Characterization of Properties and Transglycosylation Abilities of Recombinant α-Galactosidase from Cold-Adapted Marine Bacterium Pseudoalteromonas KMM 701 and Its C494N and D451A Mutants

Abstract
1. Introduction
2. Results
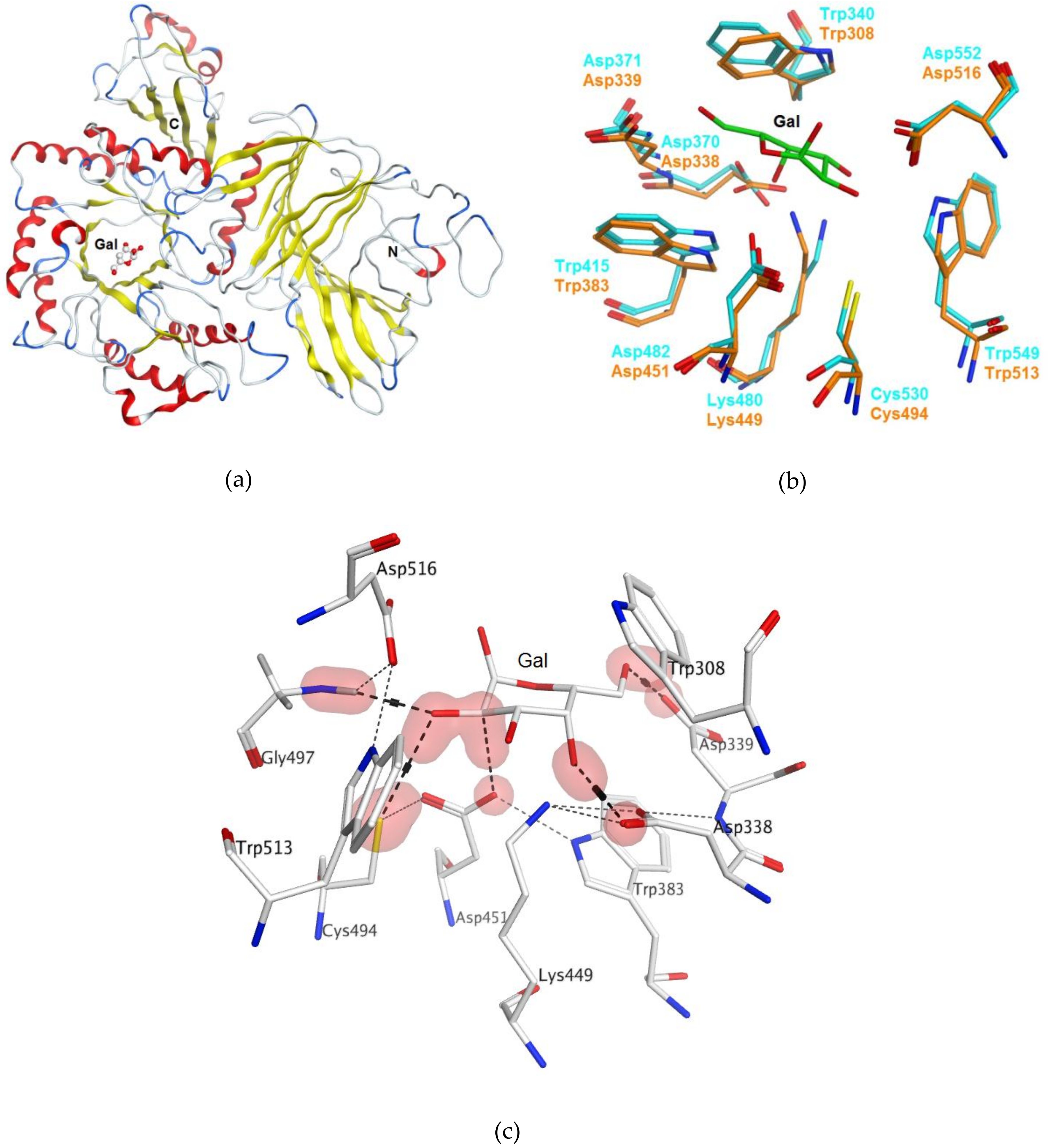
2.1. Bioinformatics Analysis and Homology Modeling of α-PsGal Protein 3D-Structure
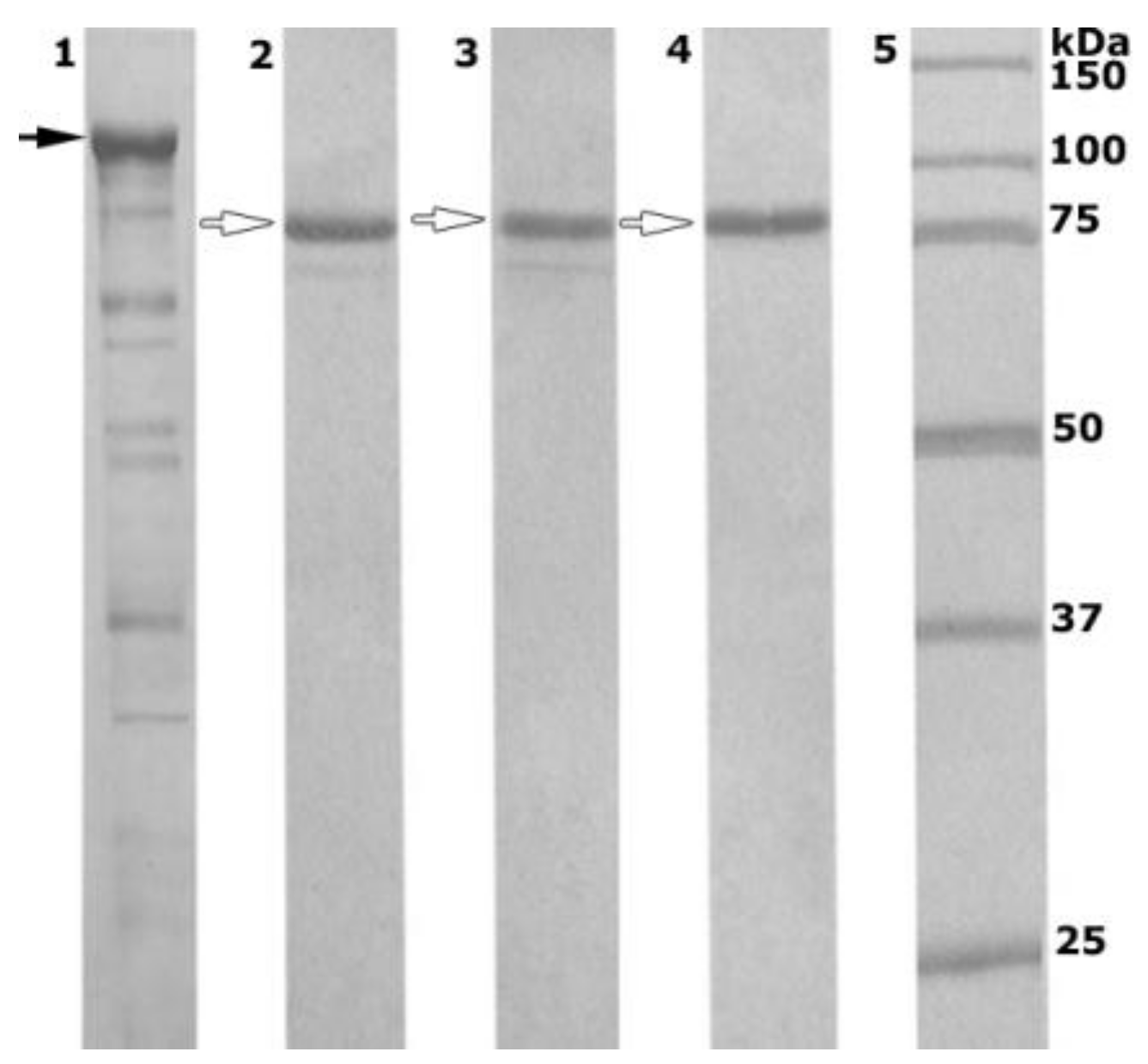
2.2. Enzyme Production and Purification
2.3. Properties of Recombinant Wild-Type α-PsGal and Mutant C494N
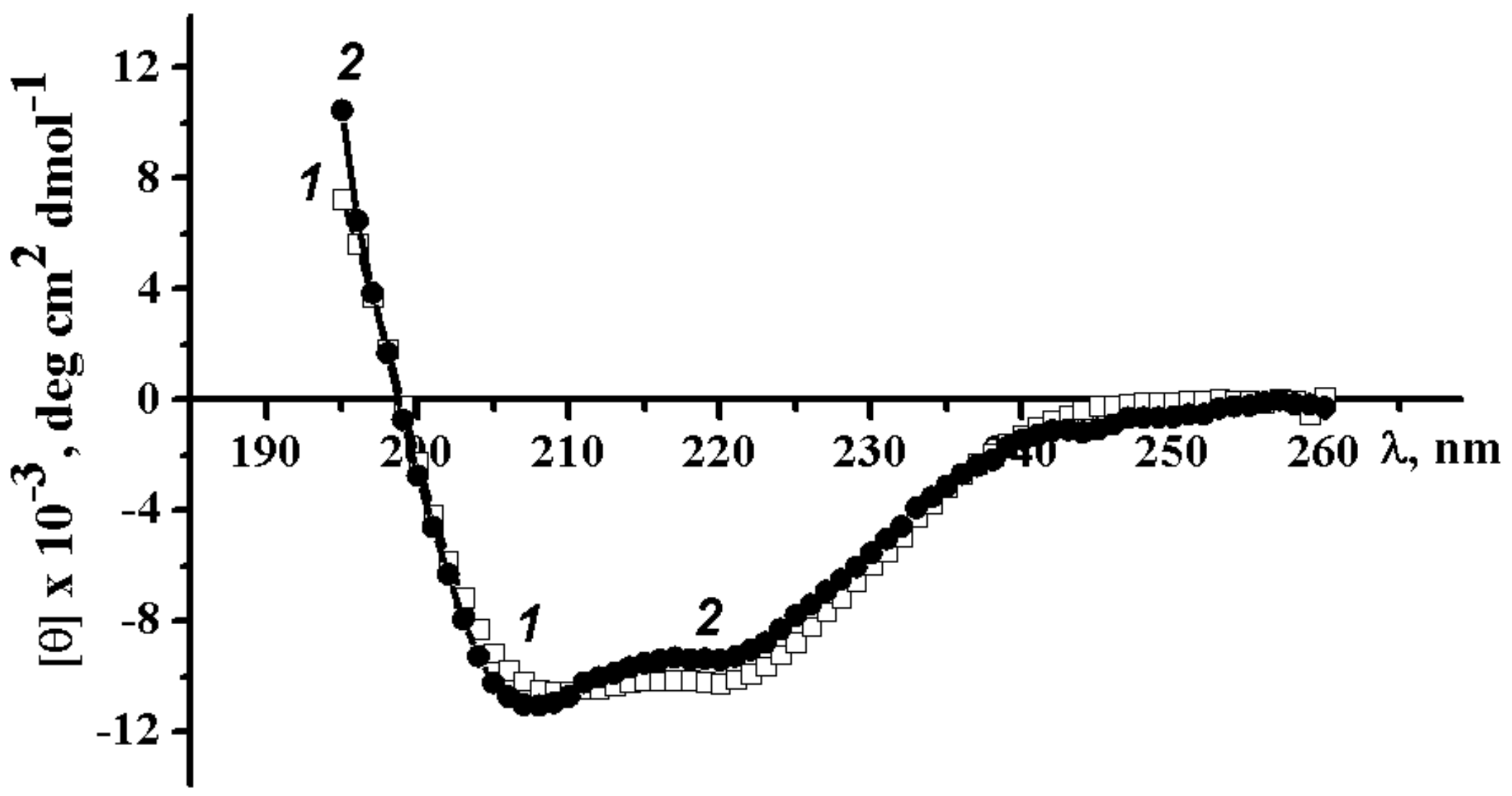
2.3.1. Circular Dichroism Spectra of Wild-Type α-PsGal and Mutant C494N
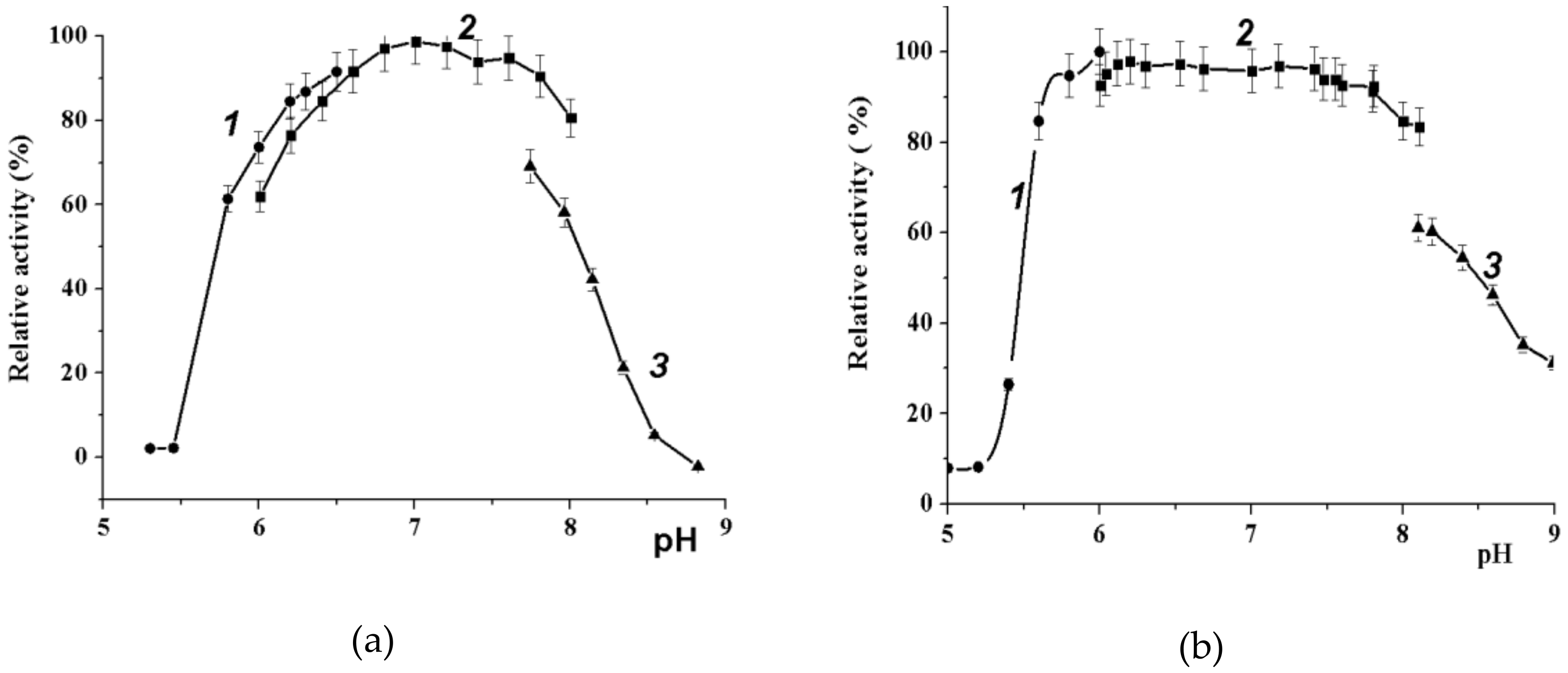
2.3.2. Effect of pH on Wild-Type α-PsGal and C494N Mutant Activities
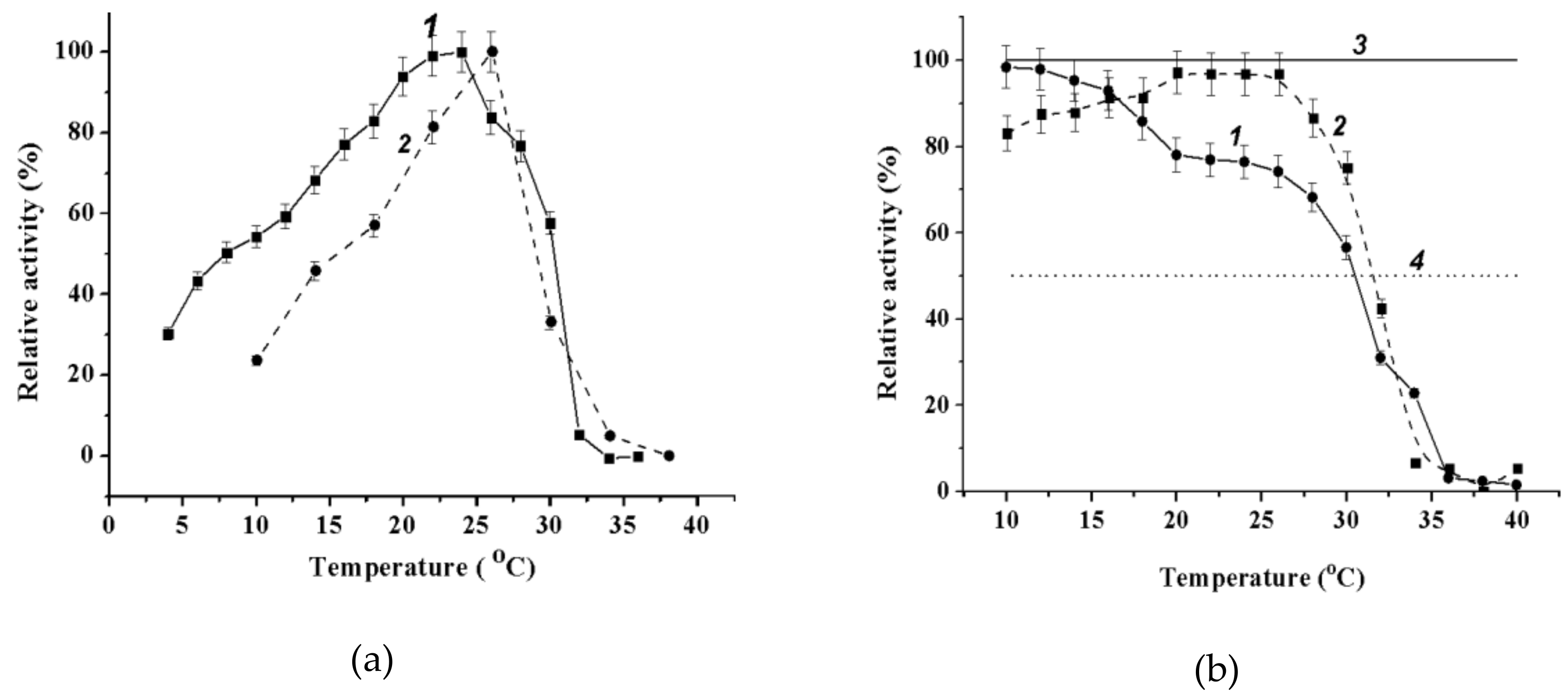
2.3.3. Effect of Temperature on Wild-Type α-PsGal and Mutant C494N Activities
2.3.4. Kinetic Parameters of Catalytic Reactions for Wild-Type α-PsGal and Mutant C494N
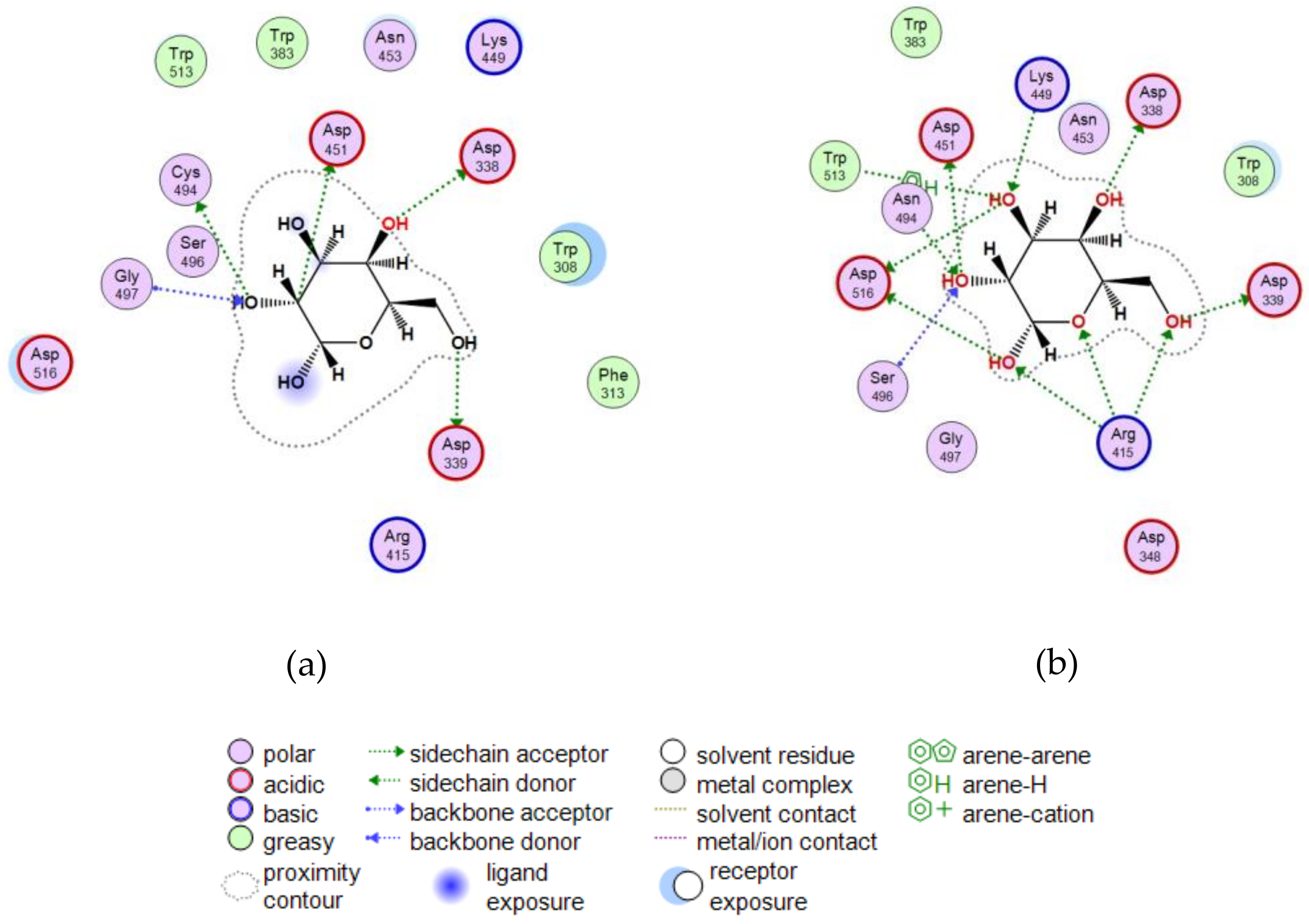
2.3.5. Theoretical Model of the D-Gal Complexes with Wild α-PsGal and Mutant C494N
2.4. Acceptor Specificity of Recombinant α-PsGal
