Ciguatoxins Evoke Potent CGRP Release by Activation of Voltage-Gated Sodium Channel Subtypes NaV1.9, NaV1.7 and NaV1.1

 and
and 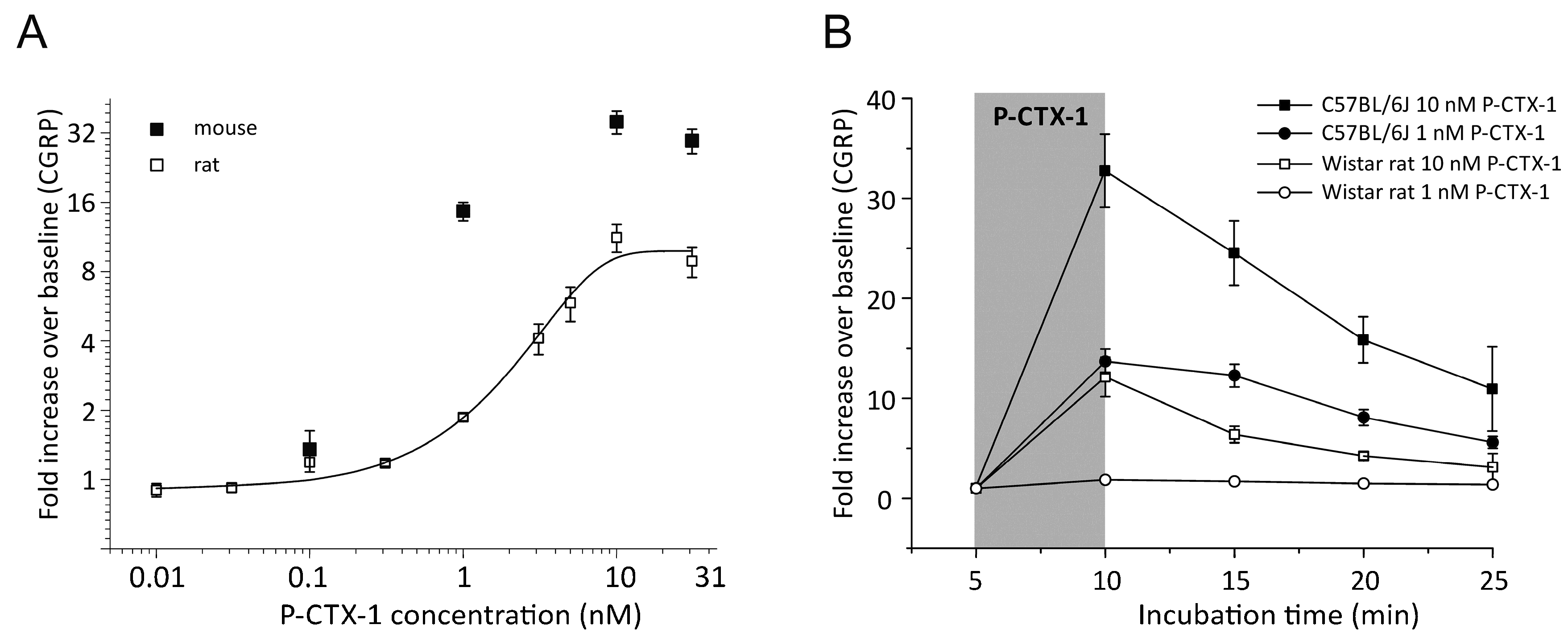{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Animals, Preparation of Skin Flaps and Solutions
2.2. Stimulation Procedure and Compounds
2.3. Enzyme Immunoassay
2.4. Data Analysis
3. Results
3.1. P-CTX-1-Induced CGRP-Release Is Greater in Mouse Than Rat Skin
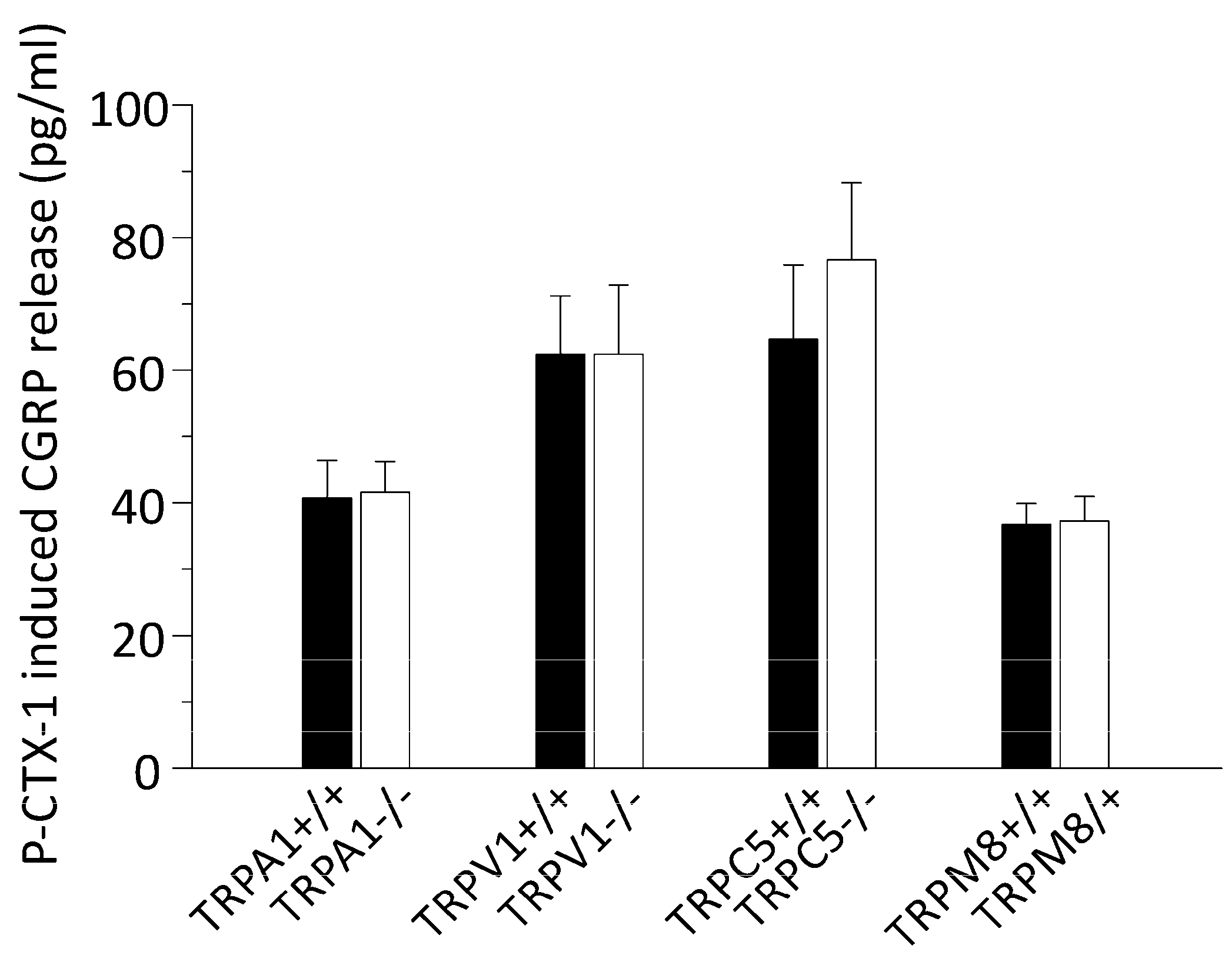
3.2. P-CTX-1-Induced CGRP Release Requires Extracellular Calcium but Is Not Reduced in Mice Deficient of TRPV1, TRPA1, TRPM8, or TRPC5
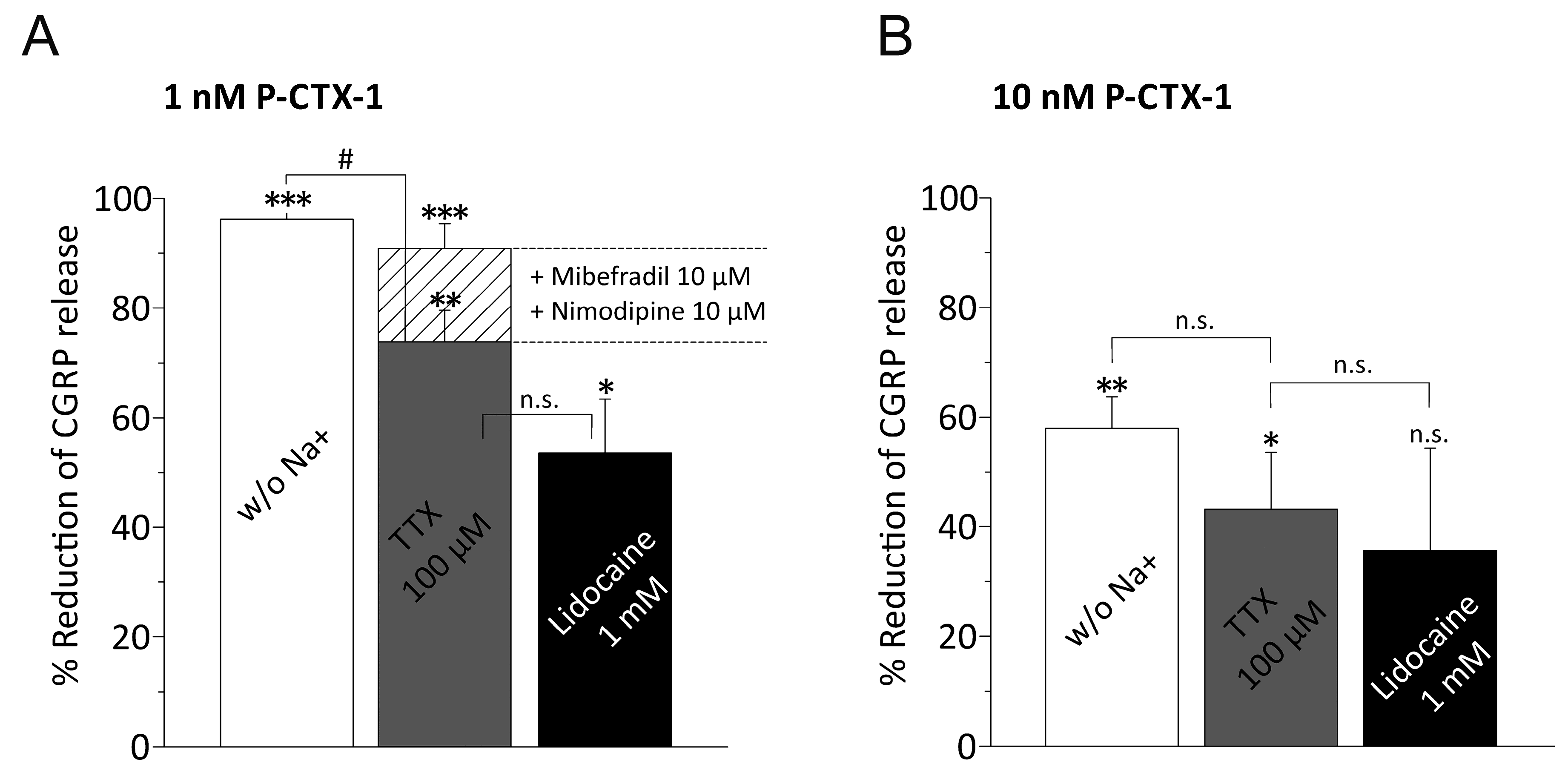
3.3. P-CTX-1-Induced CGRP Release Requires Extracellular Sodium and Activation of VGSCs
3.4. P-CTX-1-Induced VGSC Opening Triggers Activation of L- and T-Type VGCC
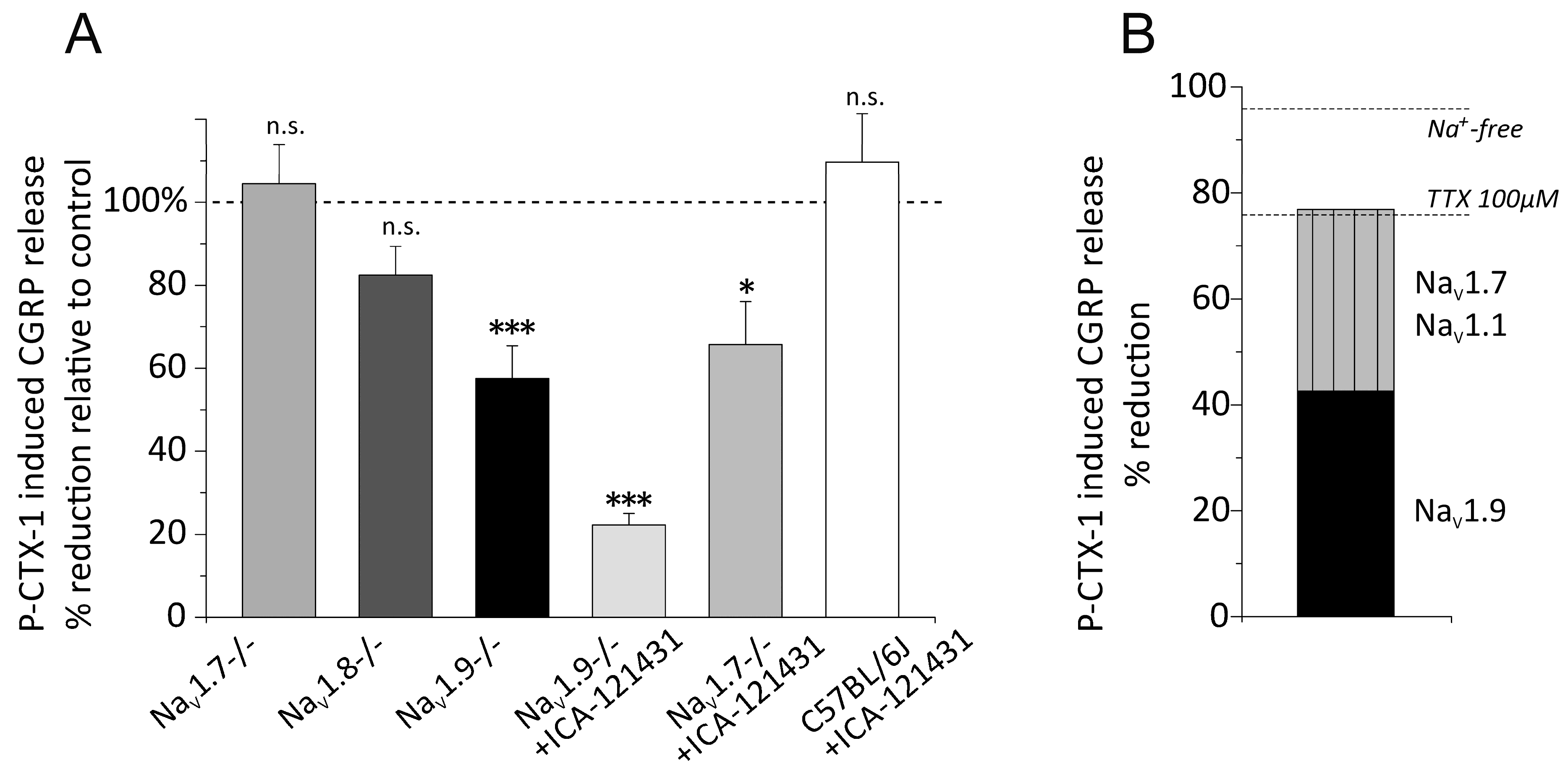
3.5. Distinct VGSC Subtypes Mediate the P-CTX-1-Induced CGRP Release
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lewis, R.J. Ciguatera: Australian perspectives on a global problem. Toxicon 2006, 48, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Bagnis, R.; Kuberski, T.; Laugier, S. Clinical Observations on 3009 Cases of Ciguatera (Fish Poisoning) in the South-Pacific. Am. J. Trop Med. Hyg. 1979, 28, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Strachan, L.C.; Lewis, R.J.; Nicholson, G.M. Differential actions of Pacific ciguatoxin-1 on sodium channel subtypes in mammalian sensory neurons. J. Pharmacol. Exp. Ther. 1999, 288, 379–388. [Google Scholar] [PubMed]
- Yamaoka, K.; Inoue, M.; Miyahara, H.; Miyazaki, K.; Hirama, M. A quantitative and comparative study of the effects of a synthetic ciguatoxin CTX3C on the kinetic properties of voltage-dependent sodium channels. Br. J. Pharmacol. 2004, 142, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Inserra, M.C.; Israel, M.R.; Caldwell, A.; Castro, J.; Deuis, J.R.; Harrington, A.M.; Keramidas, A.; Garcia-Caraballo, S.; Maddern, J.; Erickson, A.; et al. Multiple sodium channel isoforms mediate the pathological effects of Pacific ciguatoxin-1. Sci. Rep. 2017, 7, 42810. [Google Scholar] [CrossRef] [PubMed]
