Genomic Sequence and Experimental Tractability of a New Decapod Shrimp Model, Neocaridina denticulata

 ,
, 
Abstract
:
1. Introduction
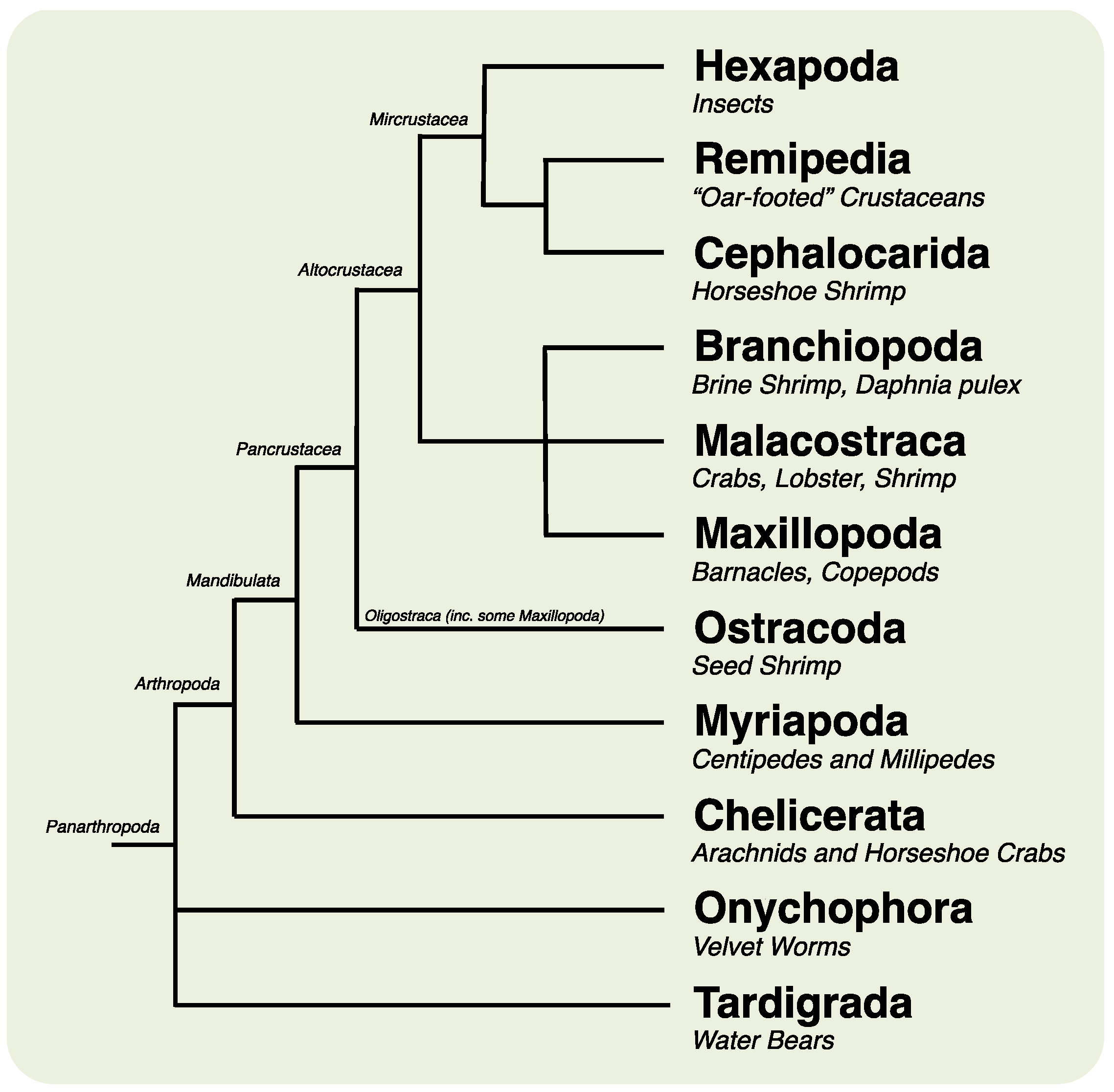
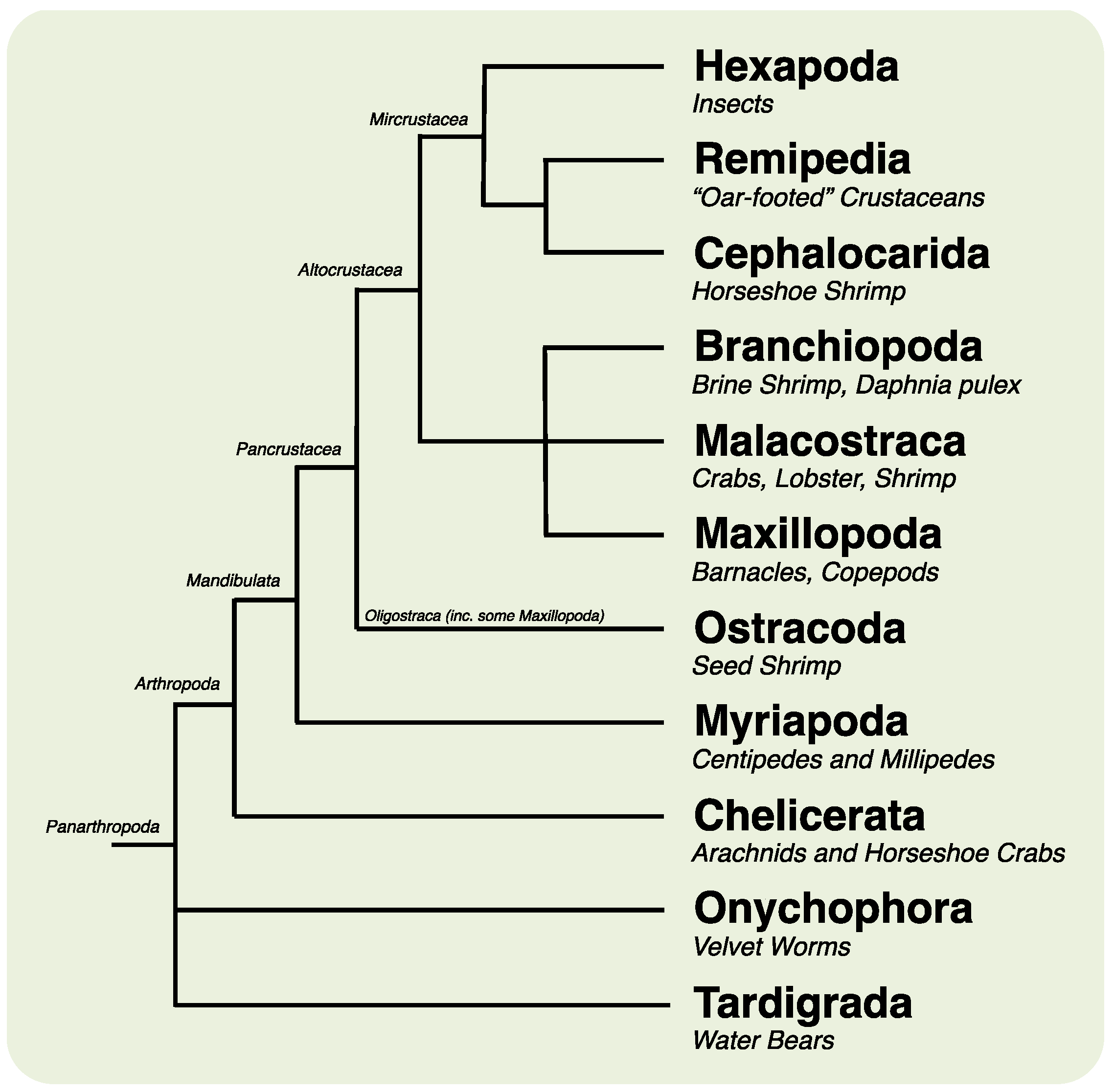
2. Results and Discussion
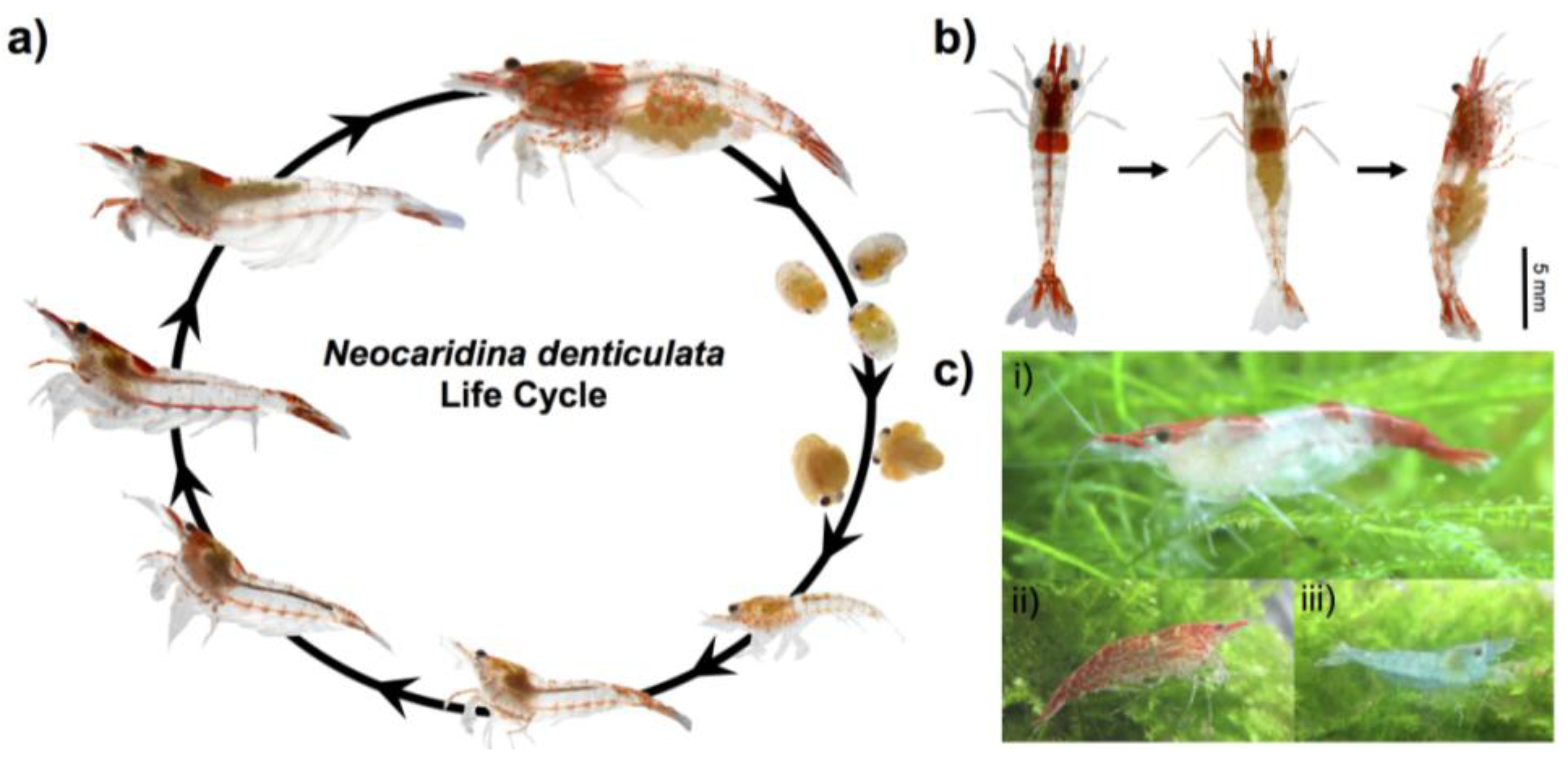
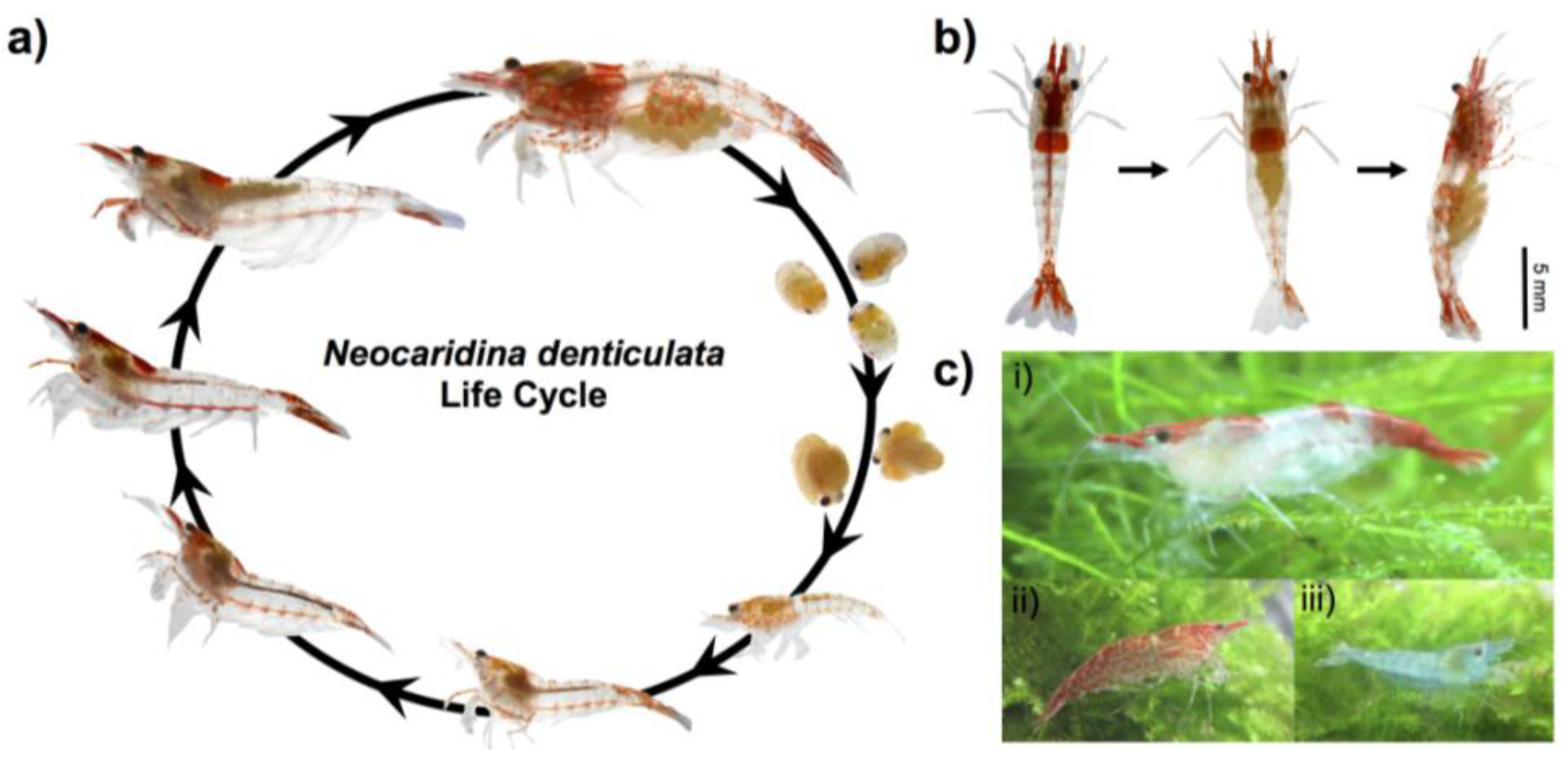
2.1. Animal Culture and Lifecycle
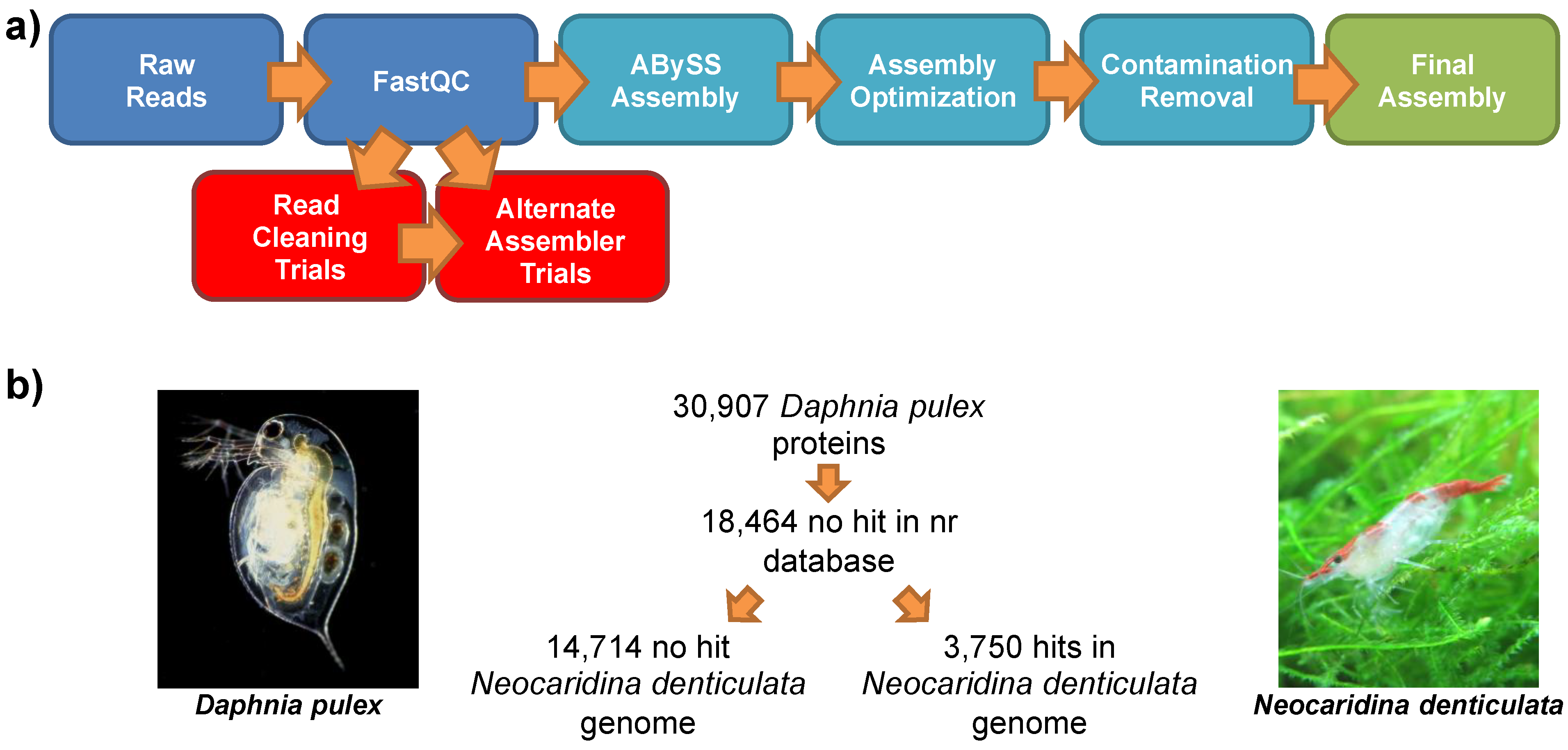
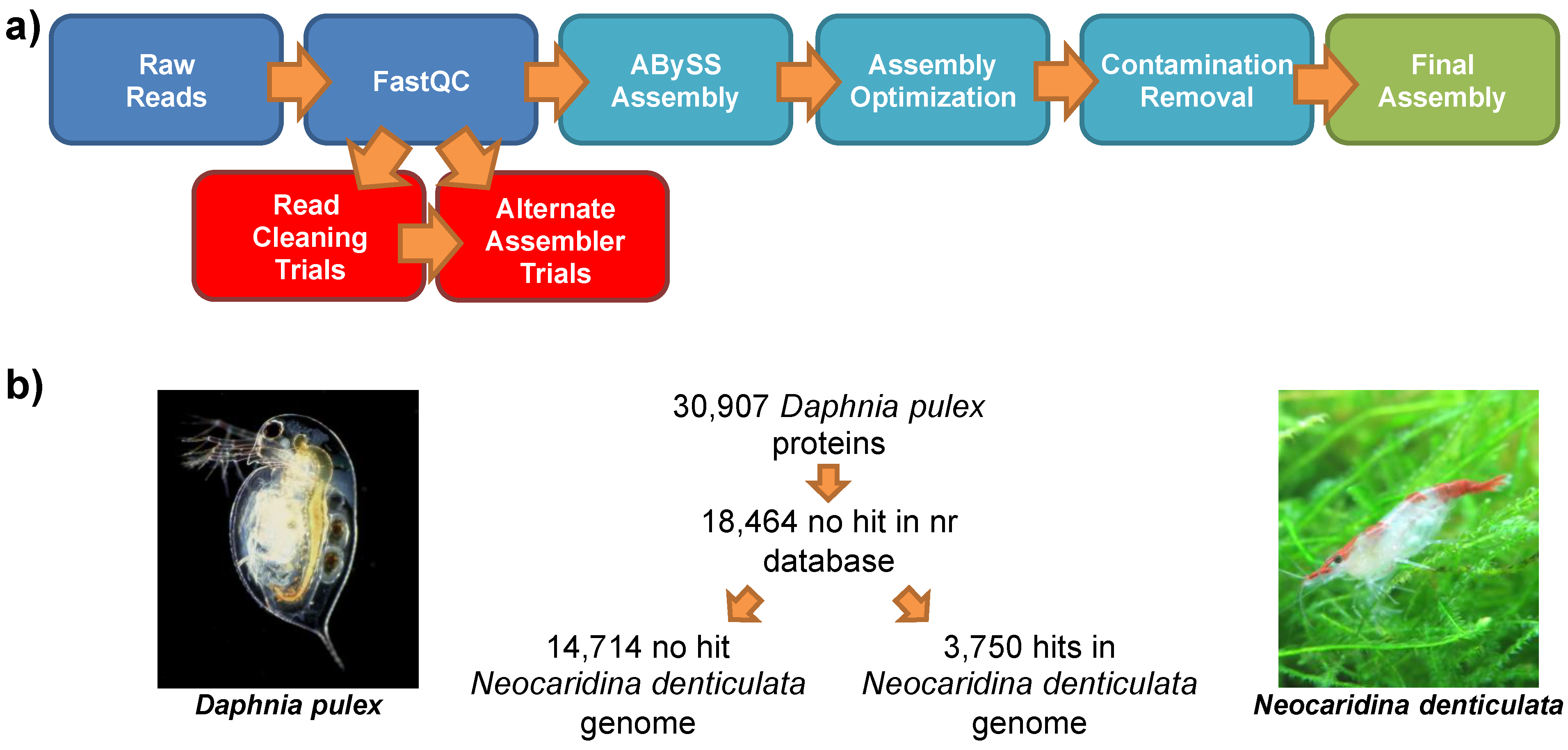
2.2. Genomic Sequencing

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Platform | Illumina HiSeq2000 |
|---|---|
| Number of Reads | 364,013,140 |
| Read Length (bp) | 100 |
| Average GC % | 36 |
| Fragment Size | 167.22 |
| Fragment Size SD (bp) | 12.01 |
2.3. Genomic Assembly
| Criteria | Value (base pairs) |
|---|---|
| Min. contig length | 200 |
| Max. contig length | 124,746 |
| Mean contig length | 383.84 |
| Standard deviation of contig length | 285.33 |
| Median contig length | 302 |
| N50 contig length | 400 |
| Number of contigs | 3,346,358 |
| Number of contigs ≥1 kb | 97,432 |
| Number of contigs in N50 | 987,201 |
| Number of bases in all contigs | 1,284,468,468 |
| Number of bases in contigs ≥1 kb | 132,397,543 |
| GC Content of contigs (%) | 35.21 |
2.4. Comparison of Core Eukaryotic Genes
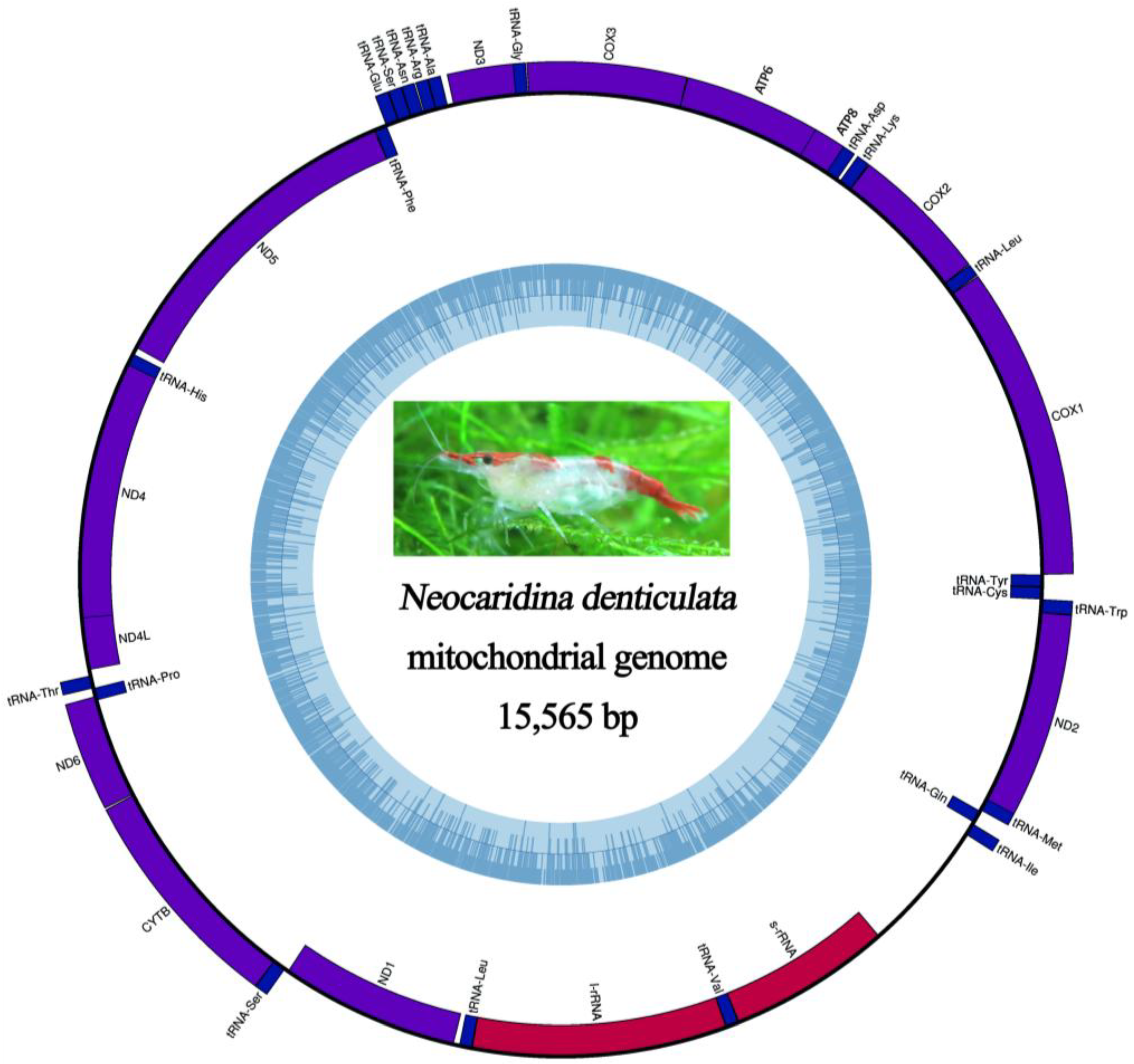
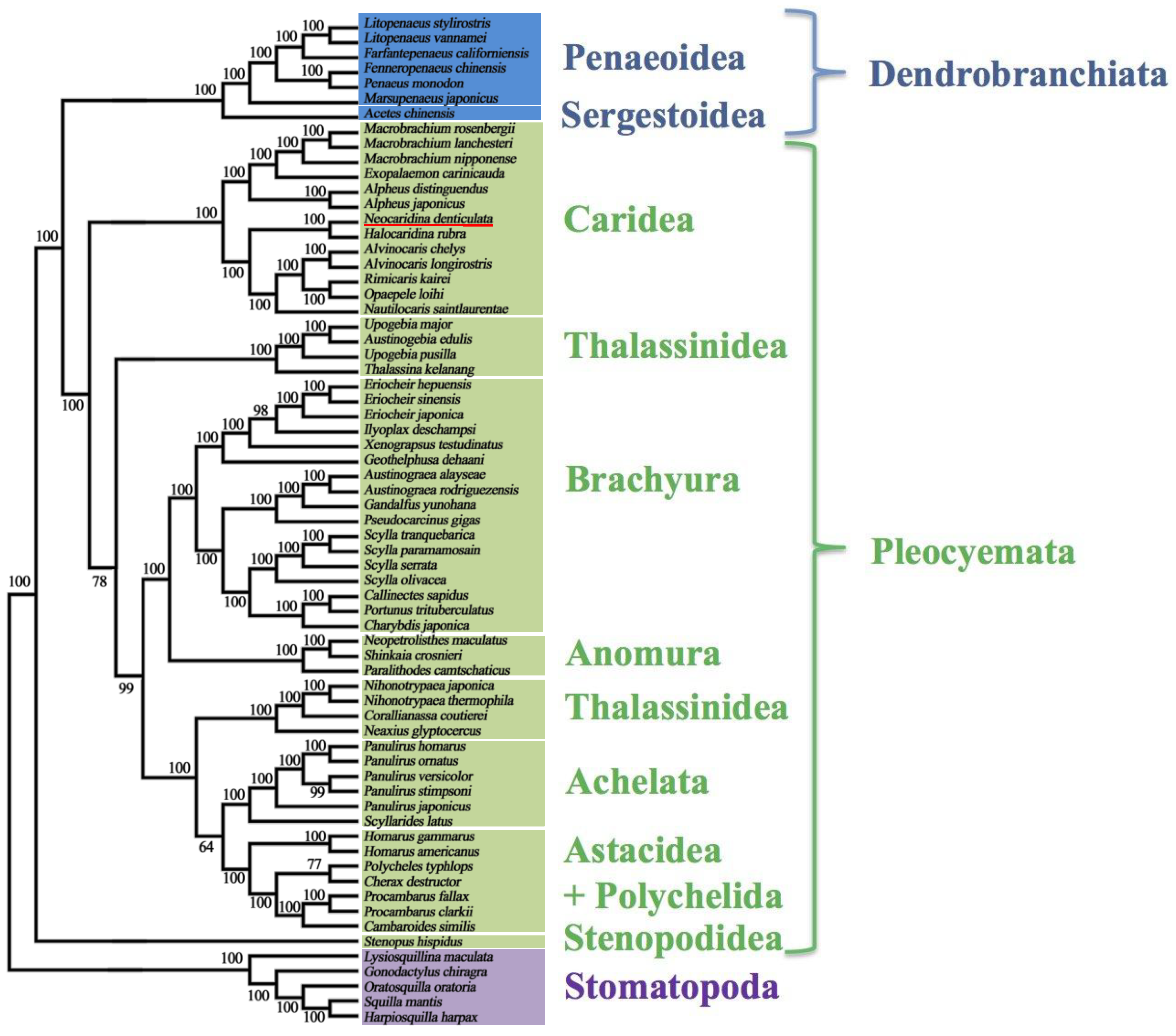
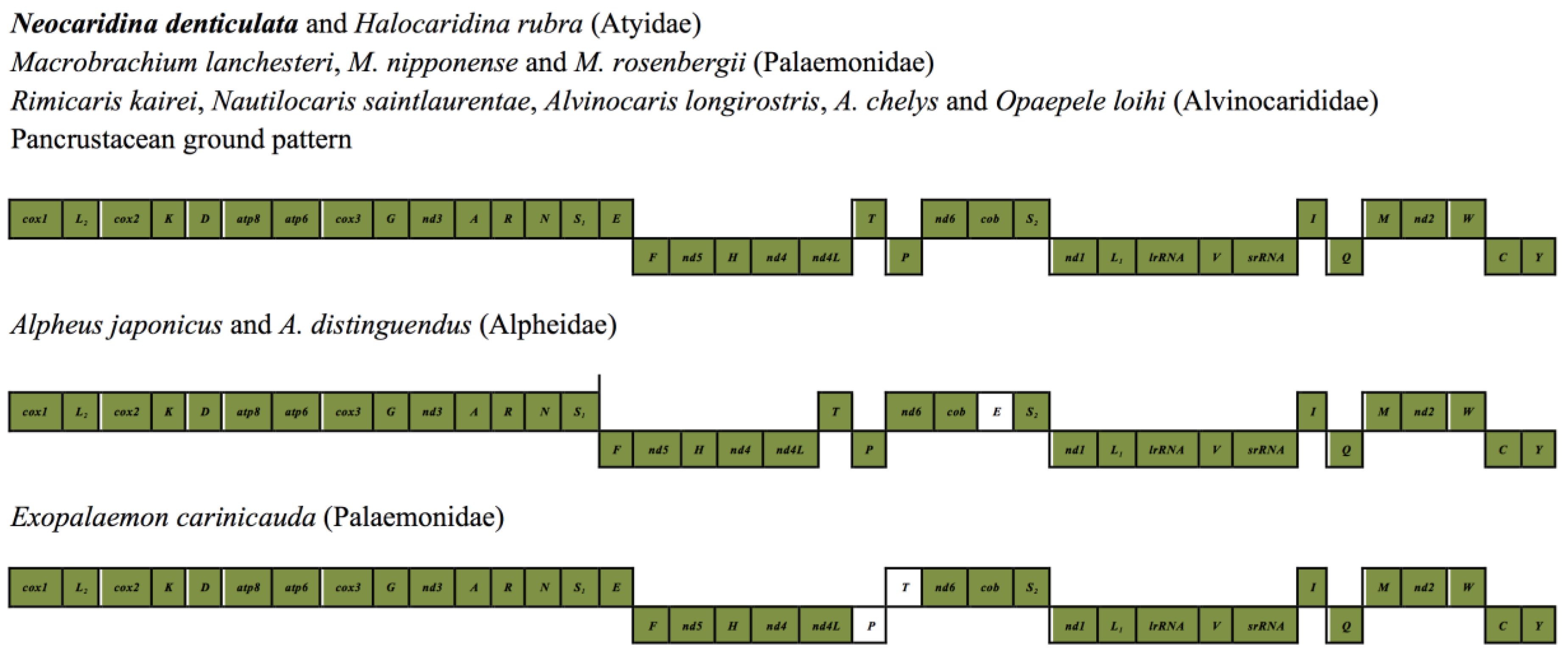
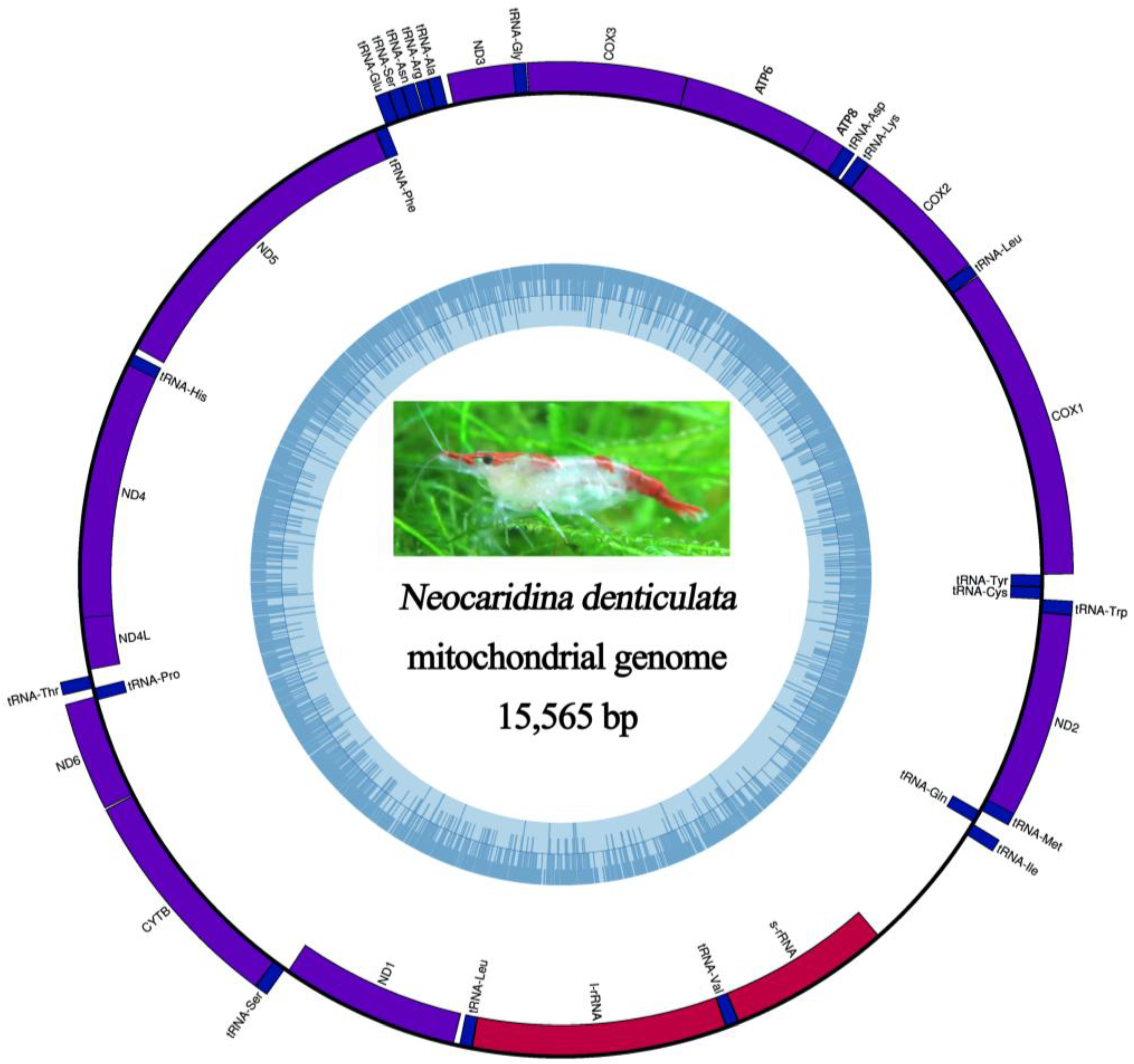
2.5. Mitochondrial Genomic Characteristics
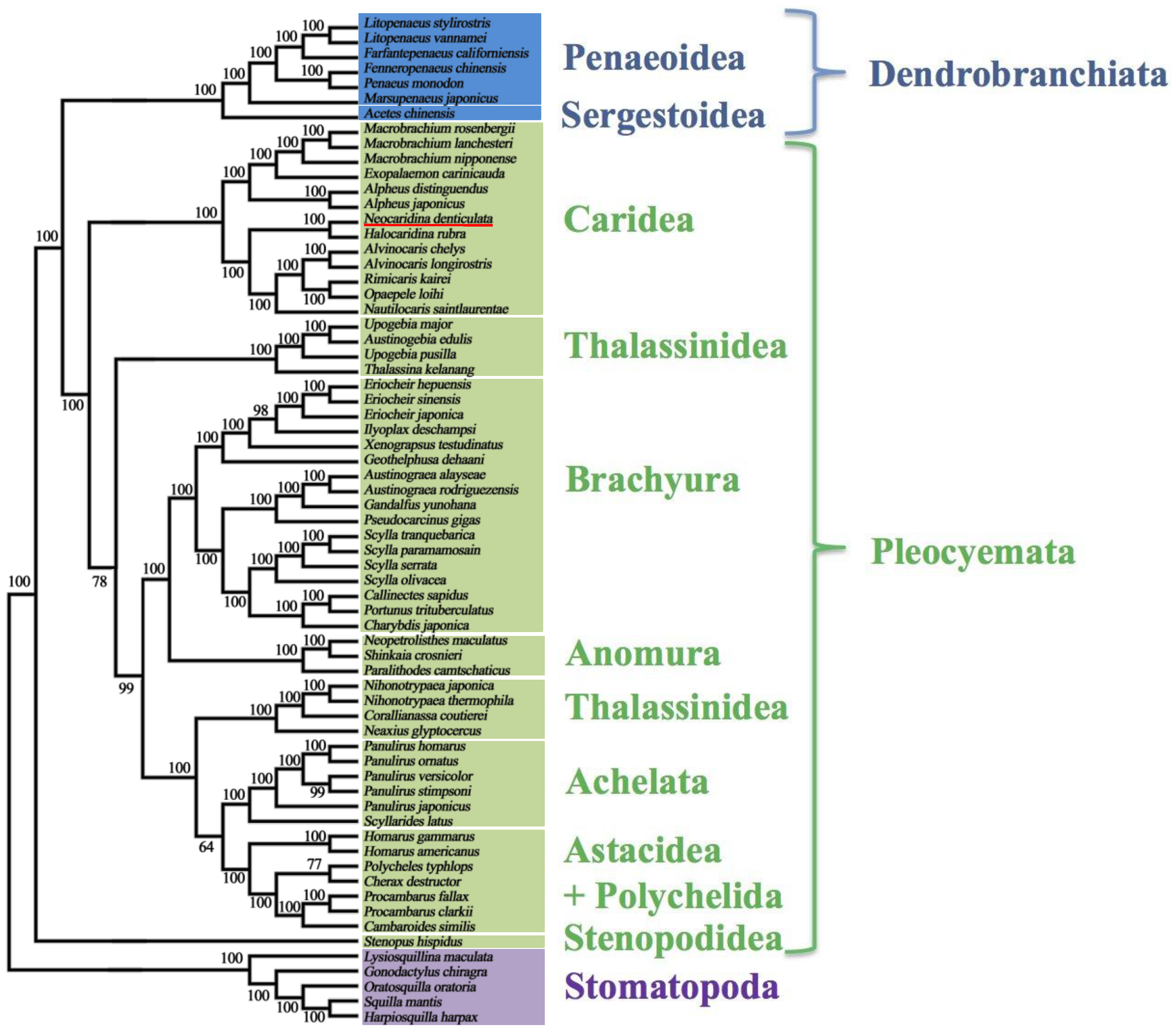
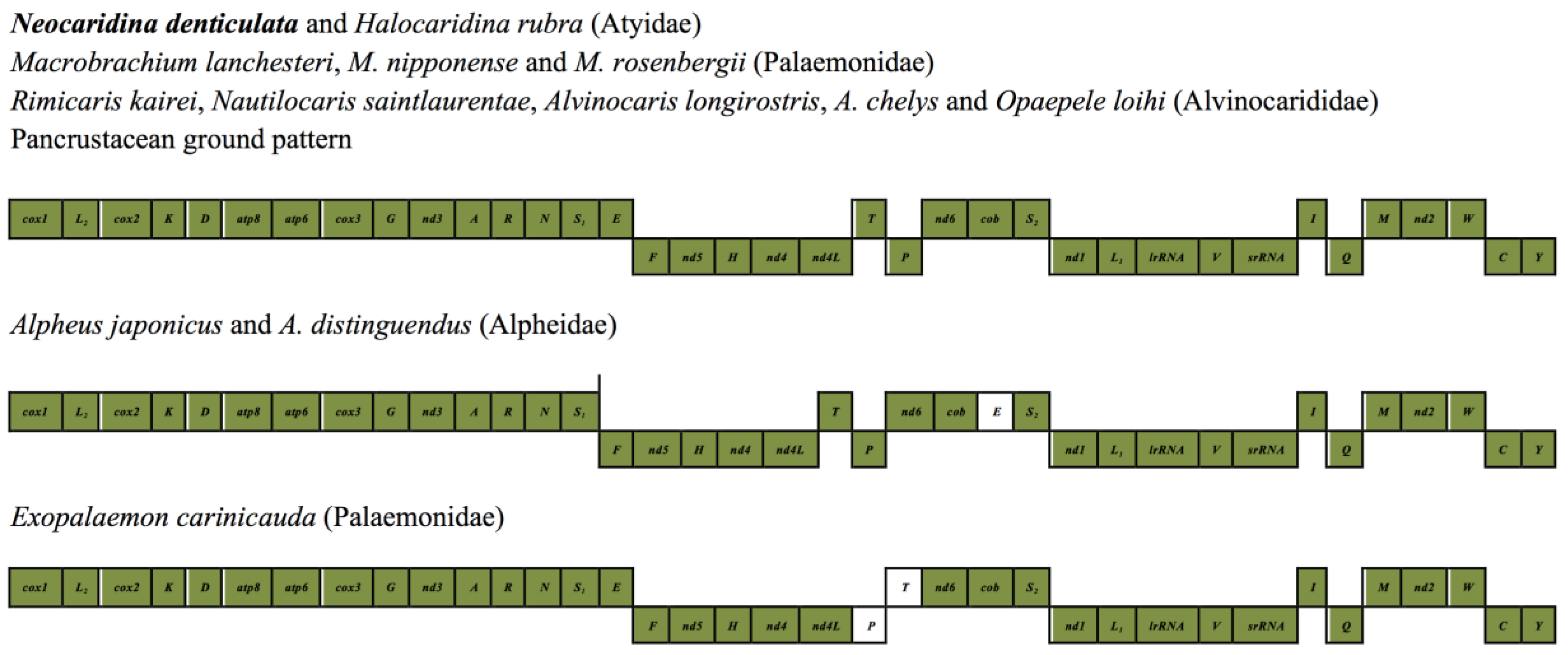
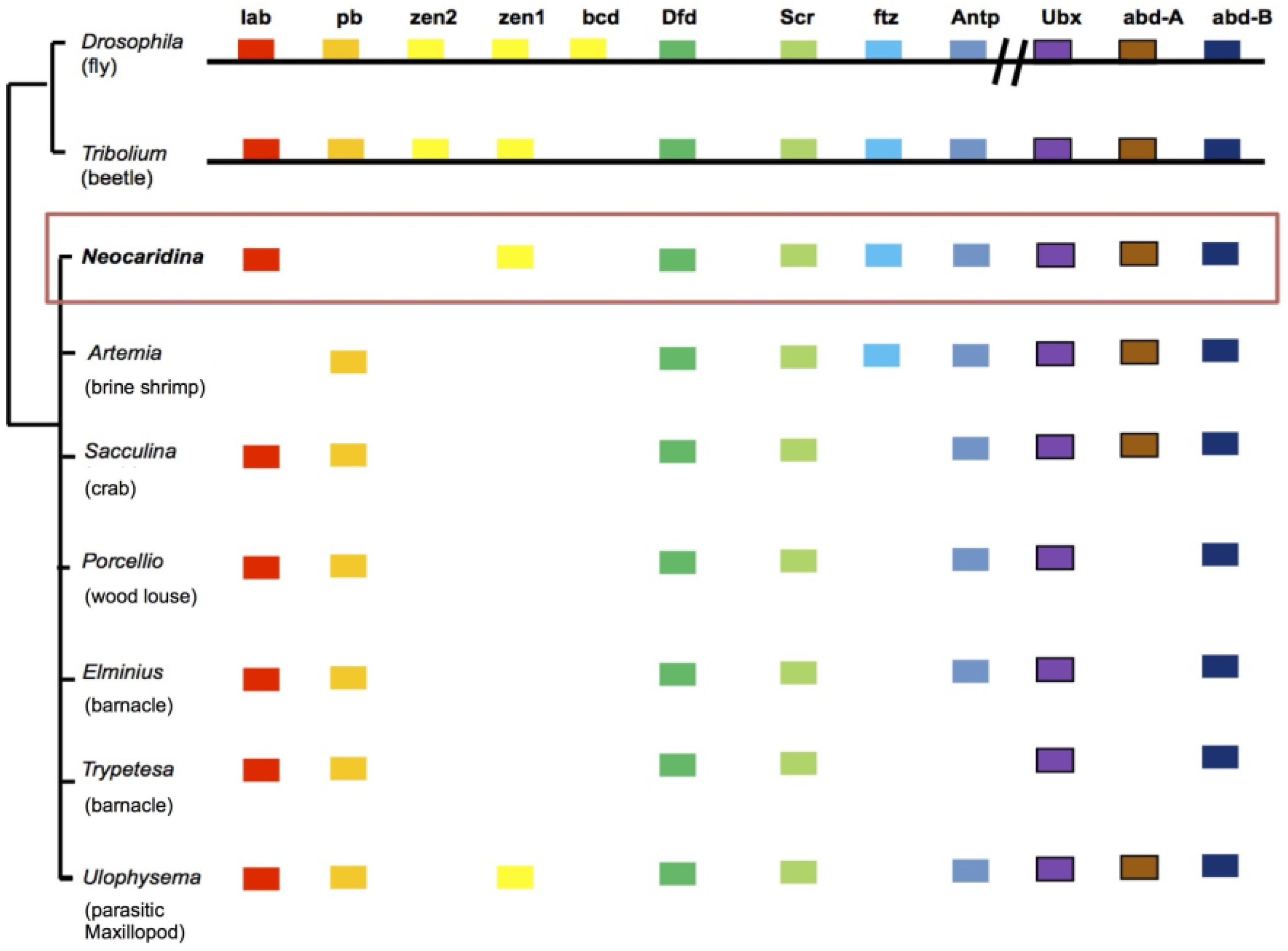
2.6. Recovery of Hox Genes and Other Families