2.5. Production of Transglysolylation Reactions
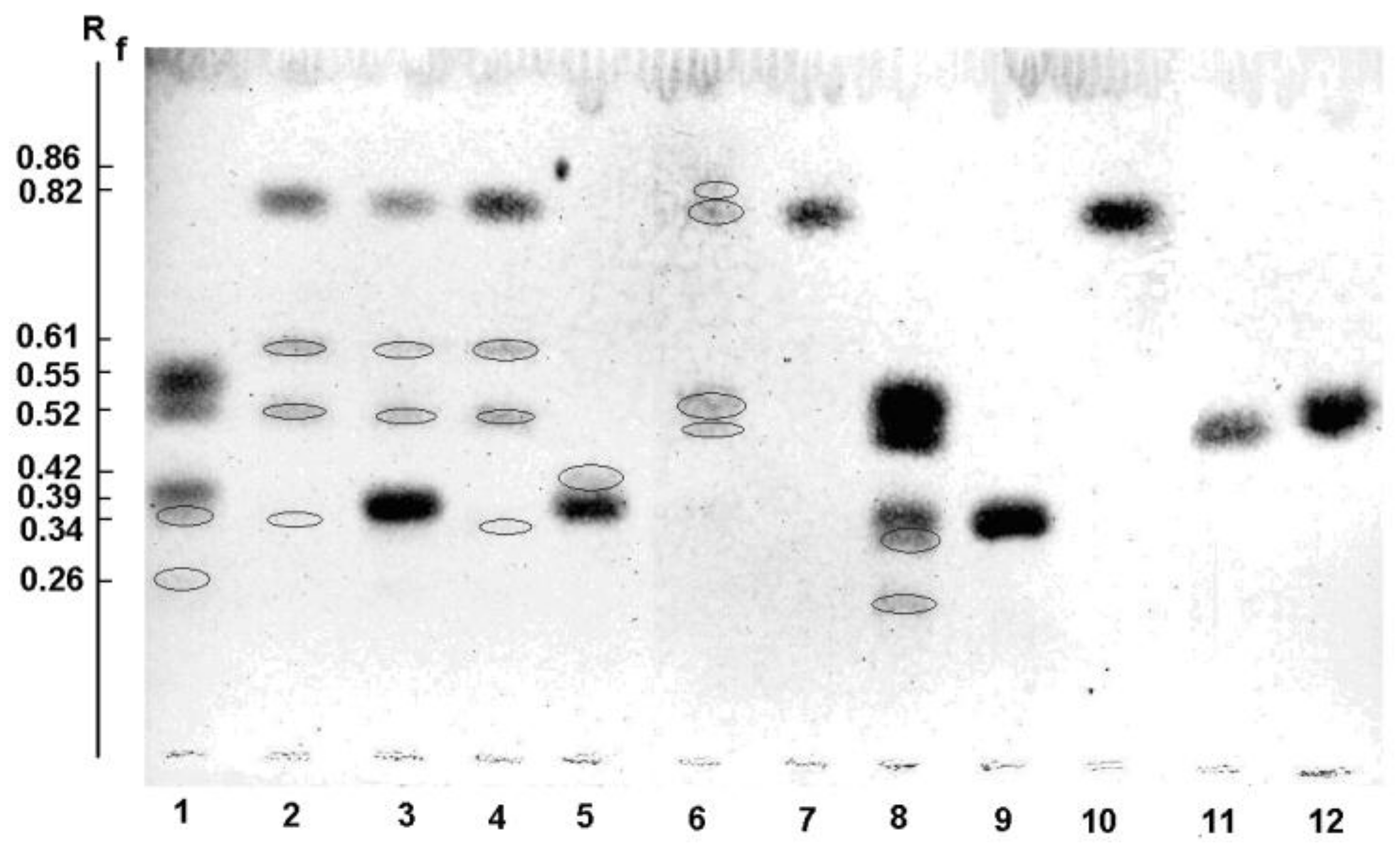
2.5.1. Thin Layer Chromatography (TLC)
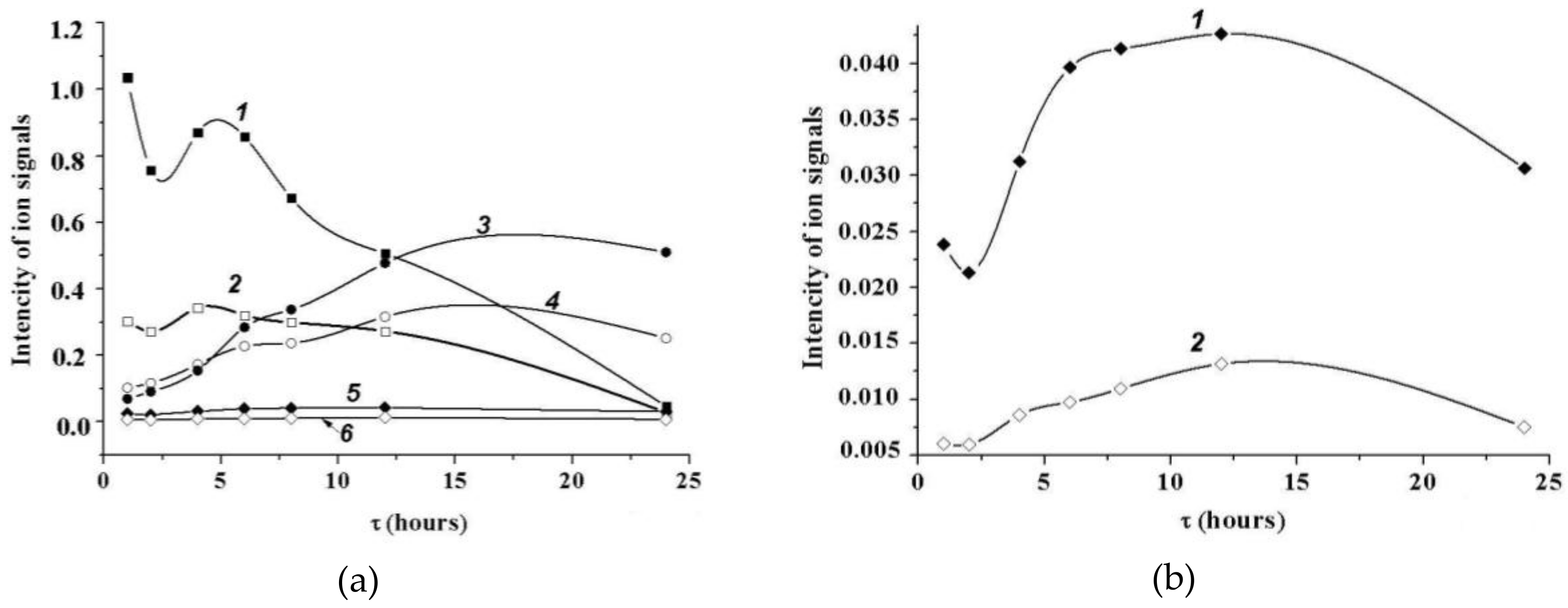
2.5.2. MALDI Mass Spectrometry
2.5.3. NMR Spectroscopy
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Homology Model of α-PsGal 3D Structure
4.3. Production of Recombinant Enzymes
- (1)
- D451A dir 5′-TTAAGTACATTAAATGGGCTATGAACCGCGA-3′ D451A rev 5′-GTTAATATCGCGGTTCATAGCCCATTTAATG-3′
- (2)
- C494N dir 5′-AGGGCTTGAAATAGAAAGCAATTCGTCAGGTGG-3′ C494N rev 5′-ACGTGCACCACCTGACGAATTGCTTTCTATTTC-3′
4.4. Enzyme and Protein Essays
4.4.1. Circular Dichroism Spectra
4.4.2. UV Absorption Spectra
4.5. Effect of pH and Temperature
4.6. Determination of Kinetic Parameters
4.7. Transglycosylation
4.7.1. Acceptor Specificity of Transglycosylation
4.7.2. Transglycosylation Using Heavy-Oxygen Water (H218O)
4.7.3. Identification of Transglycosylation Products
4.7.4. Thin-Layer Chromatography Analysis
4.7.5. Mass Spectrometry Analysis
4.7.6. NMR Spectroscopy Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef] [PubMed]
