Aurantoside C Targets and Induces Apoptosis in Triple Negative Breast Cancer Cells

 , ,
, ,
Abstract
1. Introduction
2. Results
2.1. Screening the Crude Extract of the Sponge Manihinea Lynbeazleyae
2.2. Bioassay-Guided Fractionation, Isolation and Characterisation of Aurantoside C
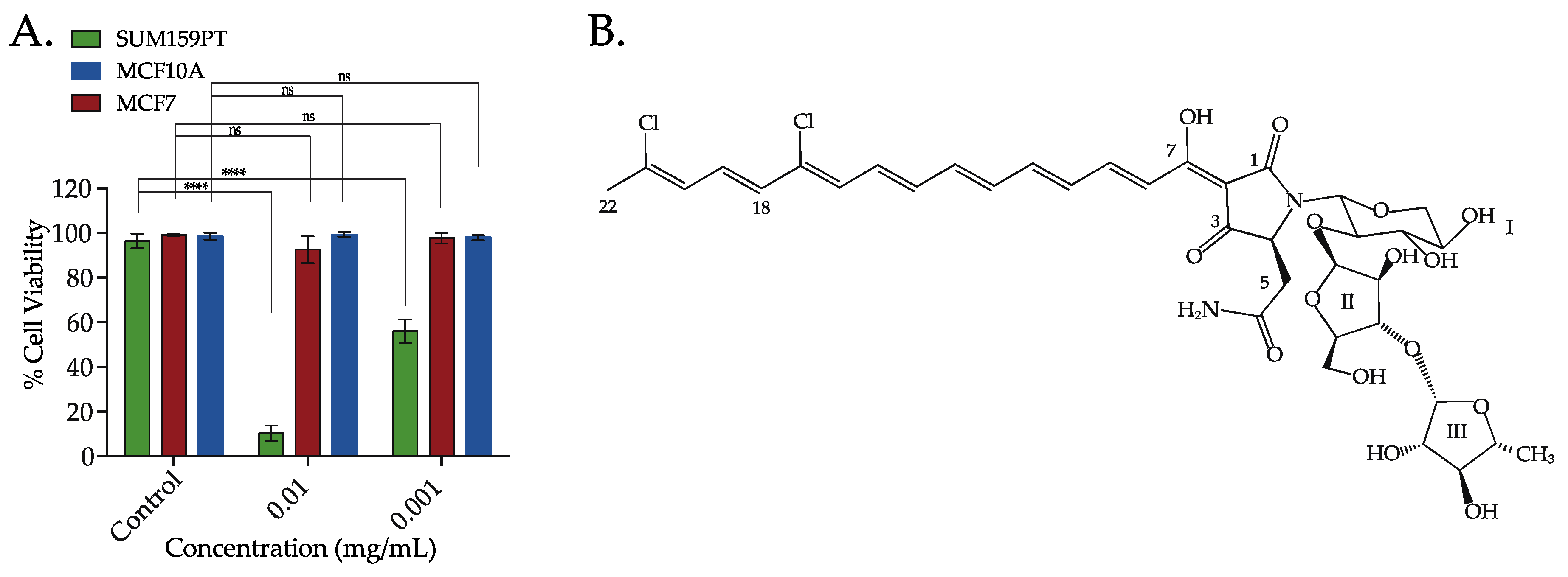
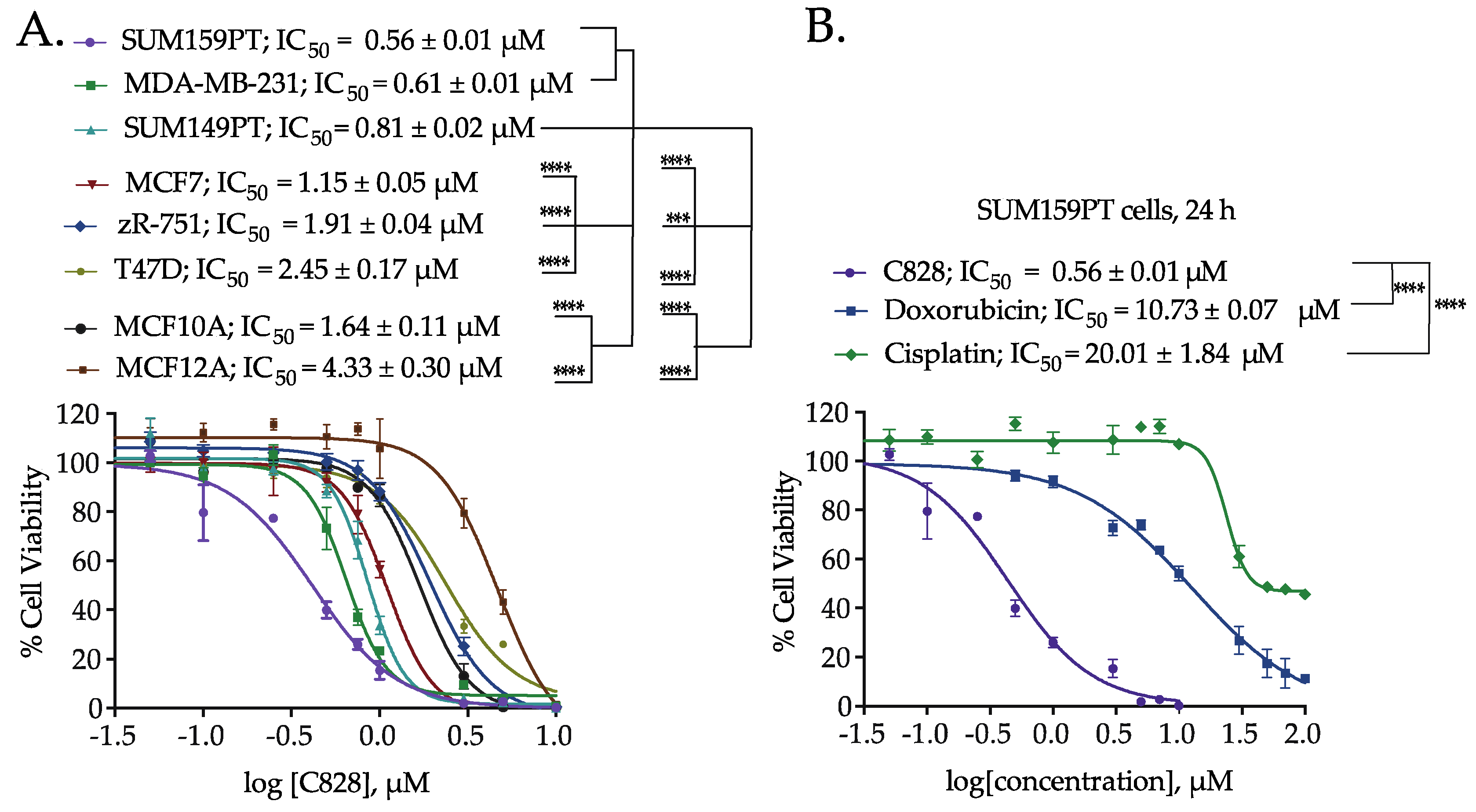
2.3. Aurantoside C Shows Preferential Cytotoxicity in TNBC Cells Compared to Non-TNBC Cells
2.4. Aurantoside C is More Efficient in Inducing Cell Death in TNBC Cells than Commonly Used Chemotherapeutic Drugs
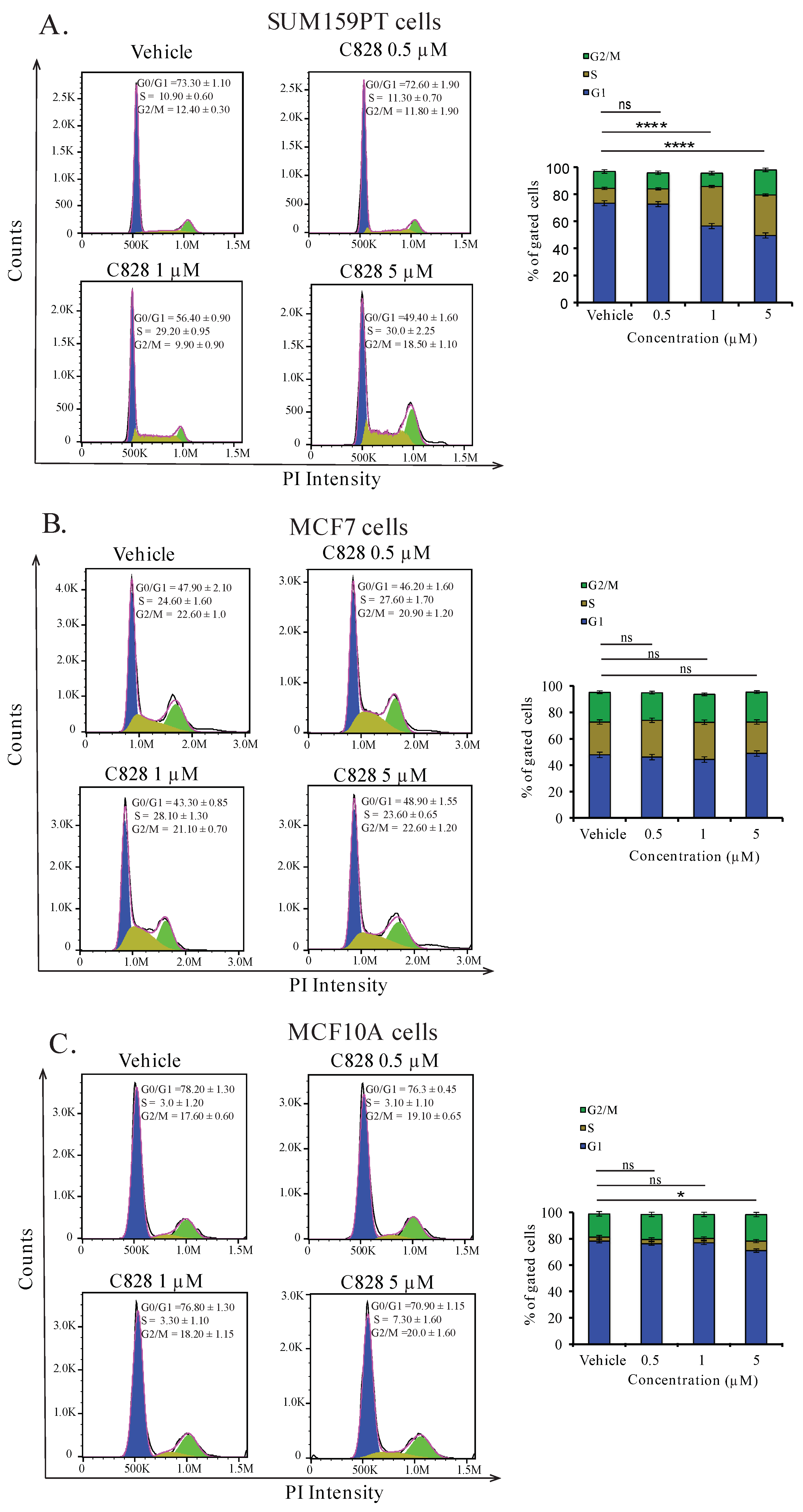
2.5. Aurantoside C Induces Accumulation of Cells at S-phase in TNBC Cells
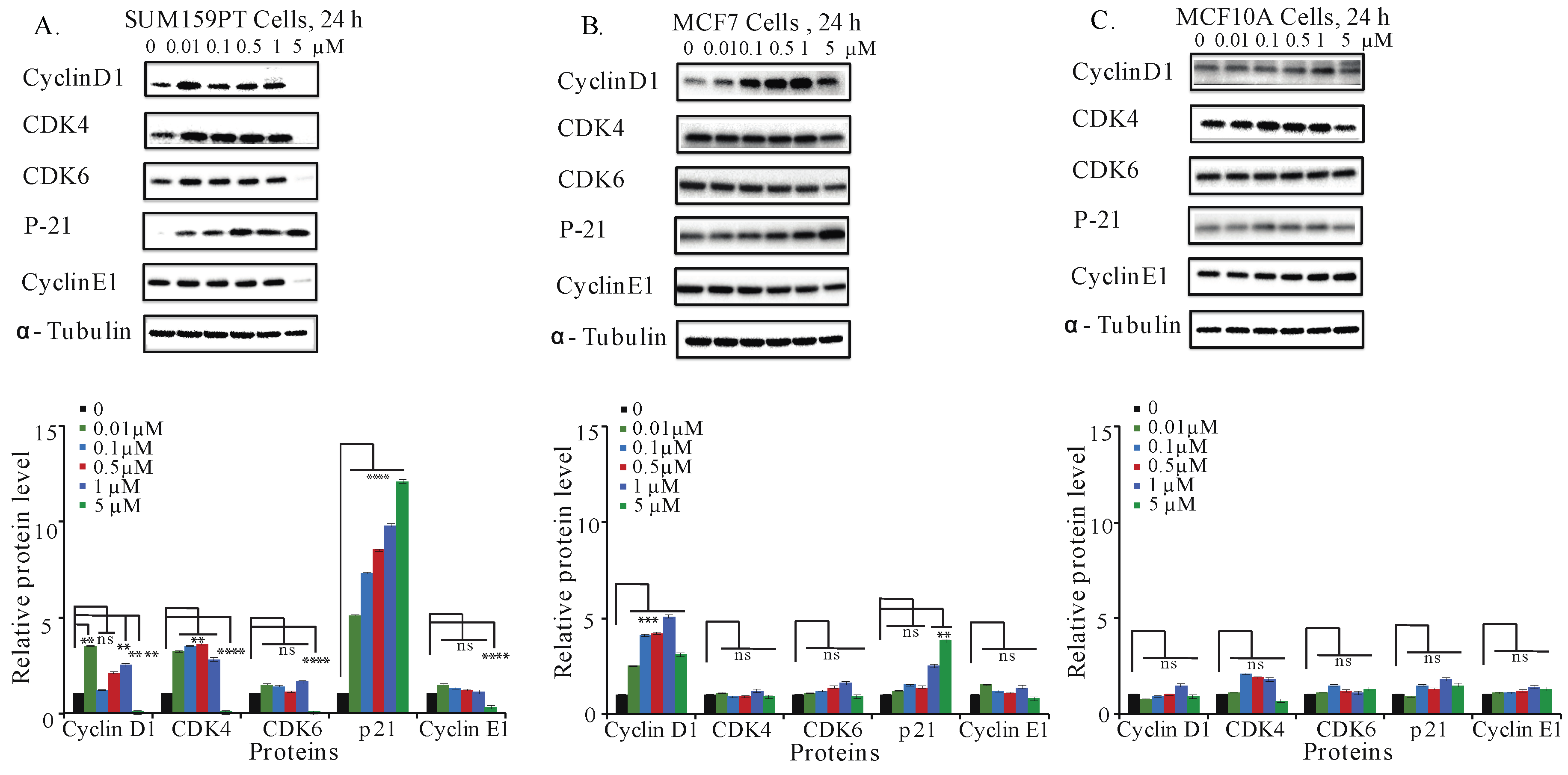
2.6. Aurantoside C Regulates Cell Cycle Related Proteins in TNBC Cells
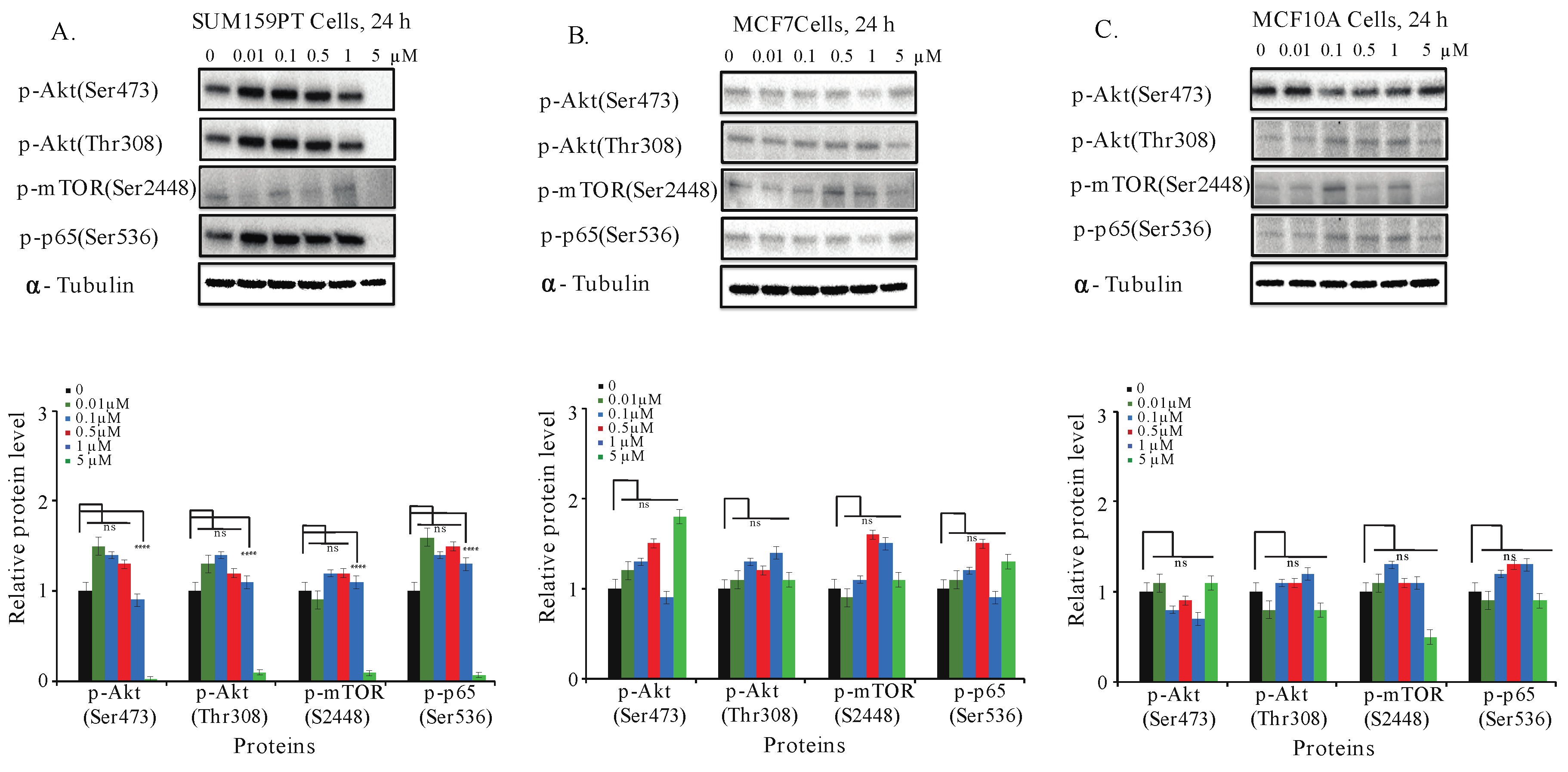
2.7. Aurantoside C Inhibits Phosphorylation of Akt/mTOR and NF-κB Signaling Pathways in TNBC Cells
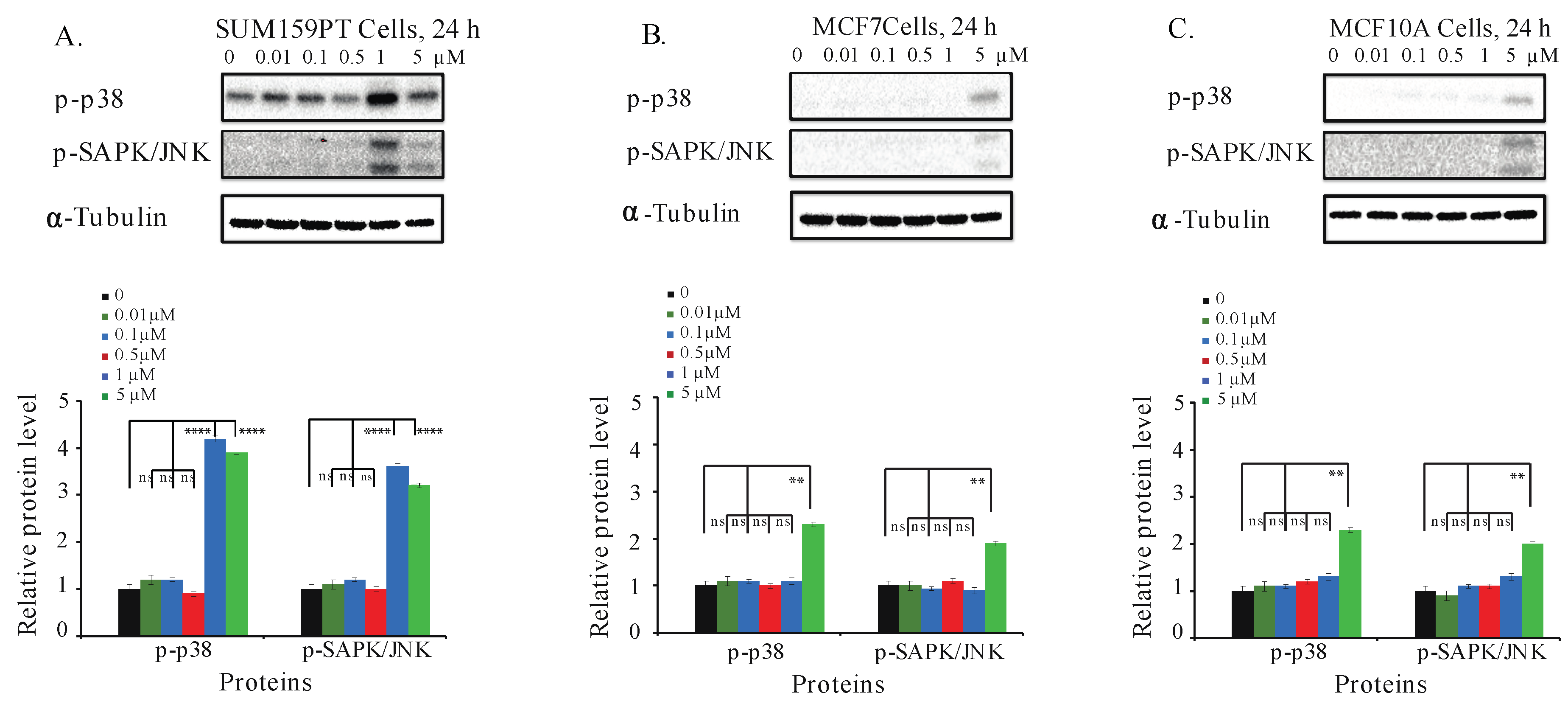
2.8. Aurantoside C Activates Phosphorylation of p38 MAPK and SAPK/JNK Signaling Pathways in TNBC Cells
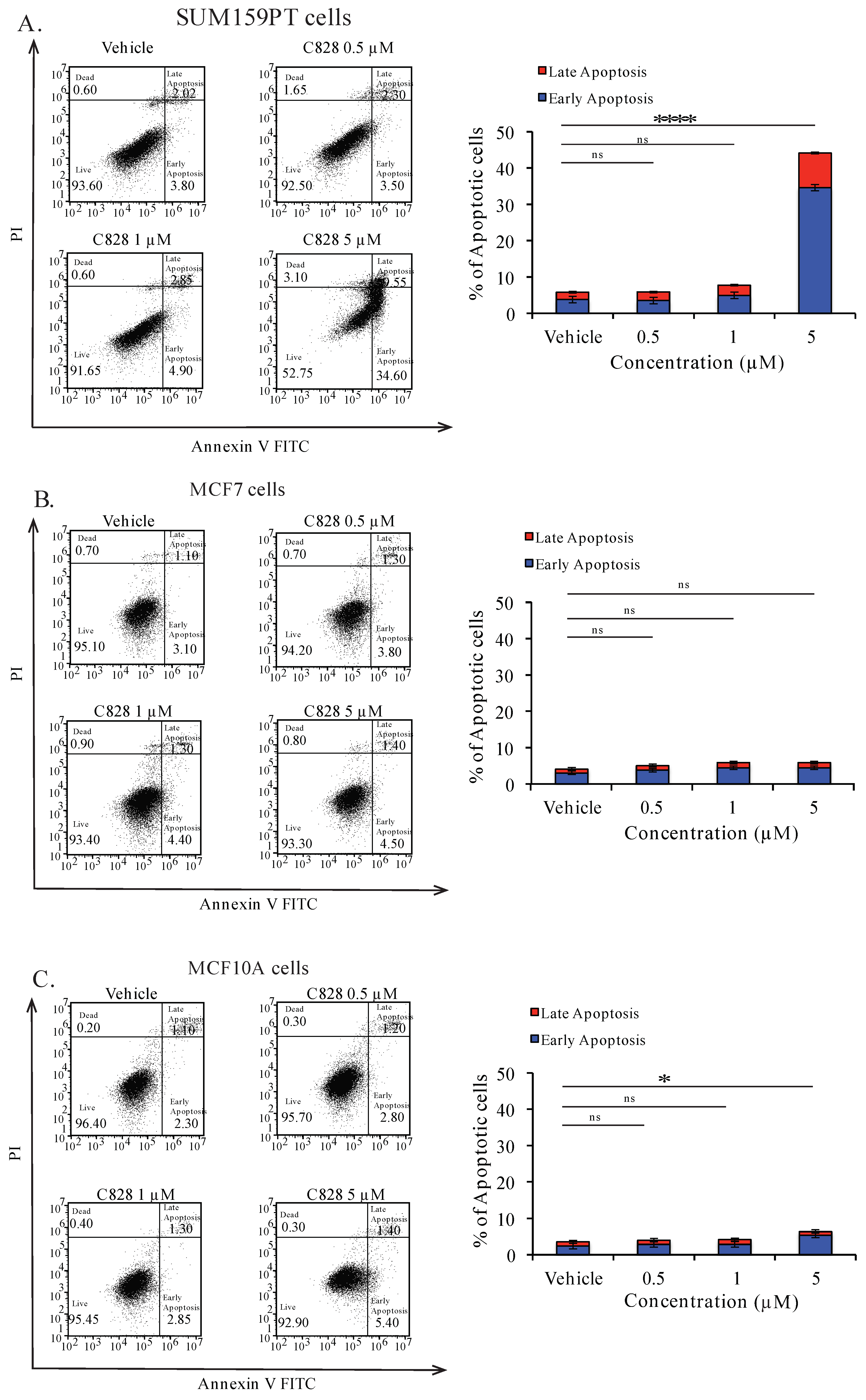
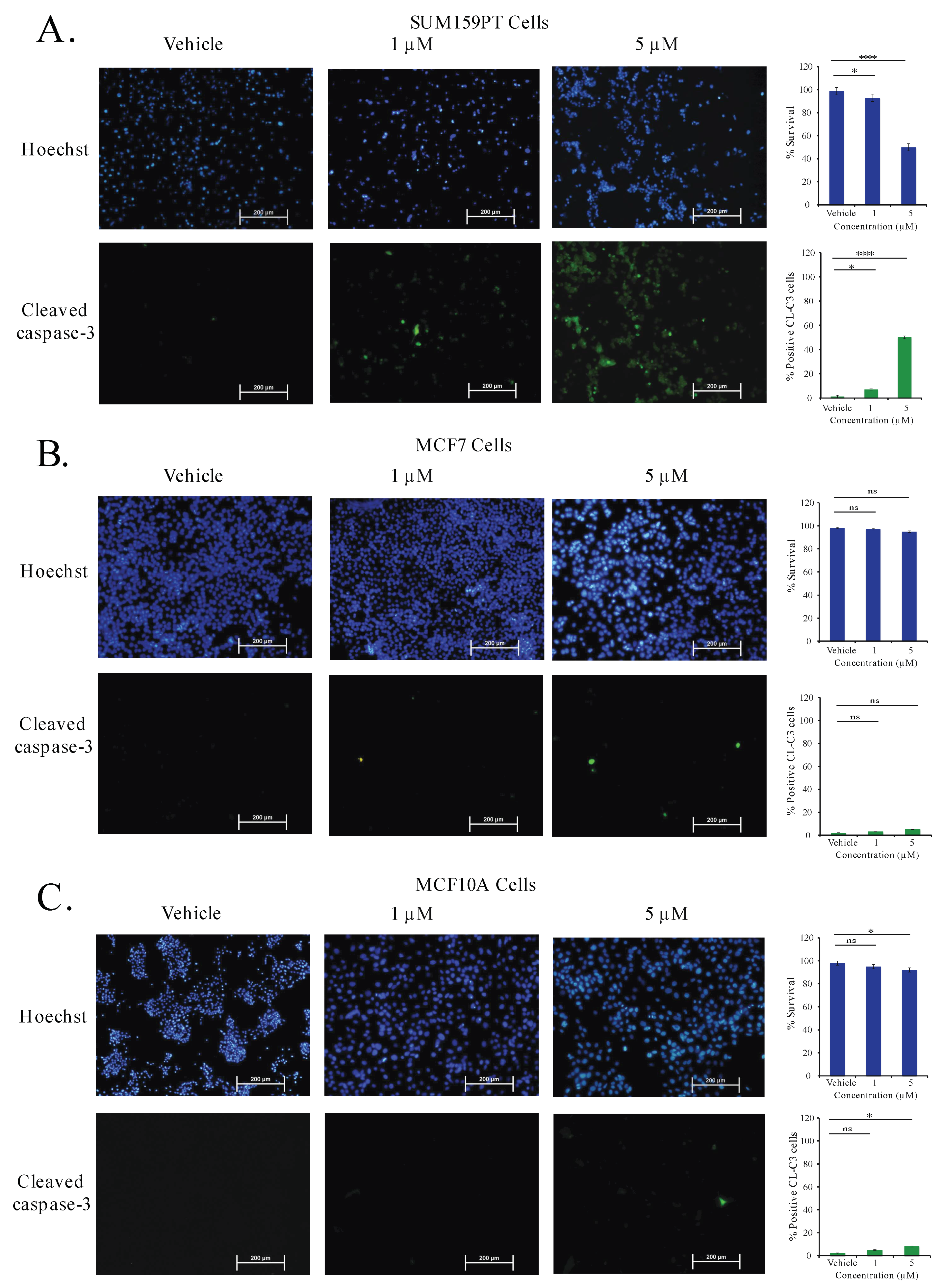
2.9. Aurantoside C Triggers Apoptosis in TNBC Cells
3. Discussion
4. Materials and Methods
4.1. General Experimental Procedures
4.2. Reagents
4.3. Details of Sponge Materials and Identification
4.4. Purification and Isolation of Aurantoside C (C828)
4.5. Cell Culture
4.6. Cell Viability Assay
4.7. Cell Cycle Analysis
4.8. Western Blot Analysis
4.9. Apoptosis Assay Using Annexin-V-PI-Binding Assay
4.10. Apoptosis Assay by Immunofluorescence
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- McArthur, H.L.; Mahoney, K.M.; Morris, P.G.; Patil, S.; Jacks, L.M.; Howard, J.; Norton, L.; Hudis, C.A. Adjuvant trastuzumab with chemotherapy is effective in women with small, node-negative, HER2-positive breast cancer. Cancer 2011, 117, 5461–5468. [Google Scholar] [CrossRef] [PubMed]
- Cheang, M.C.; Voduc, D.; Bajdik, C.; Leung, S.; McKinney, S.; Chia, S.K.; Perou, C.M.; Nielsen, T.O. Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin. Cancer Res. 2008, 14, 1368–1376. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Parker, J.S.; Karginova, O.; Fan, C.; Livasy, C.; Herschkowitz, J.I.; He, X.P.; Perou, C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010, 12, R68. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Perou, C.M. Deconstructing the molecular portraits of breast cancer. Mol. Oncol. 