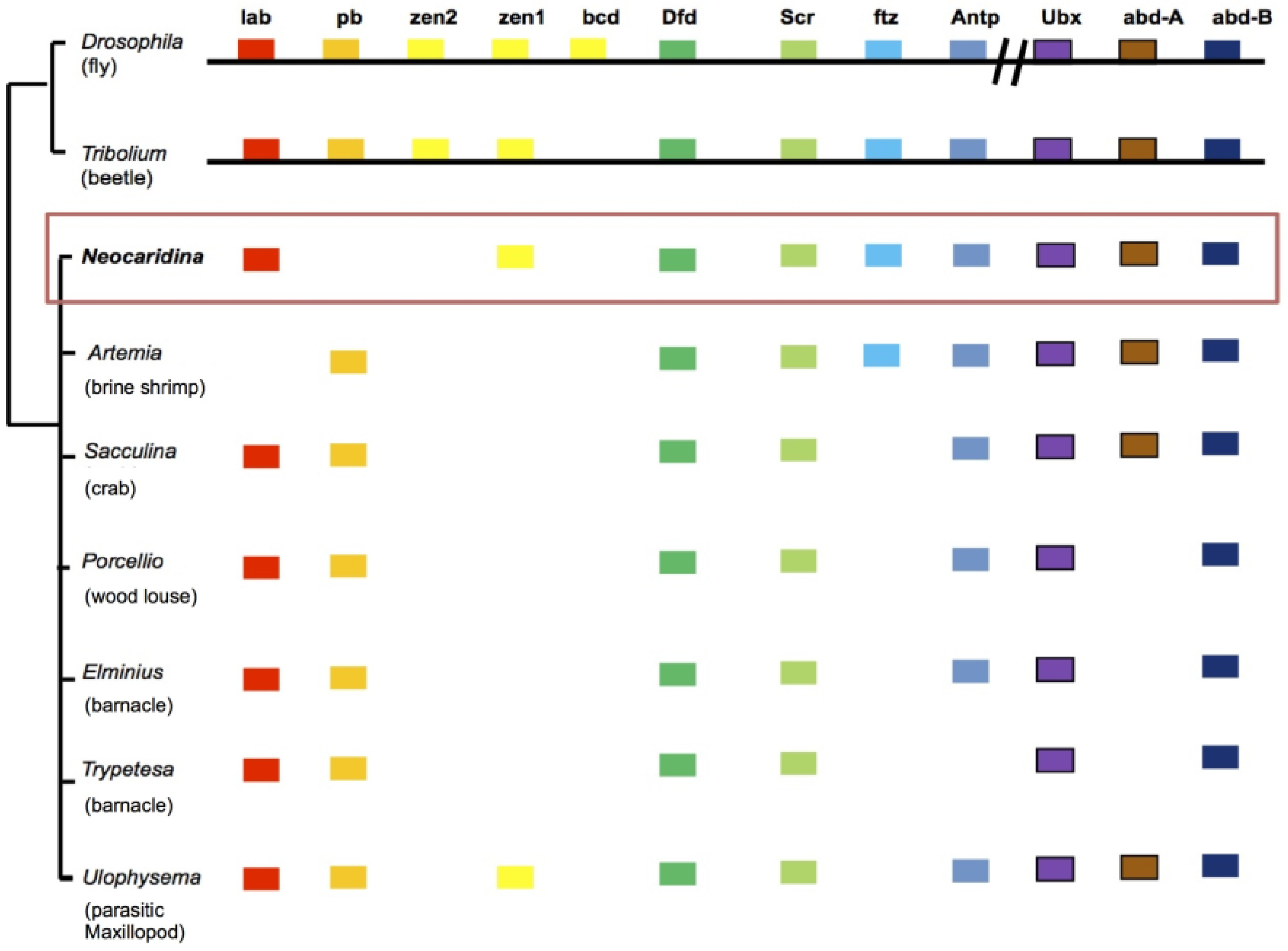
| Gene Classes | Homologues Recovered | Missing |
|---|---|---|
| Homeobox Genes (HoxL) | 20 | Pb |
| Fox Genes | 21 | Fox E, H, I, J2/3, L1, M, Q1 |
| T-box Genes | 10 | T-Brain, Tbx 4/5, Tbx 15/18/22 |
| miRNA processing genes | 8 | - |
2.7. Comparison to the Lineage-Specifically Gained Genes of Daphnia Pulex
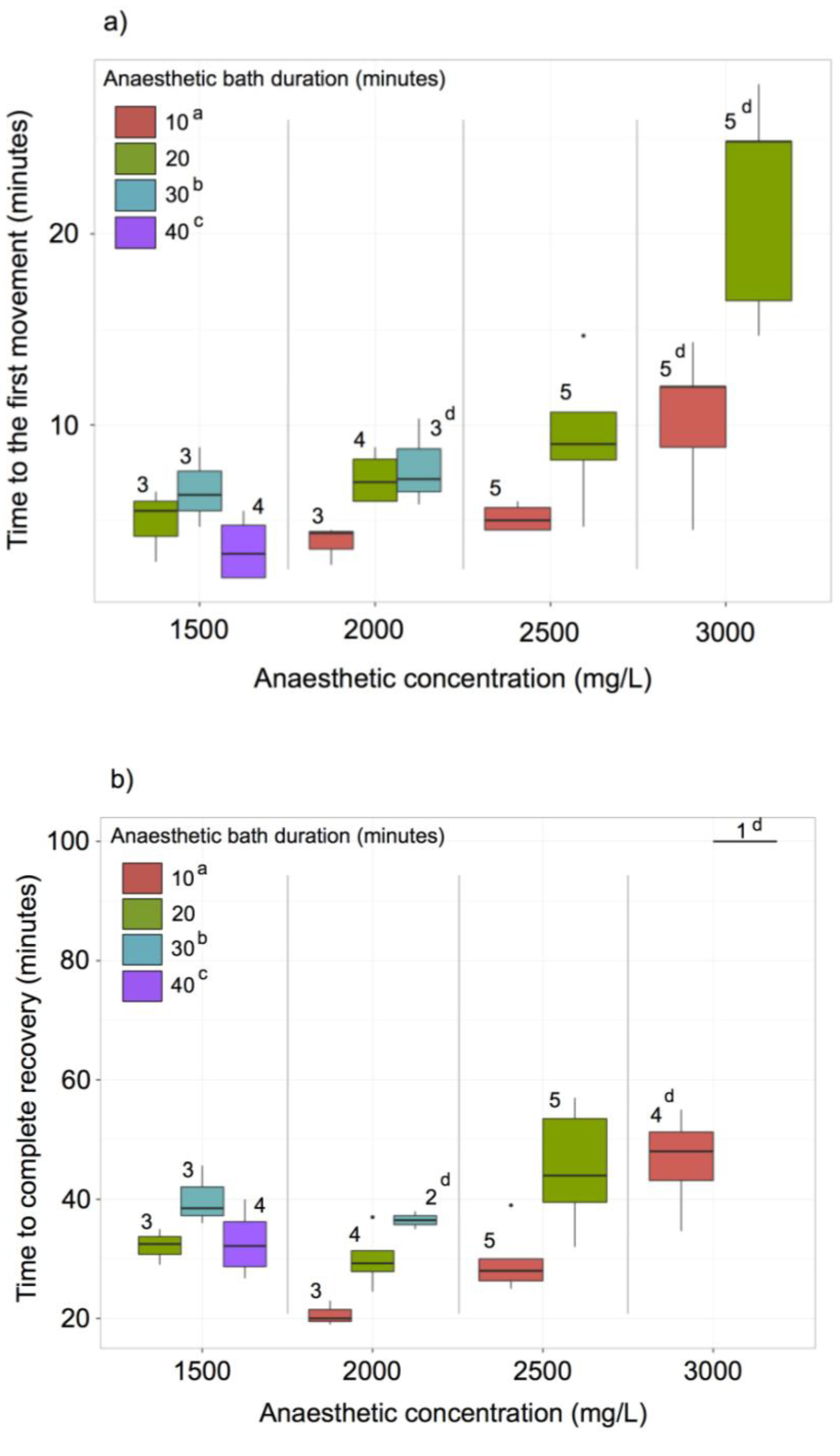
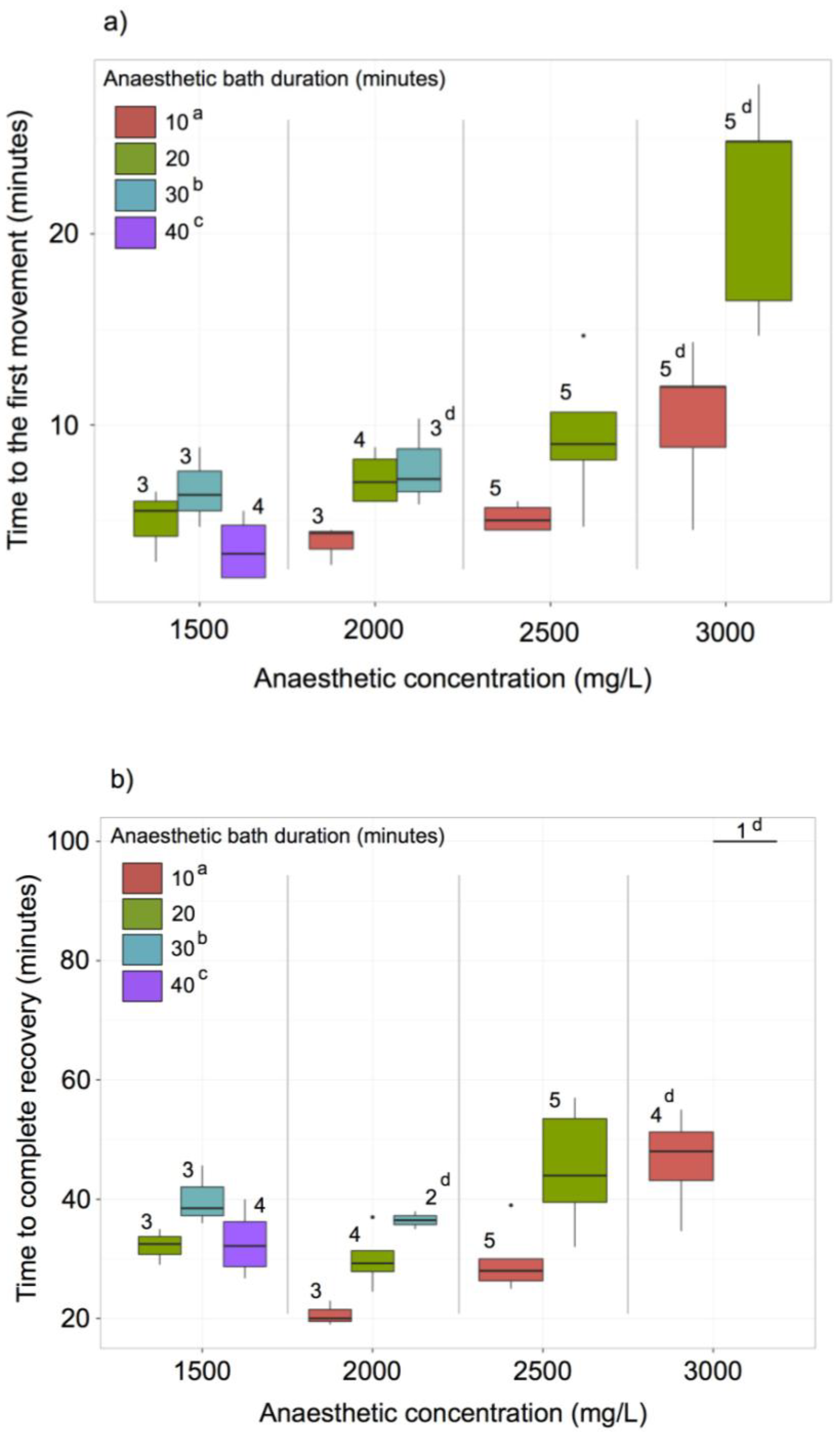
2.8. MS-222 Treatment
3. Experimental Section
3.1. Animal Husbandry and Genomic DNA Extraction
3.2. Illumina Hi-Seq and Assembly
3.3. mtDNA/ Nuclear Gene Retrieval
3.4. Gene Comparison
3.5. mtDNA Annotation and Display
3.6. MS222 Anaesthesia
4. Conclusions
Supplementary Files
Conflicts of Interest
Author Contributions
Acknowledgements
References
- Stillman, J.H.; Colbourne, J.K.; Lee, C.E.; Patel, N.H.; Phillips, M.R.; Towle, D.W.; Eads, B.D.; Gelembuik, G.W.; Henry, R.P.; Johnson, E.A.; et al. Recent advances in crustacean genomics. Integr. Comp. Biol. 2008, 48, 852–868. [Google Scholar] [CrossRef]
- Kenny, N.J.; Quah, S.; Holland, P.W.H.; Tobe, S.S.; Hui, J.H.L. How are comparative genomics and the study of microRNAs changing our views on arthropod endocrinology and adaptations to the environment? Gen. Comp. Endocrinol. 2013, 188, 16–22. [Google Scholar] [CrossRef]
- Martin, J.W.; Davis, G.E. An updated classification of the recent Crustacea. Sci. Ser. 2001, 39, 1–124. [Google Scholar]
- Zhang, Z.Q. Phylum Arthropoda von Siebold, 1848. Zootaxa 2011, 3148, 99–103. [Google Scholar]