- Vetter, I.; Touska, F.; Hess, A.; Hinsbey, R.; Sattler, S.; Lampert, A.; Sergejeva, M.; Sharov, A.; Collins, L.S.; Eberhardt, M.; et al. Ciguatoxins activate specific cold pain pathways to elicit burning pain from cooling. EMBO J. 2012, 31, 3795–3808. [Google Scholar] [CrossRef] [PubMed]
- Schlereth, T.; Breimhorst, M.; Werner, N.; Pottschmidt, K.; Drummond, P.D.; Birklein, F. Inhibition of neuropeptide degradation suppresses sweating but increases the area of the axon reflex flare. Exp. Dermatol. 2013, 22, 299–301. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, K.; Deuis, J.R.; Inserra, M.C.; Collins, L.S.; Namer, B.; Cabot, P.J.; Reeh, P.W.; Lewis, R.J.; Vetter, I. Analgesic treatment of ciguatoxin-induced cold allodynia. Pain 2013, 154, 1999–2006. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, M.; Dux, M.; Namer, B.; Miljkovic, J.; Cordasic, N.; Will, C.; Kichko, T.I.; de la Roche, J.; Fischer, M.; Suarez, S.A.; et al. H2S and NO cooperatively regulate vascular tone by activating a neuroendocrine HNO-TRPA1-CGRP signalling pathway. Nat. Commun. 2014, 5, 4381. [Google Scholar] [CrossRef] [PubMed]
- Nassini, R.; Materazzi, S.; Benemei, S.; Geppetti, P. The TRPA1 channel in inflammatory and neuropathic pain and migraine. Rev. Physiol. Biochem. Pharmacol. 2014, 167, 1–43. [Google Scholar] [PubMed]
- McCoy, E.S.; Taylor-Blake, B.; Street, S.E.; Pribisko, A.L.; Zheng, J.; Zylka, M.J. Peptidergic CGRPalpha primary sensory neurons encode heat and itch and tonically suppress sensitivity to cold. Neuron 2013, 78, 138–151. [Google Scholar] [CrossRef] [PubMed]
- Engel, M.A.; Leffler, A.; Niedermirtl, F.; Babes, A.; Zimmermann, K.; Filipovic, M.R.; Izydorczyk, I.; Eberhardt, M.; Kichko, T.I.; Mueller-Tribbensee, S.M.; et al. TRPA1 and substance P mediate colitis in mice. Gastroenterology 2011, 141, 1346–1358. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.A.; Mapp, P.I.; Kelly, S. Calcitonin gene-related peptide in the joint: Contributions to pain and inflammation. Br. J. Clin. Pharmacol. 2015, 80, 965–978. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, R.C.; House, D.; Ryan, J.C. Defining the neurotoxin derived illness chronic ciguatera using markers of chronic systemic inflammatory disturbances: A case/control study. Neurotoxicol. Teratol. 2010, 32, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Le Garrec, R.; L’Herondelle, K.; Le Gall-Ianotto, C.; Lebonvallet, N.; Leschiera, R.; Buhe, V.; Talagas, M.; Vetter, I.; Lewis, R.J.; Misery, L. Release of neuropeptides from a neuro-cutaneous co-culture model: A novel in vitro model for studying sensory effects of ciguatoxins. Toxicon 2016, 116, 4–10. [Google Scholar] [CrossRef] [PubMed]
- McCormack, K.; Santos, S.; Chapman, M.L.; Krafte, D.S.; Marron, B.E.; West, C.W.; Krambis, M.J.; Antonio, B.M.; Zellmer, S.G.; Printzenhoff, D.; et al. Voltage sensor interaction site for selective small molecule inhibitors of voltage-gated sodium channels. Proc. Natl. Acad. Sci. USA 2013, 110, E2724–E2732. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.B.; Gray, J.; Gunthorpe, M.J.; Hatcher, J.P.; Davey, P.T.; Overend, P.; Harries, M.H.; Latcham, J.; Clapham, C.; Atkinson, K.; et al. Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature 2000, 405, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Kwan, K.Y.; Allchorne, A.J.; Vollrath, M.A.; Christensen, A.P.; Zhang, D.S.; Woolf, C.J.; Corey, D.P. TRPA1 contributes to cold, mechanical, and chemical Nociception but is not essential for hair-cell transduction. Neuron 2006, 50, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Dhaka, A.; Murray, A.N.; Mathur, J.; Earley, T.J.; Petrus, M.J.; Patapoutian, A. TRPM8 is required for cold sensation in mice. Neuron 2007, 54, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Phelan, K.D.; Shwe, U.T.; Abramowitz, J.; Wu, H.; Rhee, S.W.; Howell, M.D.; Gottschall, P.E.; Freichel, M.; Flockerzi, V.; Birnbaumer, L.; et al. Canonical transient receptor channel 5 (TRPC5) and TRPC1/4 contribute to seizure and excitotoxicity by distinct cellular mechanisms. Mol. Pharmacol. 2013, 83, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Akopian, A.N.; Souslova, V.; England, S.; Okuse, K.; Ogata, N.; Ure, J.; Smith, A.; Kerr, B.J.; McMahon, S.B.; Boyce, S.; et al. The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat. Neurosci. 1999, 2, 541–548. [Google Scholar] [PubMed]
