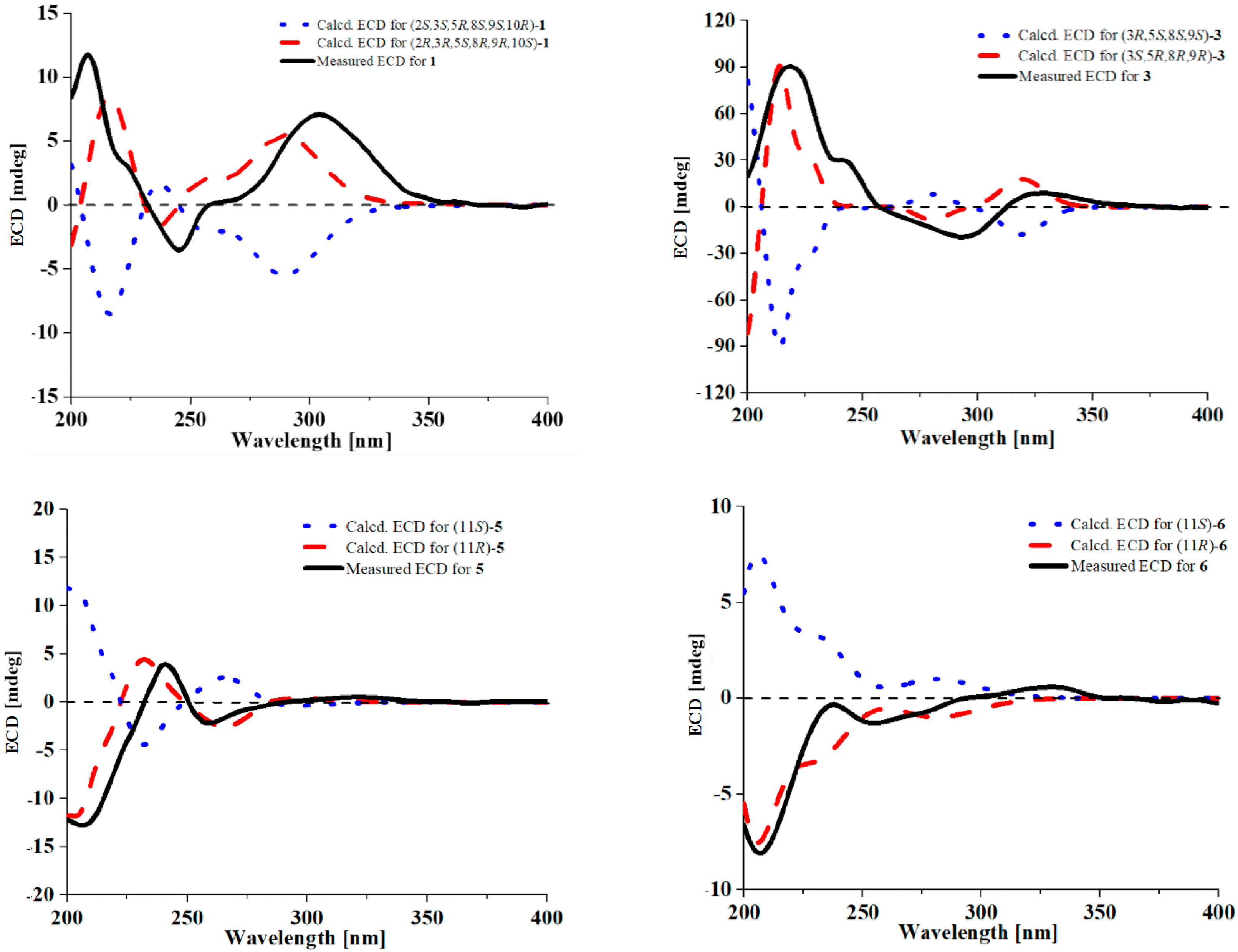
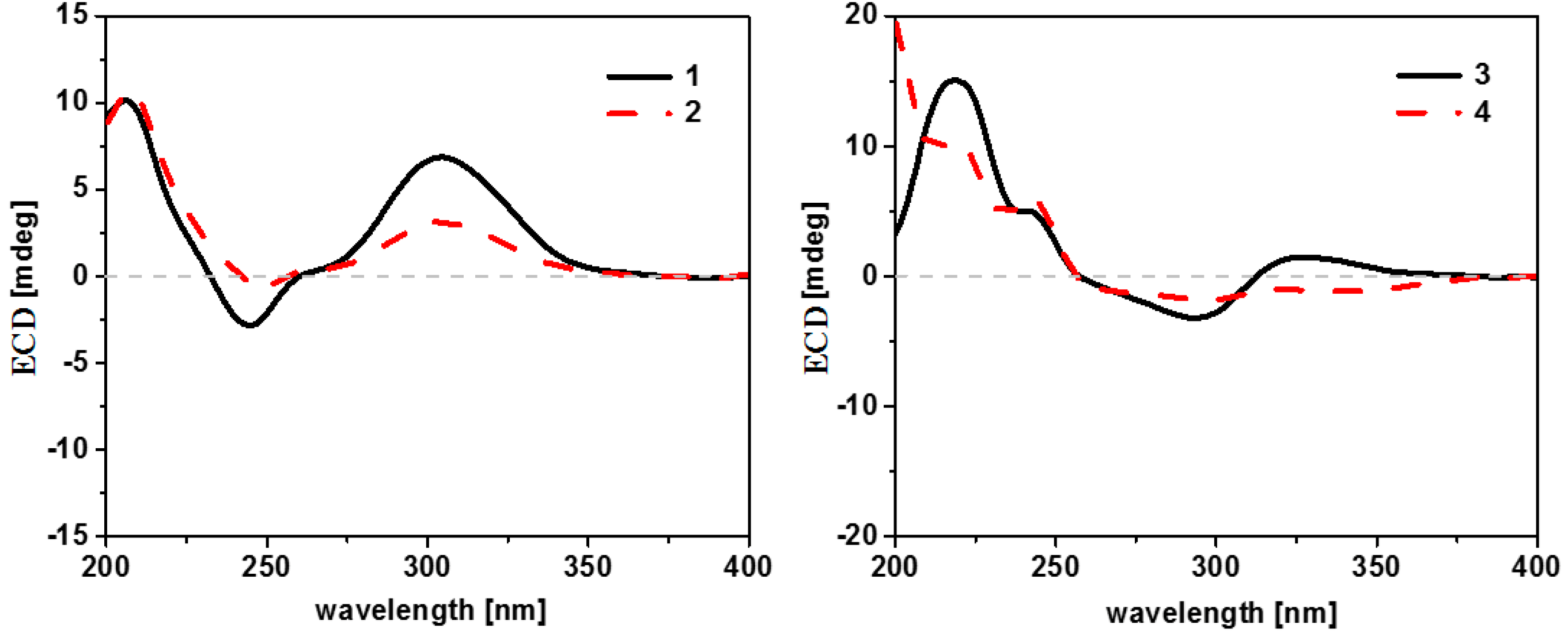
2. Results and Discussion
3. Experimental Section
3.1. General Experimental Procedures
3.2. Collection and Phylogenetic Analysis
3.3. Cultivation and Extraction
3.4. Purification
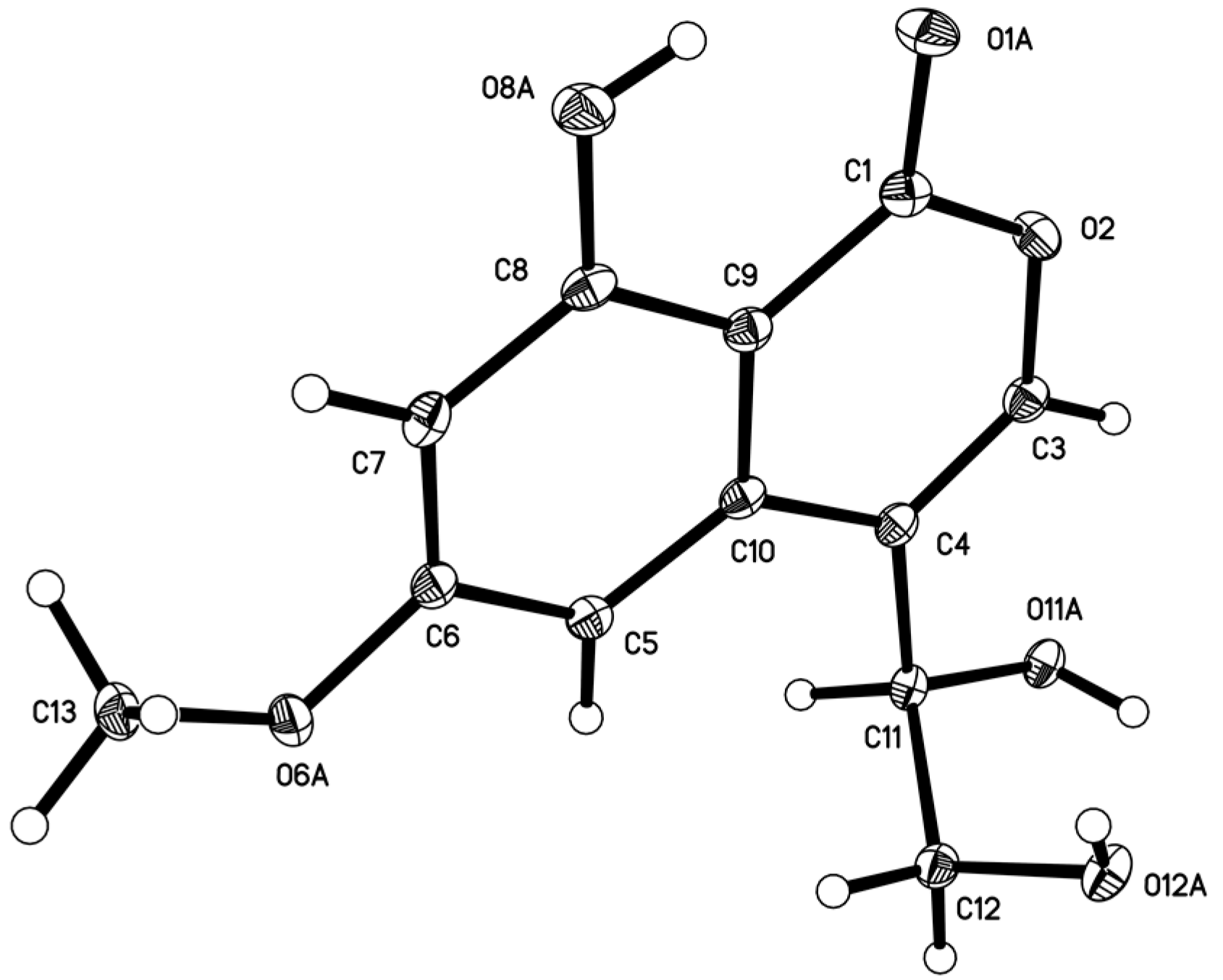
3.5. X-ray Structure Determination of Compound 5
3.6. α-Glucosidase Inhibitory Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Aly, A.H.; Debbab, A.; Proksch, P. Fifty years of drug discovery from fungi. Fungal Divers. 2011, 50, 3–19. [Google Scholar] [CrossRef]
- Bladt, T.T.; Frisvad, J.C.; Knudsen, P.B.; Larsen, T.O. Anticancer and antifungal compounds from Aspergillus, Penicillium and other filamentous fungi. Molecules 2013, 18, 11338–11376. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.A.; Bacha, N.; Ahmad, B.; Lutfullah, G.; Farooq, U.; Cox, R.J. Fungi as chemical industries and genetic engineering for the production of biologically active secondary metabolites. Asian Pac. J. Trop. Biomed. 2014, 4, 859–870. [Google Scholar] [CrossRef]
- Rateb, M.E.; Ebel, R. Secondary metabolites of fungi from marine habitats. Nat. Prod. Rep. 2011, 28, 290–344. [Google Scholar] [CrossRef] [PubMed]
- Bugnia, T.S.; Ireland, C.M. Marine-derived fungi: A chemically and biologically diverse group of microorganisms. Nat. Prod. Rep. 2004, 21, 143–163. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Meng, W.; Cao, C.; Wang, J.; Shan, W.; Wang, Q. Antibacterial and antifungal compounds from marine fungi. Mar. Drugs 2015, 13, 3479–3513. [Google Scholar] [CrossRef] [PubMed]
- Moghadamtousi, S.Z.; Nikzad, S.; Kadir, H.A.; Abubakar, S.; Zandi, K. Potential antiviral agents from marine fungi: An overview. Mar. Drugs 2015, 13, 4520–4538. [Google Scholar] [CrossRef] [PubMed]