- Regier, J.C.; Shultz, J.W.; Zwick, A.; Hussey, A.; Ball, B.; Wetzer, R.; Martin, J.W.; Cunningham, C.W. Arthropod relationships revealed by phylogenomic analysis of nuclear protein-coding sequences. Nature 2010, 463, 1079–1083. [Google Scholar] [CrossRef]
- Rota-Stabelli, O.; Campbell, L.; Brinkmann, H.; Edgecombe, G.D.; Longhorn, S.J.; Peterson, K.J.; Pisani, D.; Philippe, H.; Telford, M.J. A congruent solution to arthropod phylogeny: Phylogenomics, microRNAs and morphology support monophyletic Mandibulata. Proc. Biol. Sci. 2011, 278, 298–306. [Google Scholar] [CrossRef]
- Colbourne, J.K.; Pfrender, M.E.; Gilbert, D.; Thomas, W.K.; Tucker, A.; Oakley, T.H.; Tokishita, S.; Aerts, A.; Arnold, G.J.; Basu, M.K.; et al. The ecoresponsive genome of Daphnia pulex. Science 2011, 331, 555–561. [Google Scholar] [CrossRef]
- Rehm, E.J.; Hannibal, R.L.; Chaw, R.C.; Vargas-Vila, M.A.; Patel, N.H. The crustacean Parhyale hawaiensis: A new model for arthropod development. Cold Spring Harb. Protoc. 2009, 1, 373–404. [Google Scholar]
- Sloof, W.; de Kruijf, H.; Hopkin, S.P.; Jones, D.T.; Dietrich, D. The isopod Porcellio scaber as a monitor of the bioavailability of metals in terrestrial ecosystems: Towards a global “woodlouse watch” scheme. Sci. Total Environ. 1993, 134, 357–365. [Google Scholar] [CrossRef]
- Hart, R.C. Population dynamics and production of the tropical freshwater shrimp Caridina nilotica (Decapoda: Atyidae) in the littoral of Lake Sibaya. Freshw. Biol. 1981, 11, 531–547. [Google Scholar] [CrossRef]
- De Silva, P.K.; de Silva, K.H.G.M. Aspects of the population ecology of a tropical freshwater atyid shrimp Caridina fernandoi Arud. & Costa, 1962 (Crustacea: Decapoda: Caridea). Arch. Hydrobiol. 1962, 117, 237–253. [Google Scholar]