- Baker, M.D.; Chandra, S.Y.; Ding, Y.; Waxman, S.G.; Wood, J.N. GTP-induced tetrodotoxin-resistant Na+ current regulates excitability in mouse and rat small diameter sensory neurones. J. Physiol. 2003, 548, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Minett, M.S.; Nassar, M.A.; Clark, A.K.; Passmore, G.; Dickenson, A.H.; Wang, F.; Malcangio, M.; Wood, J.N. Distinct NaV1.7-dependent pain sensations require different sets of sensory and sympathetic neurons. Nat. Commun. 2012, 3, 791. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J.; Sellin, M.; Poli, M.A.; Norton, R.S.; MacLeod, J.K.; Sheil, M.M. Purification and characterization of ciguatoxins from moray eel (Lycodontis javanicus, Muraenidae). Toxicon 1991, 29, 1115–1127. [Google Scholar] [CrossRef]
- Zimmermann, K.; Reeh, P.W.; Averbeck, B. S+ -flurbiprofen but not 5-HT1 agonists suppress basal and stimulated CGRP and PGE2 release from isolated rat dura mater. Pain 2003, 103, 313–320. [Google Scholar] [CrossRef]
- Averbeck, B.; Reeh, P.W. Interactions of inflammatory mediators stimulating release of calcitonin gene-related peptide, substance P and prostaglandin E(2) from isolated rat skin. Neuropharmacology 2001, 40, 416–423. [Google Scholar] [CrossRef]
- Weller, K.; Reeh, P.W.; Sauer, S.K. TRPV1, TRPA1, and CB1 in the isolated vagus nerve--axonal chemosensitivity and control of neuropeptide release. Neuropeptides 2011, 45, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Kichko, T.I.; Kobal, G.; Reeh, P.W. Cigarette smoke has sensory effects through nicotinic and TRPA1 but not TRPV1 receptors on the isolated mouse trachea and larynx. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L812–L820. [Google Scholar] [CrossRef] [PubMed]
- Cuypers, E.; Yanagihara, A.; Rainier, J.D.; Tytgat, J. TRPV1 as a key determinant in ciguatera and neurotoxic shellfish poisoning. Biochem. Biophys. Res. Commun. 2007, 361, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, N.C.; Lewis, R.J.; Pearn, J.H.; Bourke, A.T.; Holmes, M.J.; Bourke, J.B.; Shields, W.J. Ciguatera in Australia. Occurrence, clinical features, pathophysiology and management. Med. J. Aust. 1986, 145, 584–590. [Google Scholar] [PubMed]