- Henrissat, B.; Callebaut, I.; Fabrega, S.; Lehn, P.; Mornon, J.P.; Davies, G. Conserved catalytic machinery and the prediction of a common fold for several families of glycosyl hydrolases. Proc. Natl. Acad. Sci. USA 1995, 92, 7090–7094. [Google Scholar] [CrossRef] [PubMed]
- Koshland, D.E., Jr. Stereochemistry and the mechanism of enzymatic reactions. Biol. Rev. Camb. Phil. Soc. 1953, 28, 416–436. [Google Scholar] [CrossRef]
- Bakunina, I.Y.; Balabanova, L.A.; Pennacchio, A.; Trincone, A. Hooked on α-d-galactosidases: from biomedicine to enzymatic synthesis. Crit. Rev. Biotechnol. 2016, 36, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, E.P.; Bakunina, I.Y.; Nedashkovskaya, O.I.; Gorshkova, N.M.; Mikhailov, V.V.; Elyakova, L.A. Incidence of marine microorganisms producing β-N-acetylglucosaminidases, α-d-galactosidases and α-N-acetylgalactosaminidases. Rus. J. Mar. Biol. 1998, 24, 365–372. [Google Scholar]
- Bakunina, I.Y.; Nedashkovskaya, O.I.; Alekseeva, S.A.; Ivanova, E.P.; Romanenko, L.A.; Gorshkova, N.M.; Isakov, V.V.; Zvyagintseva, T.N.; Mikhailov, V.V. Degradation of fucoidan by the marine proteobacterium Pseudoalteromonas citrea. Microbiology 2002, 71, 41–47. [Google Scholar] [CrossRef]