- De Grave, S.; Pentcheff, N.D.; Ahyong, S.T.; Chan, T.Y.; Crandall, K.A.; Dworschak, P.C.; Felder, D.L.; Feldmann, R.M.; Fransen, C.H.J.M.; Goulding, L.Y.D.; et al. A classification of living and fossil genera of decapod crustaceans. Raffles Bull. Zool. 2009, 21, 1–109. [Google Scholar]
- Huang, D.; Chen, H. Effects of chlordane and lindane on testosterone and vitellogenin levels in green neon shrimp (Neocaridina denticulata). Int. J. Toxicol. 2004, 23, 91–95. [Google Scholar]
- Huang, D.-J.; Chen, H.-C.; Wu, J.-P.; Wang, S.-Y. Reproduction obstacles for the female green neon shrimp (Neocaridina denticulata) after exposure to chlordane and lindane. Chemosphere 2006, 64, 11–16. [Google Scholar]
- Mizue, K.; Iwamoto, Y. On the development and growth of Neocaridina denticulata de Haan. Bull. Fac. Fish. Nagasaki Univ. 1961, 10, 15–24. [Google Scholar]
- Liang, X. Crustacea: Decapoda: Atyidae. In Fauna Sinica: Invertebrata; Science Press: Beijing, China, 2004; Volume 36. [Google Scholar]
- Bracken, H.; de Grave, S.; Felder, D. Phylogeny of the infraorder Caridea based on mitochondrial and nuclear genes (Crustacea: Decapoda). Decapod Crustac. 2009, 18, 281–305. [Google Scholar] [CrossRef]
- Englund, R.; Cai, Y. Occurrence and description of Neocaridina denticulata sinensis (Kemp, 1918) (Crustacea: Decapoda: Atyidae), a new introduction to the Hawaiian Islands. Bish. Museum Occas. Pap. 1999, 58, 58–65. [Google Scholar]
- Oh, C.-W.; Ma, C.-W.; Hartnoll, R.G.; Suh, H.-L. Reproduction and population dynamics of the temperate freshwater shrimp, Neocaridina denticulata denticulata (De Haan, 1844), in a Korean stream. Crustaceana 2003, 76, 993–1015. [Google Scholar] [CrossRef]