- Strobel, G.; Daisy, B.; Castillo, U.; Harper, J. Natural products from endophytic microorganisms. J. Nat. Prod. 2004, 67, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Borges, W.S.; Borges, K.B.; Bonato, P.S.; Said, S.; Pupo, M.T. Endophytic fungi: natural products, enzymes and biotransformation reactions. Curr. Org. Chem. 2009, 13, 1137–1163. [Google Scholar] [CrossRef]
- Nisa, H.; Kamili, A.N.; Nawchoo, I.A.; Shafi, S.; Shameem, N.; Bandh, S.A. Fungal endophytes as prolific source of phytochemicals and other bioactive natural products: A review. Microb. Pathog. 2015, 82, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.X.; Zou, W.X. Endophytes: A rich source of functional metabolites. Nat. Prod. Rep. 2001, 18, 448–459. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Li, Y.; Guo, L.; Wang, Y.; Liu, P.; Zhu, W. Indole diterpenoids and isocoumarin from the fungus, Aspergillus flavus, isolated from the prawn, Penaeus vannamei. Mar. Drugs 2014, 12, 3970–3981. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Kong, F.; Wang, Y.; Fu, P.; Zhu, W. Cladodionen, a cytotoxic hybrid polyketide from the marine-derived Cladosporium sp. OUCMDZ-1635. Mar. Drugs 2018, 16, 71. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zheng, J.; Liu, P.; Wang, W.; Zhu, W. Three new compounds from Aspergillus terreus PT06-2 grown in a high salt medium. Mar. Drugs 2011, 9, 1368–1378. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lu, Z.; Sun, K.; Zhu, W. Effects of high salt stress on secondary metabolites from marine-derived fungus Spicaria elegans. Mar. Drugs 2011, 9, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, L.; Zhuang, Y.; Kong, F.; Zhang, C.; Zhu, W. Phenolic polyketides from the co-cultivation of marine-derived Penicillium sp. WC-29-5 and Streptomyces fradiae 007. Mar. Drugs 2014, 12, 2079–2088. [Google Scholar] [CrossRef] [PubMed]
- Chinese Pharmacopeia Committee of Ministry of Public Health of the People’s Republic of China. Chinese Pharmacopeia 2000; Chemical and Technical Press: Beijing, China, 2000; p. 170. [Google Scholar]