- Bakunina, I.Y.; Ivanova, E.P.; Nedashkovskaya, O.I.; Gorshkova, N.M.; Elyakova, L.A.; Mikhailov, V.V. Search for α-d-galactosidase producers among marine bacteria of the genus Alteromonas. Prikl. Biokh. Mikrobiol. 1996, 32, 624–628. [Google Scholar]
- Bakunina, I.Y.; Nedashkovskaya, O.I.; Kim, S.B.; Zvyagintseva, T.N.; Mihailov, V.V. Diversity of glycosidase activities in the bacteria of the phylum Bacteroidetes isolated from marine algae. Microbiology 2012, 81, 688–695. [Google Scholar] [CrossRef]
- Bakunina, I.Y.; Nedashkovskaya, O.I.; Balabanova, L.A.; Zvyagintseva, T.N.; Rasskasov, V.V.; Mikhailov, V.V. Comparative analysis of glycoside hydrolases activities from phylogenetically diverse marine bacteria of the genus Arenibacter. Mar. Drugs 2013, 11, 1977–1998. [Google Scholar] [CrossRef] [PubMed]
- Bakunina, I.Y.; Sova, V.V.; Nedashkovskaya, O.I.; Kuhlmann, R.A.; Likhosherstov, L.M.; Martynova, M.D.; Mihailov, V.V.; Elyakova, L.A. α-d-galactosidase of the marine bacterium Pseudoalteromonas sp. KMM 701. Biochemisrty 1998, 63, 1209–1215. [Google Scholar]
- Balabanova, L.A.; Bakunina, I.Y.; Nedashkovskaya, O.I.; Makarenkova, I.D.; Zaporozhets, T.S.; Besednova, N.N.; Zvyagintseva, T.N.; Rasskazov, V.A. Molecular characterization and therapeutic potential of a marine bacterium Pseudoalteromonas sp. KMM 701 α-d-galactosidase. Mar. Biotechnol. 2010, 12, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Terentieva, N.A.; Timchenko, N.F.; Balabanova, L.A.; Golotin, V.A.; Belik, A.A.; Bakunina, I.Y.; Didenko, L.V.; Rasskazov, V.A. The influence of enzymes on the formation of bacterial biofilms. Health Med. Ecol. Sci. 2015, 60, 86–93. [Google Scholar]
- Bakunina, I.Y.; Balabanova, L.A.; Golotin, V.A.; Slepchenko, L.V.; Isakov, V.V.; Rasskazov, V.A. Stereochemical course of hydrolytic reaction catalyzed by alpha-galactosidase from cold adapTable marine bacterium of genus Pseudoalteromonas. Front. Chem. 2014, 2, 89. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment (MOE); 2018.01; Chemical Computing Group ULC: Montreal, QC, Canada, 2018.