- Dudgeon, D. The population dynamics of some freshwater carideans (Crustacea: Decapoda) in Hong Kong, with special reference to Neocaridina serrata (Atyidae). Hydrobiologia 1985, 120, 141–149. [Google Scholar] [CrossRef]
- Marshall, S.; Warburton, K.; Paterson, B.; Mann, D. Cannibalism in juvenile blue-swimmer crabs Portunus pelagicus (Linnaeus, 1766): Effects of body size, moult stage and refuge availability. Appl. Anim. Behav. 2005, 90, 65–82. [Google Scholar] [CrossRef]
- Hung, M.; Chan, T.; Yu, H. Atyid shrimps (Decapoda: Caridea) of Taiwan, with descriptions of three new species. J. Crustac. Biol. 1993, 13, 481–503. [Google Scholar] [CrossRef]
- Shy, J.; Ho, P.; Yu, H. Complete larval development of Neocaridina denticulata (De Haan, 1884) (Crustacean, Decapoda, Caridea) reared in the laboratory. Ann. Taiwan Mus. 1992, 35, 75–89. [Google Scholar]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef]
- Simpson, J.T.; Wong, K.; Jackman, S.D.; Schein, J.E.; Jones, S.J.; Birol, I. ABySS: A parallel assembler for short read sequence data. Genome Res. 2009, 19, 1117–1123. [Google Scholar] [CrossRef]
- Sickle Github Repository. Available online: https://github.com/najoshi/sickle (accessed on 21 April 2013).
- Liu, Y.; Schröder, J.; Schmidt, B. Musket: A multistage k-mer spectrum-based error corrector for Illumina sequence data. Bioinformatics 2013, 29, 308–315. [Google Scholar] [CrossRef]
- Neocaridina denticulata Genome Website. Available online: http://tiny.cc/shrimpgenome/ (accessed on 18 July 2013).