2011, 5, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M. Molecular stratification of triple-negative breast cancers. Oncologist 2010, 15 (Suppl. 5), 39–48. [Google Scholar] [CrossRef]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Mayer, I.A.; Abramson, V.G.; Lehmann, B.D.; Pietenpol, J.A. New strategies for triple-negative breast cancer--deciphering the heterogeneity. Clin. Cancer Res. 2014, 20, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Umemura, S.; Yoshida, S.; Ohta, Y.; Naito, K.; Osamura, R.Y.; Tokuda, Y. Increased phosphorylation of Akt in triple-negative breast cancers. Cancer Sci. 2007, 98, 1889–1892. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J. A gene transcription signature of the Akt/mTOR pathway in clinical breast tumors. Oncogene 2007, 26, 4648–4655. [Google Scholar] [CrossRef] [PubMed]
- Hay, N. The Akt-mTOR tango and its relevance to cancer. Cancer Cell 2005, 8, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Easton, J.B.; Houghton, P.J. mTOR and cancer therapy. Oncogene 2006, 25, 6436–6446. [Google Scholar] [CrossRef] [PubMed]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, B.T.; Smith, D.L.; Ram, P.T.; Lu, Y.; Mills, G.B. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discov. 2005, 4, 988–1004. [Google Scholar] [CrossRef] [PubMed]
- Biswas, D.K.; Shi, Q.; Baily, S.; Strickland, I.; Ghosh, S.; Pardee, A.B.; Iglehart, J.D. NF-KB activation in human breast cancer specimens and its role in cell proliferation and apoptosis. Proc. Natl. Acad. Sci. USA 2004, 101, 10137–10142. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.A.; Azoitei, N.; Baumann, B.; Grünert, S.; Sommer, A.; Pehamberger, H.; Kraut, N.; Beug, H.; Wirth, T. NF-κB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J. Clin. Investig. 2004, 114, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, N.; Ito, T.; Azuma, S.; Ito, E.; Honma, R.; Yanagisawa, Y.; Nishikawa, A.; Kawamura, M.; Imai, J.; Watanabe, S.; et al. Constitutive activation of nuclear factor-kappaB is preferentially involved in the proliferation of basal-like subtype breast cancer cell lines. Cancer Sci. 2009, 100, 1668–1674. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.J.; Pise-Masison, C.A.; Radonovich, M.F.; Park, H.U.; Brady, J.N. Activated AKT regulates NF-kappaB activation, p53 inhibition and cell survival in HTLV-1-transformed cells. Oncogene 2005, 24, 6719–6728. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Li, W.; Wang, Q.; Yang, H.S. AKT Activation by Pdcd4 Knockdown Up-Regulates Cyclin D1 Expression and Promotes Cell Proliferation. Genes Cancer 2011, 2, 818–828. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Giltnane, J.M.; Balko, J.M. Rationale for Targeting the Ras/MAPK Pathway in Triple-Negative Breast Cancer. Discov. Med. 2014, 17, 275–283. [Google Scholar] [PubMed]