- Shaanxi Provincial and Municipal Collaborative Group for Prevention and Treatment of Coronary Heart Diseases. Clinical observation and pharmacological experiment of Apocynum venetum root in the treatment of heart failure. Shaanxi Med. J. 1974, 5, 10–14. [Google Scholar]
- Isaka, M.; Punya, J.; Lertwerawat, Y.; Tanticharoen, M.; Thebtaranonth, Y. Antimalarial activity of macrocyclic trichothecenes isolated from the fungus Myrothecium verrucaria. J. Nat. Prod. 1999, 62, 329–331. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Wu, P.; Xue, J.; Wei, X. Cytotoxic and antibacterial quinone sesquiterpenes from a Myrothecium fungus. J. Nat. Prod. 2014, 77, 1791–1799. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Wu, P.; Xue, J.; Li, H.; Wei, X. Myrothecols G and H, two new analogues of the marine-derived quinone sesquiterpene Penicilliumin A. Mar. Drugs 2015, 13, 3360–3367. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.H.; Hirota, A.; Shima, S.; Nakagawa, M.; Nozaki, H.; Tada, T.; Nakayama, M. Structure of myrocin C, a new diterpene antibiotic produced by a strain of Myrothecium sp. Agric. Biol. Chem. 1987, 51, 3455–3457. [Google Scholar] [CrossRef]
- Zou, X.; Niu, S.; Ren, J.; Li, E.; Liu, X.; Che, Y. Verrucamides A–D, antibacterial cyclopeptides from Myrothecium verrucaria. J. Nat. Prod. 2011, 74, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Tomida, J.; Hirai, T.; Morita, Y.; Kawamura, Y.; Inoue, M. Tubakialactones A–E, new polyketides from the endophytic fungus Tubakia sp. ECN-111. Tetrahedron Lett. 2017, 58, 2248–2251. [Google Scholar] [CrossRef]
- Rukachaisirikul, V.; Rodglin, A.; Sukpondma, Y.; Phongpaichit, S.; Buatong, J.; Sakayaroj, J. Phthalide and isocoumarin derivatives produced by an Acremonium sp. isolated from a mangrove Rhizophora apiculata. J. Nat. Prod. 2012, 75, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Kornsakulkarn, J.; Thongpanchang, C.; Lapanun, S.; Srichomthong, K. Isocoumarin glucosides from the scale insect fungus Torrubiella tenuis BCC 12732. J. Nat. Prod. 2009, 72, 1341–1343. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.K.; Sato, C.; Shibata, Y.; Kobayashi, A.; Yamashita, K. Growth promoting activities of sclerotinin A and its analogs. Agric. Biol. Chem. 1974, 38, 1311–1315. [Google Scholar] [CrossRef]
- Kimura, Y.; Nakadoi, M.; Nakajima, H.; Hamasaki, T.; Nagai, T.; Kohmoto, K.; Shimada, A. Structure of sescandelin-B, a new metabolite produced by the fungus Sesquicillium candelabrum. Agric. Biol. Chem. 1991, 55, 1887–1888. [Google Scholar]
- Zhao, J.; Feng, J.; Tan, Z.; Liu, J.; Zhao, J.; Chen, R.; Xie, K.; Zhang, D.; Li, Y.; Yu, L.; et al. Stachybotrysins A−G, phenylspirodrimane derivatives from the fungus Stachybotrys chartarum. J. Nat. Prod. 2017, 80, 1819–1826. [Google Scholar] [CrossRef] [PubMed]
- Berova, N.; Bari, L.D.; Pescitelli, G. Application of electronic circular dichroism in configurational and conformational analysis of organic compounds. Chem. Soc. Rev. 2007, 36, 914–931. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; Zhao, C.; Hao, J.; Wang, C.; Wang, W.; Huang, X.; Zhu, W. New α-glucosidase inhibitors from a marine sponge-derived fungus, Aspergillus sp. OUCMDZ-1583. RSC Adv. 2015, 5, 68852–68863. [Google Scholar] [CrossRef]
- Wang, C.; Guo, L.; Hao, J.; Wang, L.; Zhu, W. α-Glucosidase inhibitors from the marine-derived fungus Aspergillus flavipes HN4-13. J. Nat. Prod. 2016, 79, 2977–2981. [Google Scholar] [CrossRef] [PubMed]
- Shim, Y.-J.; Doo, H.-K.; Ahn, S.-Y.; Kim, Y.-S.; Seong, J.-K.; Park, I.-S.; Min, B.-H. Inhibitory effect of aqueous extract from the gall of Rhus chinensis on alpha-glucosidase activity and postprandial blood glucose. J. Ethnopharmacol. 2003, 85, 283–287. [Google Scholar] [CrossRef]