- Coyle, D.E.; Sperelakis, N. Bupivacaine and lidocaine blockade of calcium-mediated slow action potentials in guinea pig ventricular muscle. J. Pharmacol. Exp. Ther. 1987, 242, 1001–1005. [Google Scholar] [PubMed]
- Spitzer, M.J.; Reeh, P.W.; Sauer, S.K. Mechanisms of potassium- and capsaicin-induced axonal calcitonin gene-related peptide release: Involvement of L- and T-type calcium channels and TRPV1 but not sodium channels. Neuroscience 2008, 151, 836–842. [Google Scholar] [CrossRef] [PubMed]
- Usoskin, D.; Furlan, A.; Islam, S.; Abdo, H.; Lonnerberg, P.; Lou, D.; Hjerling-Leffler, J.; Haeggstrom, J.; Kharchenko, O.; Kharchenko, P.V.; et al. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat. Neurosci. 2015, 18, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Osteen, J.D.; Herzig, V.; Gilchrist, J.; Emrick, J.J.; Zhang, C.; Wang, X.; Castro, J.; Garcia-Caraballo, S.; Grundy, L.; Rychkov, G.Y.; et al. Selective spider toxins reveal a role for the NaV1.1 channel in mechanical pain. Nature 2016, 534, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Hackel, D.; Krug, S.M.; Sauer, R.S.; Mousa, S.A.; Bocker, A.; Pflucke, D.; Wrede, E.J.; Kistner, K.; Hoffmann, T.; Niedermirtl, B.; et al. Transient opening of the perineurial barrier for analgesic drug delivery. Proc. Natl. Acad. Sci. USA 2012, 109, E2018–E2027. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, K.; Hein, A.; Hager, U.; Kaczmarek, J.S.; Turnquist, B.P.; Clapham, D.E.; Reeh, P.W. Phenotyping sensory nerve endings in vitro in the mouse. Nat. Protoc. 2009, 4, 174–196. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, K.; Leffler, A.; Babes, A.; Cendan, C.M.; Carr, R.W.; Kobayashi, J.; Nau, C.; Wood, J.N.; Reeh, P.W. Sensory neuron sodium channel NaV1.8 is essential for pain at low temperatures. Nature 2007, 447, 855–858. [Google Scholar] [CrossRef] [PubMed]
