Geographic Variability and Anti-Staphylococcal Activity of the Chrysophaentins and Their Synthetic Fragments

Abstract
:1. Introduction
2. Results and Discussion
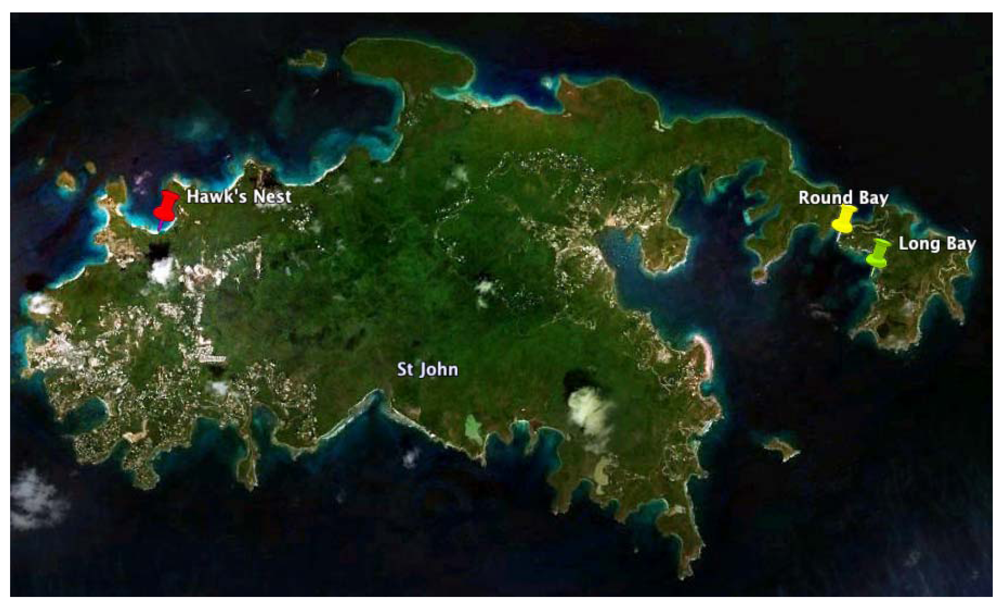
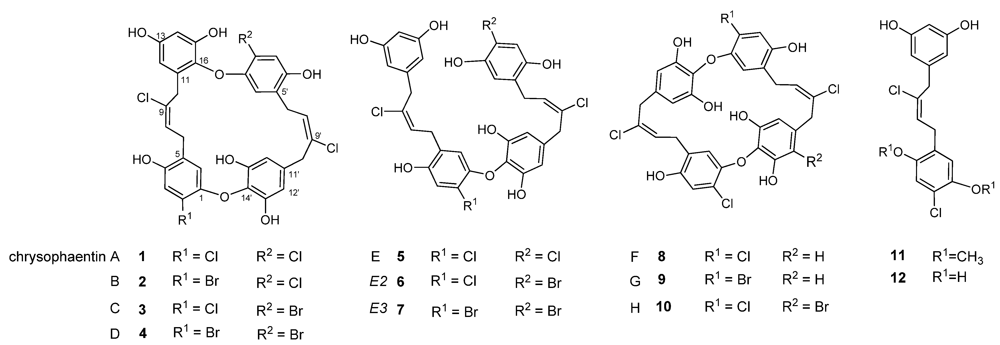
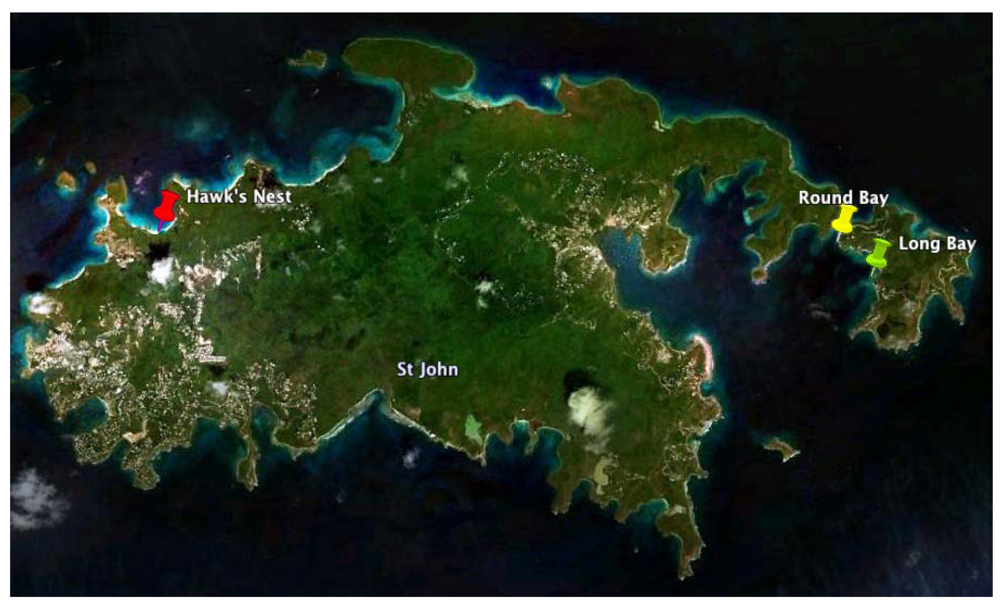
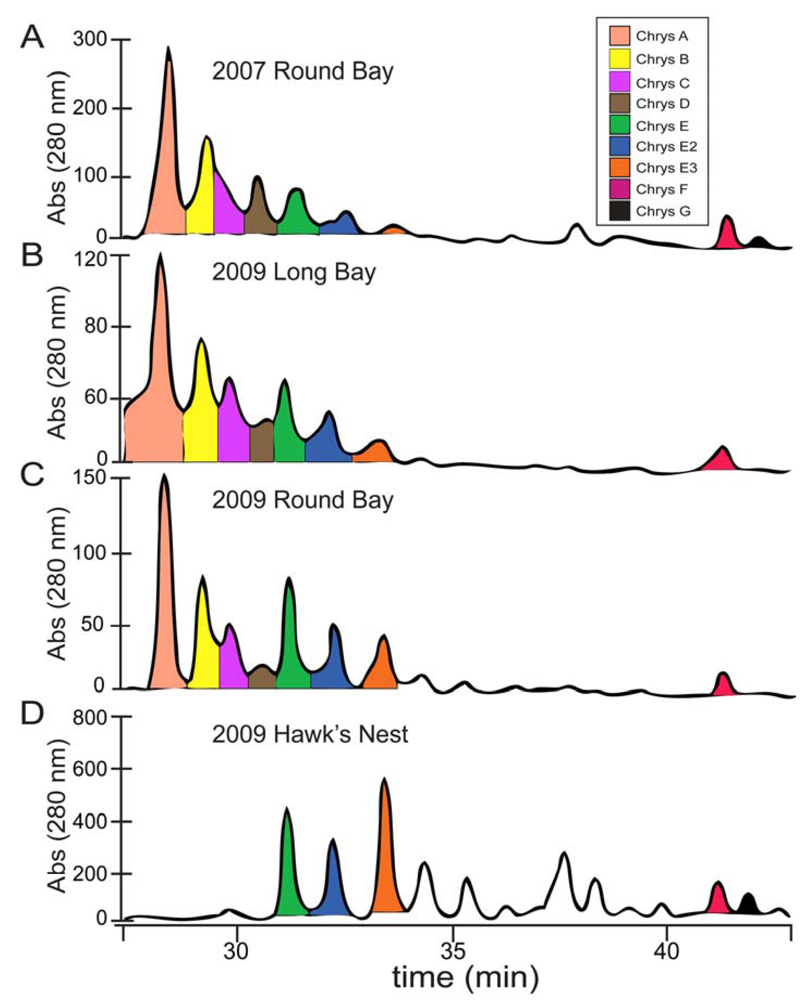
2.1. Chrysophaeum taylori Collection and Chrysophaentin Identification