- Cossa, G.; Gatti, L.; Cassinelli, G.; Lanzi, C.; Zaffaroni, N.; Perego, P. Modulation of Sensitivity to Antitumor Agents by Targeting the MAPK Survival Pathway. Curr. Pharm. Des. 2013, 19, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Penninger, J.M. Mitogen-activated protein kinases in apoptosis regulation. Oncogene 2004, 23, 2838–2849. [Google Scholar] [CrossRef] [PubMed]
- Hernandez Losa, J.; Parada Cobo, C.; Guinea Viniegra, J.; Sanchez-Arevalo Lobo, V.J.; Ramon y Cajal, S.; Sanchez-Prieto, R. Role of the p38 MAPK pathway in cisplatin-based therapy. Oncogene 2003, 22, 3998–4006. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, A.; Ridgway, L.D.; Korapati, A.L.; Zhang, Q.; Tian, L.; Wang, Y.; Siddik, Z.H.; Mills, G.B.; Claret, F.X. Sustained activation of JNK/p38 MAPK pathways in response to cisplatin leads to Fas ligand induction and cell death in ovarian carcinoma cells. J. Biol. Chem. 2003, 278, 19245–19256. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.L. Natural products in drug discovery. Drug Discov. Today 2008, 13, 894–901. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef] [PubMed]
- Simmons, T.L.; Andrianasolo, E.; McPhail, K.; Flatt, P.; Gerwick, W.H. Marine natural products as anticancer drugs. Mol. Cancer Ther. 2005, 4, 333–342. [Google Scholar] [PubMed]
- Capon, R.J. Marine Natural Products Chemistry: Past, Present, and Future. Aust. J. Chem. 2010, 63, 851–854. [Google Scholar] [CrossRef]
- Schwartsmann, G.; Brondani da Rocha, A.; Berlinck, R.G.; Jimeno, J. Marine organisms as a source of new anticancer agents. Lancet Oncol. 2001, 2, 221–225. [Google Scholar] [CrossRef]
- Hirata, Y.; Uemura, D. Halichondrins-Antitumor Polyether Macrolides from a Marine Sponge. Pure Appl. Chem. 1986, 58, 701–710. [Google Scholar] [CrossRef]
- Indumathy, S.; Dass, C.R. Finding chemo: The search for marine-based pharmaceutical drugs active against cancer. J. Pharm. Pharmacol. 2013, 65, 1280–1301. [Google Scholar] [CrossRef] [PubMed]
- Mehbub, M.F.; Lei, J.; Franco, C.; Zhang, W. Marine sponge derived natural products between 2001 and 2010: Trends and opportunities for discovery of bioactives. Mar. Drugs 2014, 12, 4539–4577. [Google Scholar] [CrossRef] [PubMed]