- Haller-Brem, S.; Muff, R.; Fischer, J.A. Calcitonin gene-related peptide and calcitonin secretion from a human medullary thyroid carcinoma cell line: Effects of ionomycin, phorbol ester and forskolin. J. Endocrinol. 1988, 119, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.Y.; Neher, E. Ca(2+)-dependent exocytosis in the somata of dorsal root ganglion neurons. Neuron 1996, 17, 135–145. [Google Scholar] [CrossRef]
- Mattei, C.; Wen, P.J.; Nguyen-Huu, T.D.; Alvarez, M.; Benoit, E.; Bourdelais, A.J.; Lewis, R.J.; Baden, D.G.; Molgo, J.; Meunier, F.A. Brevenal inhibits pacific ciguatoxin-1B-induced neurosecretion from bovine chromaffin cells. PLoS ONE 2008, 3, e3448. [Google Scholar] [CrossRef] [PubMed]
- Molgo, J.B.E. Involvement of Na+ in the actions of ciguatoxins and brevetoxins that stimulate neurotransmitter release and affect synaptic transmission. In Perspectives in Molecular Toxinology; Menez, A., Ed.; John Wiley & Sons Ltd.: London, UK, 2002; pp. 67–94. [Google Scholar]
- Ramsey, I.S.; Delling, M.; Clapham, D.E. An introduction to TRP channels. Annu. Rev. Physiol. 2006, 68, 619–647. [Google Scholar] [CrossRef] [PubMed]
- Dhaka, A.; Earley, T.J.; Watson, J.; Patapoutian, A. Visualizing cold spots: TRPM8-expressing sensory neurons and their projections. J. Neurosci. 2008, 28, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, K.; Lennerz, J.K.; Hein, A.; Link, A.S.; Kaczmarek, J.S.; Delling, M.; Uysal, S.; Pfeifer, J.D.; Riccio, A.; Clapham, D.E. Transient receptor potential cation channel, subfamily C, member 5 (TRPC5) is a cold-transducer in the peripheral nervous system. Proc. Natl. Acad. Sci. USA 2011, 108, 18114–18119. [Google Scholar] [CrossRef] [PubMed]
- Ruscheweyh, R.; Forsthuber, L.; Schoffnegger, D.; Sandkuhler, J. Modification of classical neurochemical markers in identified primary afferent neurons with Abeta-, Adelta-, and C-fibers after chronic constriction injury in mice. J. Comp. Neurol. 2007, 502, 325–336. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, P.W.; Lawson, S.N. Cell type and conduction velocity of rat primary sensory neurons with calcitonin gene-related peptide-like immunoreactivity. Neuroscience 1990, 34, 623–632. [Google Scholar] [CrossRef]