- Lin, Z.; Zhu, T.; Fang, Y.; Gu, Q.; Zhu, W. Polyketides from Penicillium sp. JP-1, an endophytic fungus associated with the mangrove plant Aegiceras corniculatum. Phytochemistry 2008, 69, 1273–1278. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Zhu, H.; Fu, P.; Wang, Y.; Zhang, Z.; Lin, H.; Liu, P.; Zhuang, Y.; Hong, K.; Zhu, W. Cytotoxic polyphenols from the marine-derived fungus Penicillium expansum. J. Nat. Prod. 2010, 73, 911–914. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lu, Z.; Liu, P.; Wang, Y.; Li, J.; Hong, K.; Zhu, W. Cytotoxic polyphenols from the fungus Penicillium expansum 091006 endogenous with the mangrove plant Excoecaria agallocha. Planta Med. 2012, 78, 1861–1866. [Google Scholar] [PubMed]
- Kong, F.; Wang, Y.; Liu, P.; Dong, T.; Zhu, W. Thiodiketopiperazines from the marine-derived fungus Phoma sp. OUCMDZ-1847. J. Nat. Prod. 2014, 77, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Han, X.; Zhu, G.; Wang, Y.; Chairoungdua, A.; Piyachaturawat, P.; Zhu, W. Polyketides from the endophytic fungus Cladosporium sp. isolated from the mangrove plant Excoecaria agallocha. Front. Chem. 2018, 6, 344. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 | 2 | 3 | 4 | ||||
|---|---|---|---|---|---|---|---|---|
| δC, Type | δH, Mult. (J in Hz) | δC, Type | δH, Mult. (J in Hz) | δC, Type | δH, Mult. (J in Hz) | δC, Type | δH, Mult. (J in Hz) | |
| 1 | 35.1, CH2 | 1.16, m; 1.55 a | 38.4, CH2 | 1.17, m; 1.57 a | 114.7, CH | 4.94, brs | 116.3, CH | 4.88, s |
| 2 | 71.4, CH | 4.83, ddd (11.5, 10.1, 4.1) | 64.4, CH | 3.65, m | 28.0, CH2 | 1.89, m; 1.23, m | 31.6, CH2 | 1.72, m |
| 3 | 78.2, CH | 2.96, dd (10.1, 4.8) | 83.3, CH | 4.29, d (9.9) | 75.6, CH | 4.62, dd (8.6, 6.5) | 71.5, CH | 3.30 a |
| 4 | 39.2, C | 39.0, C | 35.6, C | 36.9, C | ||||
| 5 | 45.6, CH | 1.56 a | 45.5, CH | 1.63 a | 43.1, CH | 2.54, m | 43.7, CH | 2.45, m |
| 6 | 20.7, CH2 | 1.54 a; 1.47, m | 20.4, CH2 | 1.56 a; 1.48, m | 26.7, CH2 | 1.79, m | 26.7, CH2 | 1.77, m |
| 7 | 30.6, CH2 | 1.55 a; 1.35, m | 30.5, CH2 | 1.57 a; 1.38, m | 30.2, CH2 | 1.54, m | 30.3, CH2 | 1.52, m |