- Sipkema, D.; Franssen, M.C.; Osinga, R.; Tramper, J.; Wijffels, R.H. Marine sponges as pharmacy. Mar. Biotechnol. 2005, 7, 142–162. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, S.; Sorolla, A.; Fromont, J.; Blancafort, P.; Flematti, G.R. Crambescidin 800, Isolated from the Marine Sponge Monanchora viridis, Induces Cell Cycle Arrest and Apoptosis in Triple-Negative Breast Cancer Cells. Mar. Drugs 2018, 16, 53. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Schmitz, F.J.; Qiu, F.; Kelly-Borges, M. Aurantoside C, a new tetramic acid glycoside from the sponge Homophymia conferta. J. Nat. Prod. 1999, 62, 170–172. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, B.L.; Nayler, S.; Demetriou, G.S.; Moodley, S.D.; Benn, C.A. Triple Negative Breast Cancer Pathologic Diagnosis and Current Chemotherapy Treatment Options. Oncol. Hematol. Rev. 2014, 10, 25–32. [Google Scholar]
- Hudis, C.A.; Gianni, L. Triple-negative breast cancer: An unmet medical need. Oncologist 2011, 16 (Suppl. 1), 1–11. [Google Scholar] [CrossRef]
- Kalimutho, M.; Parsons, K.; Mittal, D.; Lopez, J.A.; Srihari, S.; Khanna, K.K. Targeted Therapies for Triple-Negative Breast Cancer: Combating a Stubborn Disease. Trends Pharmacol. Sci. 2015, 36, 822–846. [Google Scholar] [CrossRef] [PubMed]
- Sorolla, A.; Yeramian, A.; Valls, J.; Dolcet, X.; Bergada, L.; Llombart-Cussac, A.; Marti, R.M.; Matias-Guiu, X. Blockade of NFkappaB activity by Sunitinib increases cell death in Bortezomib-treated endometrial carcinoma cells. Mol. Oncol. 2012, 6, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Wang, K.; Zhang, K.; Zhang, T.; Yin, Y.; Xu, F. Ziyuglycoside I Inhibits the Proliferation of MDA-MB-231 Breast Carcinoma Cells through Inducing p53-Mediated G2/M Cell Cycle Arrest and Intrinsic/Extrinsic Apoptosis. Int. J. Mol. Sci. 2016, 17, 1903. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.H.; Chiang, E.P.I.; Chao, C.Y.; Rodriguez, R.L.; Chou, P.Y.; Tsai, S.Y.; Pai, M.H.; Tang, F.Y. Dual Inhibition of Key Proliferation Signaling Pathways in Triple-Negative Breast Cancer Cells by a Novel Derivative of Taiwanin A. Mol. Cancer Ther. 2017, 16, 480–493. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Hannon, G.J.; Zhang, H.; Casso, D.; Kobayashi, R.; Beach, D. P21 Is a Universal Inhibitor of Cyclin Kinases. Nature 1993, 366, 701–704. [Google Scholar] [CrossRef] [PubMed]