- Fredslund, F.; Hachem, M.A.; Larsen, R.J.; Sørensen, P.G.; Coutinho, P.M.; Lo Leggio, L.; Svensson, B. Crystal structure of α-galactosidase from Lactobacillus acidophilus NCFM: Insight into tetramer formation and substrate binding. J. Mol. Biol. 2011, 412, 466–480. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Black, B.A.; Curtis, J.M.; Gänzle, M.G. Characterization of α-galacto-oligosaccharides formed via heterologous expression of α-d-galactosidases from Lactobacillus reuteri in Lactococcus lactis. Appl. Microbiol. Biotechnol. 2014, 98, 2507–2517. [Google Scholar] [CrossRef] [PubMed]
- Menshova, R.V.; Ermakova, S.P.; Anastyuk, S.D.; Isakov, V.V.; Dubrovskaya, Y.V.; Kusaykin, M.I.; Um, B.H.; Zvyagintseva, T.N. Structure, enzymatic transformation and anticancer activity of branched high molecular weight laminaran from brown alga Eisenia bicyclis. Carbohydr. Polym. 2014, 99, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Domon, B.; Costello, C.E. A Systematic nomenclature for carbohydrate fragmentations in FAB-MS/MS spectra of glycoconjugates. Glycoconj. J. 1988, 5, 397–409. [Google Scholar] [CrossRef]
- Zaia, J.; Miller, M.J.C.; Seymour, J.L.; Costello, C.E. The role of mobile protons in negative ion CID of oligosaccharides. J. Am. Soc. Mass Spectrom. 2007, 18, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Weignerova, L.; Hunkova, Z.; Kuzma, M.; Kren, V. Enzymatic synthesis of three pNP-α-galactobiopyranosides: application of the library of fungal α-d-galactosidases. J. Mol. Catal. B: Enzym. 2001, 11, 219–224. [Google Scholar] [CrossRef]
- Borisova, A.S.; Ivanen, D.R.; Bobrov, K.S.; Eneyskaya, E.V.; Rychkov, G.N.; Sandgren, M.; Kulminskaya, A.A.; Sinnott, M.L.; Shabalin, K.A. α-Galactobiosyl units: Thermodynamics and kinetics of their formation by transglycosylations catalysed by the GH36 α-d-galactosidase from Thermotoga maritima. Carbohydr. Res. 2015, 401, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Bobrov, K.S.; Borisova, A.S.; Eneyskaya, E.V.; Ivanen, D.R.; Shabalin, K.A.; Kulminskaya, A.A.; Rychkov, G.N. Improvement of efficiency of transglycosylation catalyzed by α-d-galactosidase from Thermotoga maritima by protein engineering. Biochemistry 2013, 78, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Mitfal, B.K.; Shallenberger, R.S.; Stainkraus, K.H. α-Galactosidase activity of lactobacilli. Appl. Microbiol. 1973, 26, 783–788. [Google Scholar]
- Coombs, J.; Brenchley, J.E. Characterization of two new glycosyl hydrolases from the lactic acid bacterium Carnobacterium piscicola Strain BA. Appl. Environ. Microbiol. 2001, 67, 5094–5099. [Google Scholar] [CrossRef] [PubMed]
- Garro, M.S.; Degiori, G.S.; Devaldez, G.F.; Oliver, G. Characterization of α-galactosidase from Lactobacillus fermentum. J. Appl. Bacteriol. 1993, 75, 485–488. [Google Scholar] [CrossRef]
- Carrera-Silva, E.A.; Silvestroni, A.; LeBlanc, J.G.; Piard, J.C.; de Giori, G.S.; Sesma, F. A thermostable α-galactosidase from Lactobacillus fermentum CRL722: Genetic characterization and main properties. Curr. Microbiol. 2006, 53, 374–378. [Google Scholar] [CrossRef] [PubMed]
- Fridjonsson, O.; Watzlawick, H.; Gehweiler, A.; Rohrhirsch, T.; Mattes, R. Cloning of the gene encoding a novel thermostable α-galactosidase from Thermus brockianus ITI360. Appl. Environ. Microbiol. 1999, 65, 3955–3963. [Google Scholar] [PubMed]
- Ishiguro, M.; Kaneko, S.; Kuno, A.; Koyama, Y.; Yoshida, S.; Park, G.G.; Sakakibara, Y.; Kusakabe, I.; Kobayashi, H. Purification and characterization of the recombinant Thermus sp. strain T2 α-galactosidase expressed in Escherichia coli. Appl. Environ. Microbiol. 2001, 67, 1601–1606. [Google Scholar] [PubMed]
- King, M.R.; White, B.A.; Blaschek, H.P.; Chassy, B.M.; Mackie, R.I.; Cann, I.K. Purification and characterization of a thermostable α-galactosidase from Thermoanaerobacterium polysaccharolyticum. J. Agric. Food Chem. 2002, 50, 5676–5682. [Google Scholar] [CrossRef] [PubMed]
- Liebl, W.; Wagner, B.; Schellhase, J. Properties of an α-galactosidase, and structure of its gene galA, within an α- and β-galactoside utilization gene cluster of the hyperthermophilic bacterium Thermotoga maritime. Syst. Appl. Microbiol. 1998, 21, 1–11. [Google Scholar] [CrossRef]
- Liu, Q.P.; Yuan, H.; Bennett, E.P.; Levery, S.B.; Nudelman, E.; Spence, J.; Pietz, G.; Saunders, K.; White, T.; Olsson, M.L.; et al. Identification of a GH110 subfamily of α-1,3-galactosidases, novel enzymes for removal of the a3Gal xenotransplantation antigen. J. Biol. Chem. 2008, 283, 8545–8554. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.P.; Sulzenbacher, G.; Yuan, H.; Bennett, E.P.; Pietz, G.; Saunders, K.; Spence, J.; Nudelman, E.; Levery, S.B.; White, T.; et al. Bacterial glycosidases for the production of universal red blood cells. Nat. Biotechnol. 2007, 25, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Robyt, J.F. Carbohydrates/Thin-Layer (Planar) Chromatography. In Encyclopedia of separation science; Wilson, I.D., Cooke, M., Poole, C.F., Eds.; Academic New Press: Pittsburgh, PA, USA, 2000; pp. 2235–2244. [Google Scholar]
- Van Laere, K.M.J.; Hartemink, R.; Beldman, G.; Pitson, S.; Dijkema, C.; Schols, H.A.; Voragen, A.G.J. Transglycosidase activity of Bifidobacterium adolescentis DSM 20083 α-d-galactosidase. Appl. Microbiol. Biotechnol. 1999, 52, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Lu, L.; Xiao, M.; Wang, Q.; Lu, Y.; Liu, C.; Wang, P.; Kumagai, H.; Yamamoto, K. Cloning and characterization of a novel α-d-galactosidase from Bifidobacterium breve 203 capable of synthesizing Gal-α-1,4 linkage. FEMS Microbiol. Lett. 2008, 285, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Cervera-Tison, M.; Tailford, L.E.; Fuell, C.; Bruel, L.; Sulzenbacher, G.; Henrissat, B.; Berrin, J.G.; Fons, M.; Giardina, T.; Jugea, N. Functional analysis of family GH-36 α-d-galactosidases from Ruminococcus gnavus E1: Insights into the metabolism of a plant oligosaccharide by a human gut symbiont. Appl. Environ. Microbiol. 2012, 78, 7720–7732. [Google Scholar] [CrossRef] [PubMed]