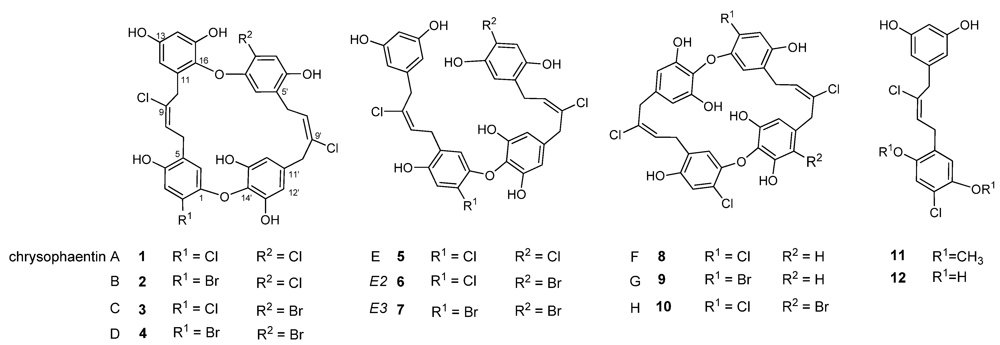
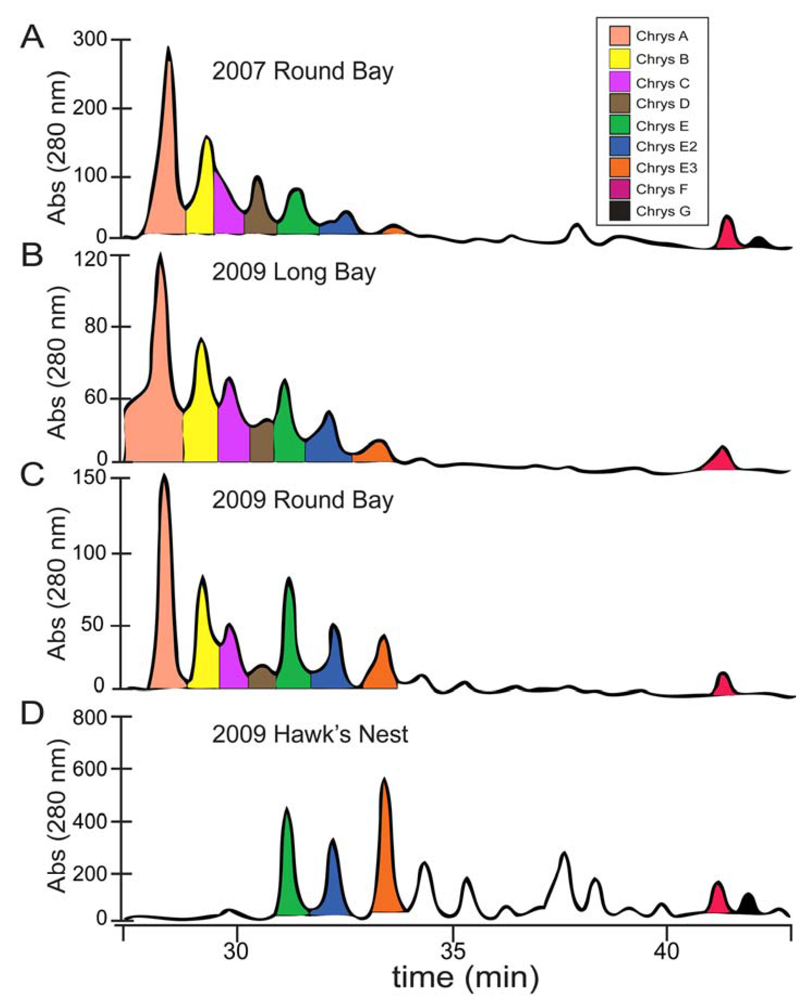
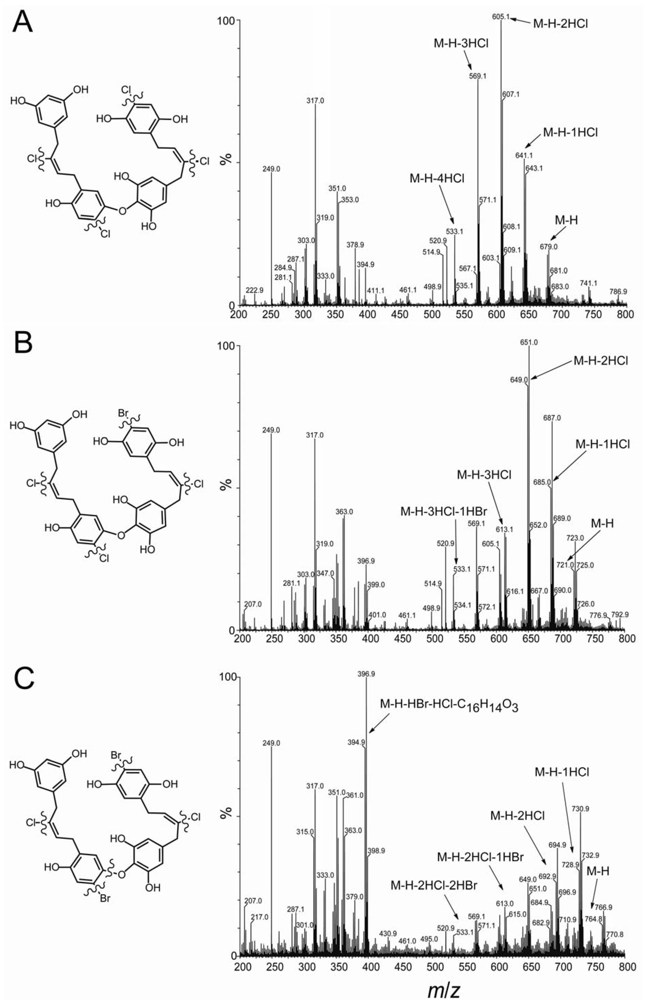
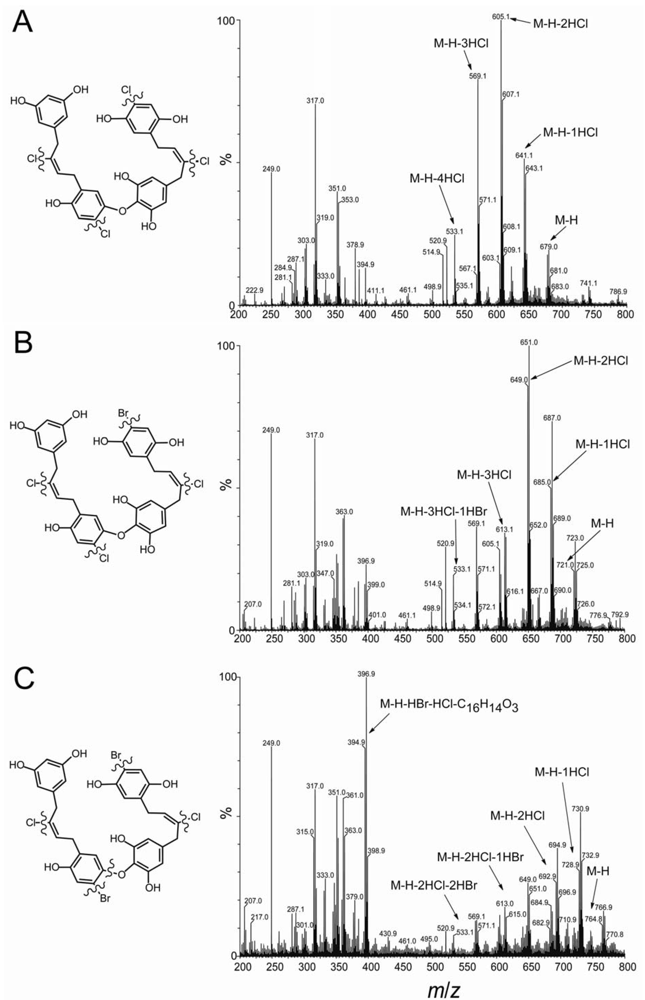
2.2. Identification of New Acyclic Chrysophaentins
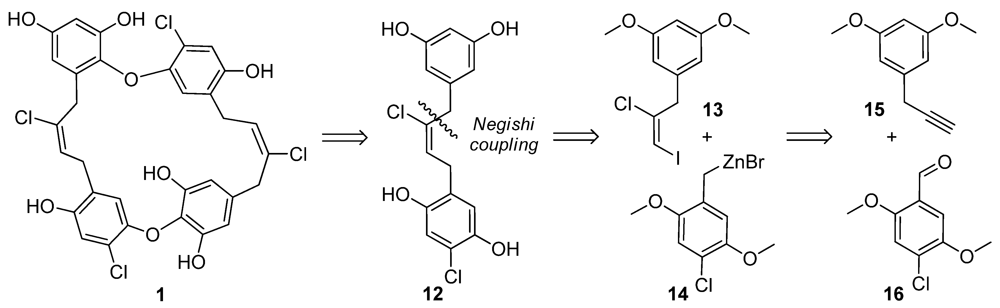
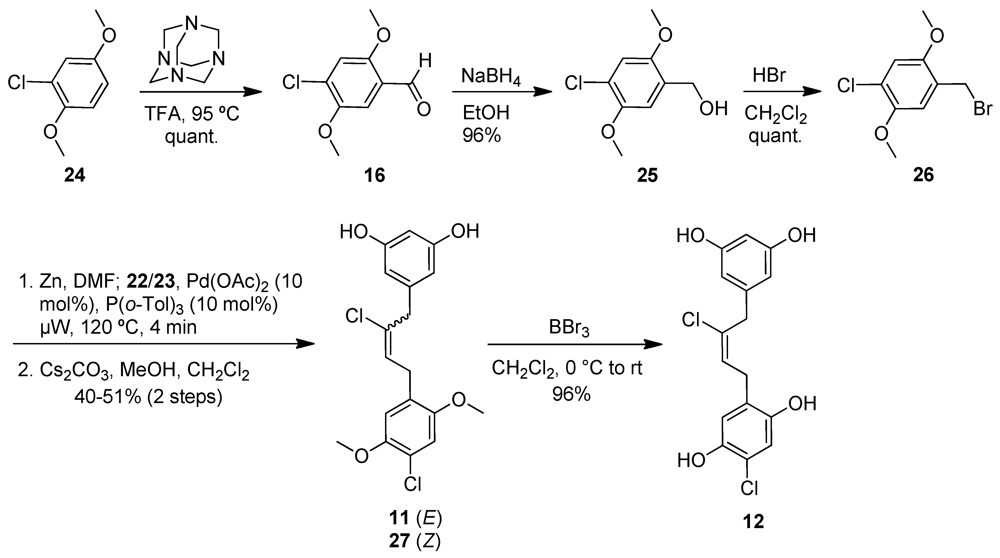
2.3. Chrysophaentin Fragment Synthesis