- Ogryzko, V.V.; Wong, P.; Howard, B.H. WAF1 retards S-phase progression primarily by inhibition of cyclin-dependent kinases. Mol. Cell. Biol. 1997, 17, 4877–4882. [Google Scholar] [CrossRef] [PubMed]
- Marzi, L.; Combes, E.; Vie, N.; Ayrolles-Torro, A.; Tosi, D.; Desigaud, D.; Perez-Gracia, E.; Larbouret, C.; Montagut, C.; Iglesias, M.; et al. FOXO3a and the MAPK p38 are activated by cetuximab to induce cell death and inhibit cell proliferation and their expression predicts cetuximab efficacy in colorectal cancer. Br. J. Cancer 2016, 115, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Dickens, M.; Raingeaud, J.; Davis, R.J.; Greenberg, M.E. Opposing Effects of Erk and Jnk-P38 Map Kinases on Apoptosis. Science 1995, 270, 1326–1331. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.M.; Feng, C.W.; Chen, S.Y.; Hsieh, H.Y.; Chen, Y.H.; Hsu, C.D. Cyproheptadine, an antihistaminic drug, inhibits proliferation of hepatocellular carcinoma cells by blocking cell cycle progression through the activation of P38 MAP kinase. BMC Cancer 2015, 15, 134. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.W.; Lin, A.W. Apoptosis in cancer. Carcinogenesis 2000, 21, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Evan, G.I.; Vousden, K.H. Proliferation, cell cycle and apoptosis in cancer. Nature 2001, 411, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Fesik, S.W. Promoting apoptosis as a strategy for cancer drug discovery. Nat. Rev. Cancer 2005, 5, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Mann, J. Natural products in cancer chemotherapy: Past, present and future. Nat. Rev. Cancer 2002, 2, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, S.; Fusetani, N.; Kato, Y.; Hirota, H. Aurantosides A and B: Cytotoxic Tetramic Acid Glycosides from the Marine Sponge Theonella sp. J. Am. Chem. Soc. 1991, 113, 9690–9692. [Google Scholar] [CrossRef]
- Sata, N.U.; Matsunaga, S.; Fusetani, N.; van Soest, R.W.M. Aurantosides D, E, and F: New antifungal tetramic acid glycosides from the marine sponge Siliquariaspongia japonica. J. Nat. Prod. 1999, 62, 969–971. [Google Scholar] [CrossRef] [PubMed]