- Dion, M.; Nisole, A.; Spangenberg, P.; Andreé, C.; Glottin-Fleury, A.; Mattes, R.; Tellier, C.; Rabiller, C. Modulation of the regioselectivity of a Bacillus α-d-galactosidase by directed evolution. Glycoconj. J. 2001, 18, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Hinz, S.W.A.; Doeswijk-Voragen, C.H.L.; Schipperus, R.; Broek, L.A.M.; Vincken, J.P.; Voragen, A.G.J. Increasing the transglycosylation activity of α-d-galactosidase from Bifidobacterium adolescentis DSM 20083 by site-directed mutagenesis. Biotechnol. Bioeng. 2006, 93, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Nakai, H.; Baumann, M.J.; Petersen, B.O.; Westphal, Y.; Hachem, M.A.; Dilokpimol, A.; Duus, J.Ø.; Schols, H.A.; Svensson, B. Aspergillus nidulans α-d-galactosidase of glycoside hydrolase family 36 catalyses the formation of α-galacto-oligosaccharides by transglycosylation. FEBS J. 2010, 277, 3538–3551. [Google Scholar] [CrossRef] [PubMed]
- Spangenberg, P.; Andre, C.; Dion, M.; Rabiller, C.; Mattes, R. Potential of new sources of α-d-galactosidases for the synthesis of disaccharides. Carbohydr. Res. 2000, 329, 65–73. [Google Scholar] [CrossRef]
- Schroder, S.; Kroger, L.; Mattes, R.; Thiem, J. Transglycosylations employing recombinant α- and β-galactosidases and novel donor substrates. Carbohydr. Res. 2015, 403, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Lu, Q.; Zhang, R.; Wang, Y.; Wu, Q.; Li, J.; Tang, X.; Xu, B.; Ding, J.; Huang, Z. Characterization of two glycoside hydrolase family 36 α-d-galactosidases: Novel transglycosylation activity, lead–zinc tolerance, alkaline and multiple pH optima, and low-temperature activity. Food Chem. 2016, 194, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, K.; Tanimoto, T.; Okada, Y.; Hara, K.; Fujita, K.; Hashimoto, H.; Kitahata, S. Isolation and characterization of novel heterogeneous branched cyclomalto-oligosaccharides (cyclodextrins) produced by transgalactosylation with α-d-galactosidase from coffee bean. Carbohydr. Res. 1995, 278, 129–142. [Google Scholar] [CrossRef]
- Spangenberg, P.; Andre, C.; Langlois, V.; Dion, M.; Rabiller, C. α-Galactosyl fluoride in transfer reactions mediated by the green coffee beans α-d-galactosidase in ice. Carbohydr. Res. 2002, 337, 221–228. [Google Scholar] [CrossRef]
- Savel’ev, A.N.; Ibatyllin, F.M.; Eneyskaya, E.V.; Kachurin, A.M.; Neustroev, K.N. Enzymatic properties of α-d-galactosidase from Trichoderma reesei. Carbohydr. Res. 1996, 296, 261–273. [Google Scholar] [CrossRef]
- Eneyskaya, E.V.; Golubev, A.M.; Kachurin, A.M.; Savel'ev, A.N.; Neustroev, K.N. Transglycosylation activity of α-d-galactosidase from Trichoderma reesei. An investigation of the active site. Carbohydr. Res. 1998, 305, 83–91. [Google Scholar] [CrossRef]
- Shabalin, K.A.; Kulminskaya, A.A.; Savel’ev, A.N.; Shishlyannikov, S.M.; Neustroev, K.N. Enzymatic properties of α-d-galactosidase from Trichoderma reesei in the hydrolysis of galactooligosaccharides. Enzyme Microb. Technol. 2002, 30, 231–239. [Google Scholar] [CrossRef]
- Shivam, K.; Mishra, S.K. Purification and characterization of a thermostable α-d-galactosidase with transglycosylation activity from Aspergillus parasiticus MTCC-2796. Process Biochem. 2010, 45, 1088–1093. [Google Scholar] [CrossRef]
- Wang, C.; Wang, H.; Ma, R.; Shi, P.; Niu, C.; Luo, H.; Yang, P.; Yao, B. Biochemical characterization of a novel thermophilic α-d-galactosidase from Talaromyces leycettanus JCM12802 with significant transglycosylation activity. J. Biosci. Bioeng. 2016, 121, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Kurakake, M.; Moriyama, Y.; Sunouchi, R.; Nakatani, S. Enzymatic properties and transglycosylation of α-d-galactosidase from Penicillium oxalicum SO. Food Chem. 2011, 126, 177–182. [Google Scholar] [CrossRef]
- Vic, G.; Scigelova, M.; Hastings, J.J.; Howarth, O.W.; Crout, D.H.G. Glycosydase catalysed synthesis of oligosaccharides: trisaccharides with the α-d-Gal-(1→3)-d-Gal terminus responsible for the hyper acute rejection response in cross-species transplant rejection from pigs to man. Chem. Commun. 1996, 12, 1473–1474. [Google Scholar] [CrossRef]
- Gong, W.; Xu, L.; Gu, G.; Lu, L.; Xiao, M. Efficient and regioselective synthesis of globotriose by a novel α-d-galactosidase from Bacteroides fragilis. Appl. Microbiol. Biotechnol. 2016, 100, 6693–6702. [Google Scholar] [CrossRef] [PubMed]
- Brouns, J.J.S.; Smits, N.; Wu, H.; Snijders, A.P.L.; Wright, P.C.; de Vos, W.M.; van der Oost, J. Identification of a novel alpha-galactosidase from the hyperthermophilic archaeon Sulfolobus solfataricus. J. Bacteriol. 2006, 188, 2392–2399. [Google Scholar] [CrossRef] [PubMed]
- Comfort, D.A.; Bobrov, K.S.; Ivanen, D.R.; Shabalin, K.A.; Harris, J.M.; Kulminskaya, A.A.; Brumer, H.; Kelly, R.M. Biochemical analysis of Thermotoga maritima GH36 α-d-galactosidase (TmGalA) confirms the mechanistic commonality of clan GH-D glycoside hydrolases. Biochemistry. 2007, 46, 3319–3330. [Google Scholar] [CrossRef] [PubMed]
- Cobucci-Ponzano, B.; Zorzetti, C.; Strazzulli, A.; Carillo, S.; Bedini, E.; Corsaro, M.M.; Comfort, D.A.; Kelly, R.M.; Rossi, M.; Moracci, M. A novel α-d-galactosynthase from Thermotoga maritima converts β-d-galactopyranosyl azide to α-galacto-oligosaccharides. Glycobiology 2011, 21, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Shared Resource Center Far Eastern Computing Resource of Institute of Automation and Control Processes Far Eastern Branch of the Russian Academy of Sciences (IACP FEB RAS). Available online: https://cc.dvo.ru (accessed on 12 January 2018).