- Renganathan, M.; Cummins, T.R.; Waxman, S.G. Contribution of Nav1.8 sodium channels to action potential electrogenesis in DRG neurons. J. Neurophysiol. 2001, 86, 629–640. [Google Scholar] [PubMed]
- Blair, N.T.; Bean, B.P. Roles of tetrodotoxin (TTX)-sensitive Na+ current, TTX-resistant Na+ current, and Ca2+ current in the action potentials of nociceptive sensory neurons. J. Neurosci. 2002, 22, 10277–10290. [Google Scholar] [PubMed]
- Abrahamsen, B.; Zhao, J.; Asante, C.O.; Cendan, C.M.; Marsh, S.; Martinez-Barbera, J.P.; Nassar, M.A.; Dickenson, A.H.; Wood, J.N. The cell and molecular basis of mechanical, cold, and inflammatory pain. Science 2008, 321, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Dib-Hajj, S.D.; Cummins, T.R.; Black, J.A.; Waxman, S.G. Sodium channels in normal and pathological pain. Annu. Rev. Neurosci. 2010, 33, 325–347. [Google Scholar] [CrossRef] [PubMed]
- Hockley, J.R.; Boundouki, G.; Cibert-Goton, V.; McGuire, C.; Yip, P.K.; Chan, C.; Tranter, M.; Wood, J.N.; Nassar, M.A.; Blackshaw, L.A.; et al. Multiple roles for NaV1.9 in the activation of visceral afferents by noxious inflammatory, mechanical, and human disease-derived stimuli. Pain 2014, 155, 1962–1975. [Google Scholar] [CrossRef] [PubMed]
- Dib-Hajj, S.D.; Black, J.A.; Waxman, S.G. NaV1.9: A sodium channel linked to human pain. Nat. Rev. Neurosci. 2015, 16, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Maingret, F.; Coste, B.; Padilla, F.; Clerc, N.; Crest, M.; Korogod, S.M.; Delmas, P. Inflammatory mediators increase NaV1.9 current and excitability in nociceptors through a coincident detection mechanism. J. Gen. Physiol. 2008, 131, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Hogg, R.C.; Lewis, R.J.; Adams, D.J. Ciguatoxin-induced oscillations in membrane potential and action potential firing in rat parasympathetic neurons. Eur. J. Neurosci. 2002, 16, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Alonso, E.; Vale, C.; Sasaki, M.; Fuwa, H.; Konno, Y.; Perez, S.; Vieytes, M.R.; Botana, L.M. Calcium Oscillations Induced by Gambierol in Cerebellar Granule Cells. J. Cell. Biochem. 2010, 110, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Cuypers, E.; Abdel-Mottaleb, Y.; Kopljar, I.; Rainier, J.D.; Raes, A.L.; Snyders, D.J.; Tytgat, J. Gambierol, a toxin produced by the dinoflagellate Gambierdiscus toxicus, is a potent blocker of voltage-gated potassium channels. Toxicon 2008, 51, 974–983. [Google Scholar] [CrossRef] [PubMed]