- Ratnayake, A.S.; Davis, R.A.; Harper, M.K.; Veltri, C.A.; Andjelic, C.D.; Barrows, L.R.; Ireland, C.M. Aurantosides G, H, and I: Three new tetramic acid glycosides from a Papua New Guinea Theonella swinhoei. J. Nat. Prod. 2005, 68, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Angawi, R.F.; Bavestrello, G.; Calcinai, B.; Dien, H.A.; Donnarumma, G.; Tufano, M.A.; Paoletti, I.; Grimaldi, E.; Chianese, G.; Fattorusso, E.; et al. Aurantoside J: A New Tetramic Acid Glycoside from Theonella swinhoei. Insights into the Antifungal Potential of Aurantosides. Mar. Drugs 2011, 9, 2809–2817. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Subramani, R.; Feussner, K.D.; Aalbersberg, W. Aurantoside K, a New Antifungal Tetramic Acid Glycoside from a Fijian Marine Sponge of the Genus Melophlus. Mar. Drugs 2012, 10, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Podo, F.; Buydens, L.M.; Degani, H.; Hilhorst, R.; Klipp, E.; Gribbestad, I.S.; Van Huffel, S.; van Laarhoven, H.W.; Luts, J.; Monleon, D.; et al. Triple-negative breast cancer: Present challenges and new perspectives. Mol. Oncol. 2010, 4, 209–229. [Google Scholar] [CrossRef] [PubMed]
- El Guerrab, A.; Bamdad, M.; Kwiatkowski, F.; Bignon, Y.J.; Penault-Llorca, F.; Aubel, C. Anti-EGFR monoclonal antibodies and EGFR tyrosine kinase inhibitors as combination therapy for triple-negative breast cancer. Oncotarget 2016, 7, 73618–73637. [Google Scholar] [CrossRef] [PubMed]
- Tanei, T.; Choi, D.S.; Rodriguez, A.A.; Liang, D.H.; Dobrolecki, L.; Ghosh, M.; Landis, M.D.; Chang, J.C. Antitumor activity of Cetuximab in combination with Ixabepilone on triple negative breast cancer stem cells. Breast Cancer Res. 2016, 18, 6. [Google Scholar] [CrossRef] [PubMed]
- Corkery, B.; Crown, J.; Clynes, M.; O‘Donovan, N. Epidermal growth factor receptor as a potential therapeutic target in triple-negative breast cancer. Ann. Oncol. 2009, 20, 862–867. [Google Scholar] [CrossRef] [PubMed]
- Hydbring, P.; Malumbres, M.; Sicinski, P. Non-canonical functions of cell cycle cyclins and cyclin-dependent kinases. Nat. Rev. Mol. Cell Biol. 2016, 17, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Koff, A.; Giordano, A.; Desai, D.; Yamashita, K.; Harper, J.W.; Elledge, S.; Nishimoto, T.; Morgan, D.O.; Franza, B.R.; Roberts, J.M. Formation and Activation of a Cyclin E-Cdk2 Complex during the G(1)-Phase of the Human Cell-Cycle. Science 1992, 257, 1689–1694. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Choi, K.M.; Kim, W.; Jeon, Y.S.; Lee, Y.M.; Hong, J.T.; Yun, Y.P.; Yoo, H.S. Hinokitiol inhibits cell growth through induction of S-phase arrest and apoptosis in human colon cancer cells and suppresses tumor growth in a mouse xenograft experiment. J. Nat. Prod. 2013, 76, 2195–2202. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhang, Q.; Peng, X.; Zhou, C.; Zhong, Y.; Chen, X.; Qiu, Y.; Jin, M.; Gong, M.; Kong, D. Stellettin B Induces G1 Arrest, Apoptosis and Autophagy in Human Non-small Cell Lung Cancer A549 Cells via Blocking PI3K/Akt/mTOR Pathway. Sci. Rep. 2016, 6, 27071. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Zhou, Q.X.; Zhang, L.; Zhong, Y.X.; Fan, G.W.; Zhang, Z.; Wang, R.; Jin, M.H.; Qiu, Y.L.; Kong, D.X. Stellettin B induces apoptosis in human chronic myeloid leukemia cells via targeting PI3K and Stat5. Oncotarget 2017, 8, 28906–28921. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.A.; Zhou, Q.; Guo, W.Z.; Qiu, Y.; Wang, R.; Jin, M.; Zhang, W.; Li, K.; Yamori, T.; Dan, S.; Kong, D. In vitro antitumor activity of stellettin B, a triterpene from marine sponge Jaspis stellifera, on human glioblastoma cancer SF295 cells. Mar. Drugs 2014, 12, 4200–4213. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, W.; Ma, Q.; Li, J.; Zhang, D.; Liu, Z.G.; Rustgi, A.K.; Huang, C. Cyclin D1 induction through IkappaB kinase beta/nuclear factor-kappaB pathway is responsible for arsenite-induced increased cell cycle G1-S phase transition in human keratinocytes. Cancer Res. 2005, 65, 9287–9293. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.K.; Yeh, S.Y.; Kang, H.Y.; Chang, C.S. Akt suppresses androgen-induced apoptosis by phosphorylating and inhibiting androgen receptor. Proc. Natl. Acad. Sci. USA 2001, 98, 7200–7205. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and regulation of apoptosis. Biochim. Biophys. Acta 2011, 1813, 1978–1986. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Cao, X.X.; Chen, Q.; Zhu, T.F.; Zhu, H.G.; Zheng, L. DIXDC1 targets p21 and cyclin D1 via PI3K pathway activation to promote colon cancer cell proliferation. Cancer Sci. 2009, 100, 1801–1808. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.H.P.; Liao, Y.; Xia, W.Y.; Spohn, B.; Lee, N.H.; Hung, M.C. Cytoplasmic localization of p21(Cip1/WAF1) by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat. Cell Biol. 2001, 3, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Spurlock, C.F.; Tossberg, J.T.; Matlock, B.K.; Olsen, N.J.; Aune, T.M. Methotrexate Inhibits NF-kappa B Activity Via Long Intergenic (Noncoding) RNA-p21 Induction. Arthritis Rheumatol. 2014, 66, 2947–2957. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cheng, Y.Y.; Qu, W.X.; Sun, Y.; Wang, Z.P.; Wang, H.Z.; Tian, B.Q. Fisetin, a Dietary Flavonoid, Induces Cell Cycle Arrest and Apoptosis through Activation of p53 and Inhibition of NF-Kappa B Pathways in Bladder Cancer Cells. Basic Clin. Pharmacol. Toxicol. 