- Bachmann, K.; Rheinsmith, E.L. Nuclear DNA amounts in Pacific Crustacea. Chromosoma 1973, 43, 225–236. [Google Scholar] [CrossRef]
- Gewin, V. Functional genomics thickens the biological plot. PLoS Biol. 2005, 3, e219. [Google Scholar] [CrossRef]
- Parra, G.; Bradnam, K.; Korf, I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 2007, 23, 1061–1067. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar]
- Yu, Y.-Q.; Yang, W.-J.; Yang, J.-S. The complete mitogenome of the Chinese swamp shrimp Neocaridina denticulata sinensis Kemp 1918 (Crustacea: Decapoda: Atyidae). Mitochondrial DNA 2013. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef]
- Shen, X.; Li, X.; Sha, Z.; Yan, B.; Xu, Q. Complete mitochondrial genome of the Japanese snapping shrimp Alpheus japonicus (Crustacea: Decapoda: Caridea): Gene rearrangement and phylogeny within Caridea. Sci. China Life Sci. 2012, 55, 591–598. [Google Scholar] [CrossRef]
- Lavrov, D.V.; Boore, J.L.; Brown, W.M. The complete mitochondrial DNA sequence of the horseshoe crab Limulus polyphemus. Mol. Biol. Evol. 2000, 17, 813–824. [Google Scholar] [CrossRef]
- Cook, C.E.; Smith, M.L.; Telford, M.J.; Bastianello, A.; Akam, M. Hox genes and the phylogeny of the arthropods. Curr. Biol. 2001, 11, 759–763. [Google Scholar] [CrossRef]
- Kaestner, K.H.; Knoechel, W.; Martinez, D.E. Unified nomenclature for the winged helix/forkhead transcription factors. Genes Dev. 2000, 14, 142–146. [Google Scholar]
- Shimeld, S.M.; Degnan, B.; Luke, G.N. Evolutionary genomics of the Fox genes: Origin of gene families and the ancestry of gene clusters. Genomics 2010, 95, 256–260. [Google Scholar] [CrossRef]