| 8 | 36.0, CH | 1.83, m | 35.6, CH | 1.85, m | 43.3, CH | 1.30, m | 42.9, CH | 1.29, m |
| 9 | 98.8, C | 98.8, C | 43.7, C | 43.7, C | ||||
| 10 | 42.8, C | 42.5, C | 143.9, C | 143.0, C | ||||
| 11 | 30.4, CH2 | 3.03, d (16.1); 2.79, d (16.0) | 30.2, CH2 | 3.11, d (16.5); 2.81, d (16.5) | 23.5, CH2 | 2.68, d (12.9); 2.53, d (12.9) | 23.3, CH2 | 2.63, d (12.7); 2.50, d (12.7) |
| 12 | 15.2, CH3 | 0.64, d (6.3) | 15.1, CH3 | 0.68, d (6.3) | 17.1, CH3 | 1.03, d (6.6) | 17.2, CH3 | 1.02, d (6.7) |
| 13 | 28.7, CH3 | 0.97, s | 28.4, CH3 | 0.79, s | 25.0, CH3 | 0.88, s | 25.2, CH3 | 0.95, s |
| 14 | 17.0, CH3 | 0.78, s | 17.5, CH3 | 0.81, s | 17.1, CH3 | 0.73, s | 14.8, CH3 | 0.59, s |
| 15 | 16.6, CH3 | 1.04, s | 16.6, CH3 | 1.03, s | 23.0, CH3 | 0.96, s | 22.9, CH3 | 0.91, s |
| 16 | 170.3, C | 170.3, C | 169.8, C | |||||
| 17 | 21.3, CH3 | 1.91, s | 21.0, CH3 | 2.02, s | 21.0, CH3 | 1.98, s | ||
| 1′ | 111.2, C | 111.2, C | 111.4, C | 111.3, C | ||||
| 2′ | 159.8, C | 159.8, C | 164.5, C | 164.8, C | ||||
| 3′ | 112.3, C | 112.2, C | 110.2, C | 110.2, C | ||||
| 4′ | 149.5, C | 149.4, C | 145.4, C | 145.4, C | ||||
| 5′ | 101.5, CH | 6.56, s | 101.6, CH | 6.61, s | 107.5, CH | 6.51, s | 107.3, CH | 6.50, s |
| 6′ | 167.3, C | 167.3, C | 164.6, C | 164.6, C | ||||
| 7′ | 193.8, CH | 10.06, s | 193.8, CH | 10.07, s | 193.3, CH | 9.96, s | 193.4, CH | 9.96, s |
| 8′ | 60.5, CH2 | 4.74, s | 60.4, CH2 | 4.74, s | 59.9, CH2 | 4.70, s | 59.8, CH2 | 4.70, s |
| 2/3-OH | 4.94, d (4.7) | 4.62, d (4.9) | 4.13, s | |||||
| 2′-OH | 12.16, s | 12.24, s | 12.84, s | 12.78, s | ||||
| 6′-OH | 10.55, brs | |||||||
| 8′-OH | 5.44, s | 5.39, s | 5.35, s | 5.36, s | ||||
| No. | 5 a | 6 b | 7 b | 8 b | ||||
|---|---|---|---|---|---|---|---|---|
| δC, Type | δH, mult. (J in Hz) | δC, Type | δH, mult. (J in Hz) | δC, Type | δH, mult. (J in Hz) | δC, Type | δH, mult. (J in Hz) | |
| 1 | 165.4, C | 164.9, C | 163.4, C | 165.2, C | ||||
| 3 | 143.1, CH | 7.45, s | 144.4, CH | 7.62, s | 153.1, CH | 8.47, s | 151.0, C | |