2011, 108, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Zhang, C.; Gao, C.Y.; Ma, T.; Zhang, H.; Zhou, M.M.; Yang, Y.W.; Yang, L.; Kong, L.Y. Anti-proliferation of triple-negative breast cancer cells with physagulide P: ROS/JNK signaling pathway induces apoptosis and autophagic cell death. Oncotarget 2017, 8, 64032–64049. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, X.; Wu, T.; Li, B.; Liu, T.; Wang, R.; Liu, Q.; Liu, Z.; Gong, Y.; Shao, C. Isoliensinine induces apoptosis in triple-negative human breast cancer cells through ROS generation and p38 MAPK/JNK activation. Sci. Rep. 2015, 5, 12579. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.Y.; Mercer, S.E.; Ewton, D.Z.; Yan, Z.; Jin, K.; Friedman, E. The stress-activated protein kinases p38 alpha and JNK1 stabilize p21(Cip1) by phosphorylation. J. Biol. Chem. 2002, 277, 29792–29802. [Google Scholar] [CrossRef] [PubMed]
- Perego, P.; Hempel, G.; Linder, S.; Bradshaw, T.D.; Larsen, A.K.; Peters, G.J.; Phillips, R.M. Cellular pharmacology studies of anticancer agents: Recommendations from the EORTC-PAMM group. Cancer Chemother. Pharmacol. 2018, 81, 427–441. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Cell Lines | IC50 (µM) |
|---|---|---|
| C828 | SUM159PT | 0.56 ± 0.01 |
| MDA-MB-231 | 0.61 ± 0.01 | |
| SUM149PT | 0.81 ± 0.02 | |
| C800 | SUM159PT | 3.42 ± 0.07 |
| MDA-MB-231 | 5.00 ± 0.57 | |
| SUM149PT | 6.02 ± 0.11 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shrestha, S.; Sorolla, A.; Fromont, J.; Blancafort, P.; Flematti, G.R. Aurantoside C Targets and Induces Apoptosis in Triple Negative Breast Cancer Cells. Mar. Drugs 2018, 16, 361. https://doi.org/10.3390/md16100361
Shrestha S, Sorolla A, Fromont J, Blancafort P, Flematti GR. Aurantoside C Targets and Induces Apoptosis in Triple Negative Breast Cancer Cells. Marine Drugs. 2018; 16(10):361. https://doi.org/10.3390/md16100361
Chicago/Turabian StyleShrestha, Sumi, Anabel Sorolla, Jane Fromont, Pilar Blancafort, and Gavin R. Flematti. 2018. "Aurantoside C Targets and Induces Apoptosis in Triple Negative Breast Cancer Cells" Marine Drugs 16, no. 10: 361. https://doi.org/10.3390/md16100361
APA StyleShrestha, S., Sorolla, A., Fromont, J., Blancafort, P., & Flematti, G. R. (2018). Aurantoside C Targets and Induces Apoptosis in Triple Negative Breast Cancer Cells. Marine Drugs, 16(10), 361. https://doi.org/10.3390/md16100361