- Papaioannou, V.E.; Silver, L.M. The T-box gene family. Bioessays 1998, 20, 9–19. [Google Scholar] [CrossRef]
- Tagawa, K.; Humphreys, T.; Satoh, N. T-brain expression in the apical organ of hemichordate tornaria larvae suggests its evolutionary link to the vertebrate forebrain. J. Exp. Zool. 2000, 288, 23–31. [Google Scholar] [CrossRef]
- Kenny, N.J.; Shimeld, S.M. Additive multiple k-mer transcriptome of the keelworm Pomatoceros lamarckii (Annelida; Serpulidae) reveals annelid trochophore transcription factor cassette. Dev. Genes Evol. 2012, 222, 325–339. [Google Scholar] [CrossRef]
- Winter, J.; Jung, S.; Keller, S.; Gregory, R.I.; Diederichs, S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat. Cell Biol. 2009, 11, 228–34. [Google Scholar] [CrossRef]
- Sneddon, L.U. Clinical anesthesia and analgesia in fish. J. Exotic Pet. Med. 2012, 21, 32–43. [Google Scholar] [CrossRef]
- Schmit, O.; Mezquita, F. Experimental test on the use of MS-222 for ostracod anaesthesia: concentration, immersion period and recovery time. J. Limnol. 2010, 69, 350–352. [Google Scholar]
- Coyle, S.D.; Dasgupta, S.; Tidwell, J.H.; Beavers, T.; Bright, L.A.; Yasharian, D.K. Comparative efficacy of anesthetics for the freshwater prawn Macrobrachiurn rosenbergii. J. World Aquac. Soc. 2007, 36, 282–290. [Google Scholar] [CrossRef]
- Hajek, G.; Choczewski, M.; Dziaman, R.; Klyszejko, B. Evaluation of immobilizing methods for the Chinese mitten crab, Eriocheir sinensis. Electron. J. Pol. Agric. Univ. 2009, 12, 1. [Google Scholar]