- Birinyi-Strachan, L.C.; Gunning, S.J.; Lewis, R.J.; Nicholson, G.M. Block of voltage-gated potassium channels by Pacific ciguatoxin-1 contributes to increased neuronal excitability in rat sensory neurons. Toxicol. Appl. Pharm. 2005, 204, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Bayliss, W.M. On the origin from the spinal cord of the vaso-dilator fibres of the hind-limb, and on the nature of these fibres. J. Physiol. 1901, 26, 173–209. [Google Scholar] [CrossRef] [PubMed]
- Kress, M.; Guthmann, C.; Averbeck, B.; Reeh, P.W. Calcitonin gene-related peptide and prostaglandin E2 but not substance P release induced by antidromic nerve stimulation from rat skin in vitro. Neuroscience 1999, 89, 303–310. [Google Scholar] [CrossRef]




© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Touska, F.; Sattler, S.; Malsch, P.; Lewis, R.J.; Reeh, P.W.; Zimmermann, K. Ciguatoxins Evoke Potent CGRP Release by Activation of Voltage-Gated Sodium Channel Subtypes NaV1.9, NaV1.7 and NaV1.1. Mar. Drugs 2017, 15, 269. https://doi.org/10.3390/md15090269
Touska F, Sattler S, Malsch P, Lewis RJ, Reeh PW, Zimmermann K. Ciguatoxins Evoke Potent CGRP Release by Activation of Voltage-Gated Sodium Channel Subtypes NaV1.9, NaV1.7 and NaV1.1. Marine Drugs. 2017; 15(9):269. https://doi.org/10.3390/md15090269
Chicago/Turabian StyleTouska, Filip, Simon Sattler, Philipp Malsch, Richard J. Lewis, Peter W. Reeh, and Katharina Zimmermann. 2017. "Ciguatoxins Evoke Potent CGRP Release by Activation of Voltage-Gated Sodium Channel Subtypes NaV1.9, NaV1.7 and NaV1.1" Marine Drugs 15, no. 9: 269. https://doi.org/10.3390/md15090269
APA StyleTouska, F., Sattler, S., Malsch, P., Lewis, R. J., Reeh, P. W., & Zimmermann, K. (2017). Ciguatoxins Evoke Potent CGRP Release by Activation of Voltage-Gated Sodium Channel Subtypes NaV1.9, NaV1.7 and NaV1.1. Marine Drugs, 15(9), 269. https://doi.org/10.3390/md15090269