- Golotin, V.A.; Balabanova, L.A.; Noskova, Y.A.; Slepchenko, L.V.; Bakunina, I.Y.; Vorobieva, N.S.; Terentieva, N.A.; Rasskazov, V.A. Optimization of cold-adapted α-d-galactosidase expression in Escherichia coli. Protein Expr. Purif. 2016, 123, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Provencher, S.W.; Glockner, J. Estimation of globular protein secondary structure from circular dichroism. Biochemistry 1981, 20, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Sreerama, N.; Woody, R.W. Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 2000, 287, 252–260. [Google Scholar] [CrossRef] [PubMed]
- ExPASy (Expert Protein Analysis System) Proteomics Server. Available online: http://cn.expasy.org/tools/protparam.html (accessed on 15 September 2015).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Km (mM) | Vmax (μmol min−1 ml−1) | kcat (s−1) | kcat/Km (mM−1 s−1) |
|---|---|---|---|---|
| Wild-type α-PsGal | 0.40 ± 0.030 | 0.32 ± 0.003 | 324.4 ± 3.5 | 820 |
| C494N mutant | 0.30 ± 0.005 | 0.024 ± 0.0003 | 2.84 ± 0.02 | 3.86 |
| Substrate | Acceptor | New Products |
|---|---|---|
| pNP-α-Gal | d-Glucose | − |
| pNP-α-Gal | d-Fructose | − |
| pNP-α-Gal | d-Xylose | − |
| pNP-α-Gal | l-Fucose | − |
| pNP-α-Gal | d-Galactose | + |
| Gal-α(1→6)-Glcα,β | d-Xylose | − |
| Gal-α(1→6)-Glcα,β | d-Fructose | − |
| Gal-α(1→6)-Glcα,β | l-Fucose | − |
| Enzyme | Substrate | Acceptor | Substrate (mol) | Acceptor (mol) | V (mL) | U or mg | t (°C) | τ (h) |
|---|---|---|---|---|---|---|---|---|
| Wild | Gal-α(1→6)-Glcα,β | - | 0.11 | 0 | 0.1 | 2.2 | 20 | 48 |
| Wild | Gal-α(1→6)-Glcα,β | - | 0.31 | 0 | 0.3 | 4.5 | 8 | 7 days |
| Wild | pNP-α-Gal | - | 0.05 | 0 | 0.2 | 1.0 | 20 | 48 |
| Wild | pNP-α-Gal | - | 0.07 | 0 | 0.2 | 0.015 | 8 | 7 days |
| Wild | Gal-α(1→6)-Glcα,β | pNP-α-Gal | 0.06 | 0.13 | 0.3 | 2.6 | 20 | 48 |
| C494N | pNP-α-Gal | - | 0.07 | 0 | 0.3 | 0.012 | 8 | 7 days |
| D451A | Gal-α(1→6)-Glcα,β | NaN3 | 0.06 | 0.3 | 0.1 | (0.008) | 20 | 75 |
| D451A | pNP-α-Gal | NaN3 | 0.07 | 0.3 | 0.3 | (0.024) | 20 | 75 |
| D451A | pNP-α-Gal | - | 0.07 | 0 | 0.2 | (0.016) | 20 | 75 |
| Enzyme | Substrate | Product Number | TLC (Rf) | MALDI-MS (m/z) |
|---|---|---|---|---|
| Wild α-PsGal | Gal-α(1→6)-Glcα,β | 0.39 | 365 | |
| 1 | 0.52 | 203 | ||
| 2 | 0.55 | 203 | ||
| 3 | 0.35 | 365 | ||
| 4 | 0.35 | 365 | ||
| 5 | 0.26 | 527 | ||
| pNP-α-Gal | 0.83 | 324 | ||
| 1 | 0.52 | 203 | ||
| 6 | 0.61 | 486 | ||
| 4 | 0.34 | 365 | ||
| Mutant C494N | pNP-α-Gal | – | 324 | |
| 1 | – | 203 | ||
| 3 | – | 365 | ||
| 6 | – | 486 |
| Sugars | Product Number | δH (J in Hz) | δC | |||||
|---|---|---|---|---|---|---|---|---|
| A | B | A | B | |||||
| αH1 | αH1 | βH1 | αC1 | αC1 | βC1 | |||
| Gal-α(1→6)-Glcα,β | 5.00 (3.7) | 5.24 | 4.67 (7.8) | 98.2 | 92.2 | 96.1 | ||
| A | B | |||||||
| Galα,β | 1 | 5.26 (3.7) | 4.64 (7.9) | 97.7 | 93.6 | |||
| Glcα,β | 2 | 5.23 (3.7) | 4.58 (7.8) | 97.6 | 93.6 | |||
| Gal-α(1→6)-Galα,β | 3 | 4.98 (4.8) | 5.27 | 4.59 (7.9) | 97.7 | 99.8 | ||
| A | B | |||||||
| Gal-α(1→4)-Glcα,β | 4 | 5.22 (3.2) | N/D * | 102.2 | N/D | N/D | ||
| A | B | |||||||
| Gal-(1→4)-Gal-(1→6)-Glc | 5 | undetected | ||||||
| pNP-α-Gal | 5.85 (3.74) | 98.1 | ||||||
| Gal-α(1→6)-Gal-α-pNP | 6 | 4.82 (3.85) | 5.91 (3.82) | 98.8 | 97.6 | |||
| A | B | |||||||
| Gal-α(1→3)-Gal-α-pNP | 7 | 5.87 (3.6) | N/D | N/D | N/D | |||
| A | B | |||||||
| Enzyme | Substrate | T (°C) | Substrate Conversion (%) | Structure of the Hydrolysis and Transglycosylation Products | Yield of Products (%) | |
|---|---|---|---|---|---|---|
| Wild | Gal-α(1→6)-Glcα,β | 20 | 88.5 | Gal | 1 | 45.627.35.50.6 |
| Glc | 2 | |||||
| Gal-α(1→6)-Galα,β | 3 | |||||
| Gal-α(1→4)-Galα,β | 4 | |||||
| Wild | Gal-α(1→6)-Glcα,β | 8 | 90.2 | Gal | 1 | 46.035.07.81.4 |
| Glc | 2 | |||||
| Gal-α(1→6)-Galα,β | 3 | |||||
| Gal-α(1→4)-Galα,β | 4 | |||||
| Wild | pNP-α-Gal | 20 | 67.2 | Gal | 1 | 21.58.03.01.21.2 |
| Gal-α(1→6)-Gal-α-pNP | 6 | |||||
| Gal-α(1→6)-Galα,β | 3 | |||||
| Gal-α(1→4)-Galα,β | 4 | |||||
| Gal-α(1→3)-Gal-α-pNP | 7 | |||||
| Wild | pNP-α-Gal | 8 | 15.9 | Gal | 1 | 9.84.11.20.78<1 |
| Gal-α(1→6)-Gal-α-pNP | 6 | |||||
| Gal-α(1→6)-Galα,β | 3 | |||||
| Gal-α(1→4)-Galα,β | 4 | |||||
| Gal-α(1→3)-Gal-α-pNP | 7 | |||||
| Wild | Gal-α(1→6)-Glcα,β + pNP-α-Gal | 20 | 32.0 | Gal | 1 | 30.09.01.60.8 |
| Gal-α(1→6)-Gal-α-pNP | 6 | |||||
| Gal-α(1→4)-Galα,β | 4 | |||||
| Gal-α(1→6)-Galα,β | 3 | |||||
| C494N | pNP-α-Gal | 8 | 19.0 | Gal | 1 | 5.02.82.01.2 |
| Gal-α(1→6)-Galα,β | 3 | |||||
| Gal-α(1→6)-Gal-α-pNP | 6 | |||||
| Gal-α(1→4)-Galα,β | 4 | |||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bakunina, I.; Slepchenko, L.; Anastyuk, S.; Isakov, V.; Likhatskaya, G.; Kim, N.; Tekutyeva, L.; Son, O.; Balabanova, L. Characterization of Properties and Transglycosylation Abilities of Recombinant α-Galactosidase from Cold-Adapted Marine Bacterium Pseudoalteromonas KMM 701 and Its C494N and D451A Mutants. Mar. Drugs 2018, 16, 349. https://doi.org/10.3390/md16100349
Bakunina I, Slepchenko L, Anastyuk S, Isakov V, Likhatskaya G, Kim N, Tekutyeva L, Son O, Balabanova L. Characterization of Properties and Transglycosylation Abilities of Recombinant α-Galactosidase from Cold-Adapted Marine Bacterium Pseudoalteromonas KMM 701 and Its C494N and D451A Mutants. Marine Drugs. 2018; 16(10):349. https://doi.org/10.3390/md16100349
Chicago/Turabian StyleBakunina, Irina, Lubov Slepchenko, Stanislav Anastyuk, Vladimir Isakov, Galina Likhatskaya, Natalya Kim, Liudmila Tekutyeva, Oksana Son, and Larissa Balabanova. 2018. "Characterization of Properties and Transglycosylation Abilities of Recombinant α-Galactosidase from Cold-Adapted Marine Bacterium Pseudoalteromonas KMM 701 and Its C494N and D451A Mutants" Marine Drugs 16, no. 10: 349. https://doi.org/10.3390/md16100349
APA StyleBakunina, I., Slepchenko, L., Anastyuk, S., Isakov, V., Likhatskaya, G., Kim, N., Tekutyeva, L., Son, O., & Balabanova, L. (2018). Characterization of Properties and Transglycosylation Abilities of Recombinant α-Galactosidase from Cold-Adapted Marine Bacterium Pseudoalteromonas KMM 701 and Its C494N and D451A Mutants. Marine Drugs, 16(10), 349. https://doi.org/10.3390/md16100349