- Brown, P.; White, M. Evaluation of three anesthetic agents for crayfish (Orconectes virilis). J. Shellfish Res. 1996, 15, 433–436. [Google Scholar]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- JGI Daphnia pulex Genome Resources. Available online: http://genome.jgi-psf.org/Dappu1/Dappu1.home.html (accessed on 25 September 2013).
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. Clustal-W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kenny, N.J.; Sin, Y.W.; Shen, X.; Zhe, Q.; Wang, W.; Chan, T.F.; Tobe, S.S.; Shimeld, S.M.; Chu, K.H.; Hui, J.H.L. Genomic Sequence and Experimental Tractability of a New Decapod Shrimp Model, Neocaridina denticulata. Mar. Drugs 2014, 12, 1419-1437. https://doi.org/10.3390/md12031419
Kenny NJ, Sin YW, Shen X, Zhe Q, Wang W, Chan TF, Tobe SS, Shimeld SM, Chu KH, Hui JHL. Genomic Sequence and Experimental Tractability of a New Decapod Shrimp Model, Neocaridina denticulata. Marine Drugs. 2014; 12(3):1419-1437. https://doi.org/10.3390/md12031419
Chicago/Turabian StyleKenny, Nathan J., Yung Wa Sin, Xin Shen, Qu Zhe, Wei Wang, Ting Fung Chan, Stephen S. Tobe, Sebastian M. Shimeld, Ka Hou Chu, and Jerome H. L. Hui. 2014. "Genomic Sequence and Experimental Tractability of a New Decapod Shrimp Model, Neocaridina denticulata" Marine Drugs 12, no. 3: 1419-1437. https://doi.org/10.3390/md12031419
APA StyleKenny, N. J., Sin, Y. W., Shen, X., Zhe, Q., Wang, W., Chan, T. F., Tobe, S. S., Shimeld, S. M., Chu, K. H., & Hui, J. H. L. (2014). Genomic Sequence and Experimental Tractability of a New Decapod Shrimp Model, Neocaridina denticulata. Marine Drugs, 12(3), 1419-1437. https://doi.org/10.3390/md12031419