| 4 | 118.6, C | 117.7, C | 114.7, C | 118.7, C | ||||
| 5 | 100.5, CH | 6.78, d (2.2) | 101.6, CH | 6.84, s | 102.3, CH | 7.46, s | 101.6, CH | 6.86, s |
| 6 | 166.4, C | 166.4, C | 166.8, C | 166.3, C | ||||
| 7 | 100.4, CH | 6.63 (d, 2.2) | 100.6, CH | 6.63, s | 101.2, CH | 6.68, s | 99.8, CH | 6.48, s |
| 8 | 163.4, C | 163.3, C | 163.2, C | 162.6, C | ||||
| 9 | 99.9, C | 99.9, C | 99.9, C | 99.2, C | ||||
| 10 | 137.6, C | 136.8, C | 134.8, C | 138.6, C | ||||
| 11 | 68.8, CH | 4.66, td (5.2, 5.2) | 68.5, CH | 5.01, s | 198.3, C | 173.9, C | ||
| 12 | 64.8, CH2 | 3.62, ddd (11.3, 5.3, 5.3); 3.51, ddd (11.3, 5.3, 5.3) | 173.4, C | 65.9, CH2 | 4.57, d (4.7) | 17.9, CH3 | 2.29, s | |
| 6-OCH3 | 56.0, CH3 | 3.88, s | 56.0, CH3 | 3.84, s | 56.0, CH3 | 3.87, s | 55.8, CH3 | 3.80, s |
| 8-OH | 11.37, s | 11.22, s | 11.03, s | 11.21, s | ||||
| 11-OH | 5.50, d (4.7) | |||||||
| 12-OH | 4.81, t (5.2) | 5.28, t (5.0) | ||||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.; Wang, C.; Liu, H.; Zhu, G.; Fu, P.; Wang, L.; Zhu, W. Meroterpenoids and Isocoumarinoids from a Myrothecium Fungus Associated with Apocynum venetum. Mar. Drugs 2018, 16, 363. https://doi.org/10.3390/md16100363
Xu Y, Wang C, Liu H, Zhu G, Fu P, Wang L, Zhu W. Meroterpenoids and Isocoumarinoids from a Myrothecium Fungus Associated with Apocynum venetum. Marine Drugs. 2018; 16(10):363. https://doi.org/10.3390/md16100363
Chicago/Turabian StyleXu, Yanchao, Cong Wang, Haishan Liu, Guoliang Zhu, Peng Fu, Liping Wang, and Weiming Zhu. 2018. "Meroterpenoids and Isocoumarinoids from a Myrothecium Fungus Associated with Apocynum venetum" Marine Drugs 16, no. 10: 363. https://doi.org/10.3390/md16100363
APA StyleXu, Y., Wang, C., Liu, H., Zhu, G., Fu, P., Wang, L., & Zhu, W. (2018). Meroterpenoids and Isocoumarinoids from a Myrothecium Fungus Associated with Apocynum venetum. Marine Drugs, 16(10), 363. https://doi.org/10.3390/md16100363