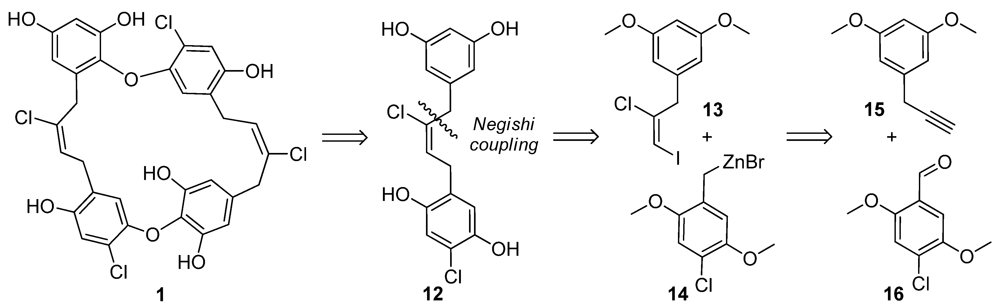
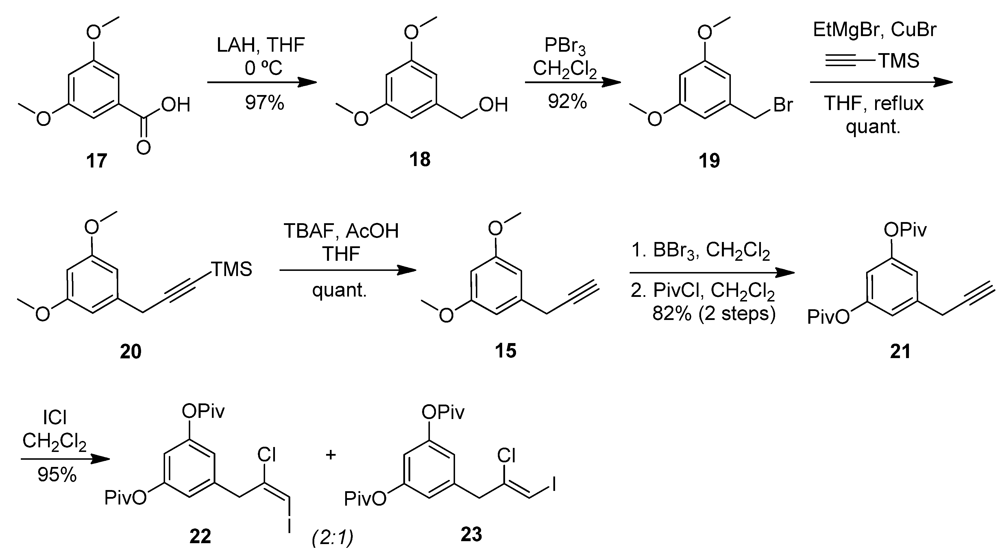
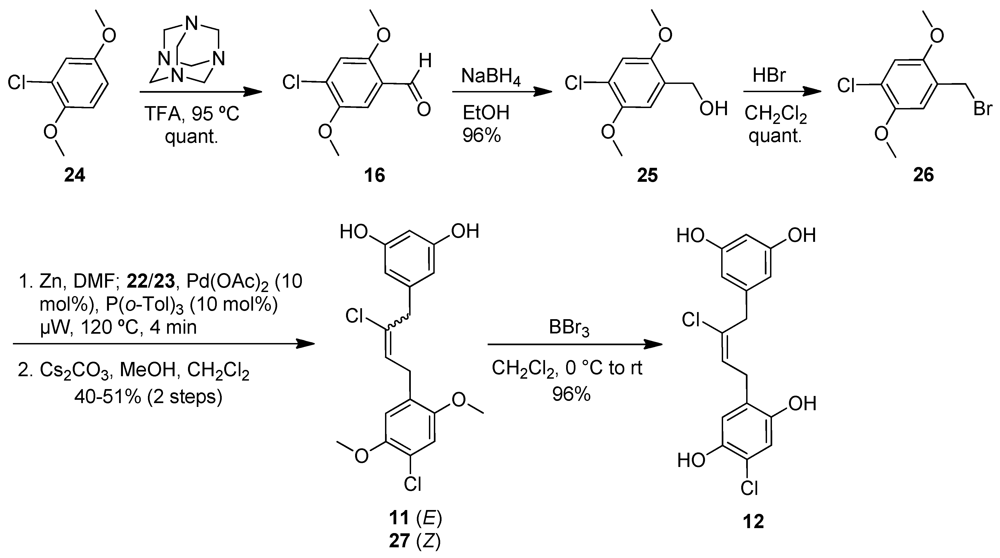
2.4. Antibacterial Evaluation
2.4.1. Minimum Bactericidal Concentrations for Select Natural Chrysophaentins

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. aureus 25923 | MRSA BAA-41 | MDRSA BAA-44 | S. aureus UAMS-1 | CA-MRSA USA300-LAC | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MIC50 | MIC90 | MBC | MIC50 | MIC90 | MBC | MIC50 | MIC90 | MIC50 | MIC90 | MBC | MIC50 | MIC90 | MBC | |
| 1 | 2.7 ± 0.9 | 9.2 | 37 | 2.3 ± 1.0 | 4.6 | 37 | 1.8 ± 0.5 | 9.2 | 5.1 ± 2.1 | 9.2 | 19 | 5.0 ± 2.4 | 9.2 | 19 |
| 4 | 37 ± 16 | 65 | n.d. | 26 ± 8.5 | 65 | n.d. | n.d. | n.d. | 49 ± 23 | 65 | >65 | 41 ± 20 | 65 | >65 |
| 5 | 16 ± 5.3 | 37 | 74 | 14 ± 4.1 | 37 | 74 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| 8 | 7.9 ± 2.9 | 19 | 74 | 6.3 ± 1.9 | 19 | 74 | n.d. | n.d. | 12 ± 5.6 | 37 | >74 | 12 ± 5.1 | 37 | 74 |
| 9 | 23 ± 7.5 | 69 | n.d. | 17 ± 4.3 | 69 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| 10 | 5.9 ± 1.8 | 17 | n.d. | 6.2 ± 1.9 | 17 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| 11 | 12 ± 4.3 | 34 | 68 | 11 ± 5.4 | 34 | 68 | 13 ± 4.7 | 34 | 20 ± 10 | 34 | 68 | 18 ± 8.4 | 34 | 68 |
| 12 | 20 ± 5.2 | 74 | 150 | 23 ± 9.9 | 74 | 150 | 27 ± 9.4 | 74 | 31 ± 13 | 74 | 150 | 29 ± 9.9 | 74 | 150 |
2.4.2. Antibacterial Activity of Synthetic Chrysophaentin Fragments
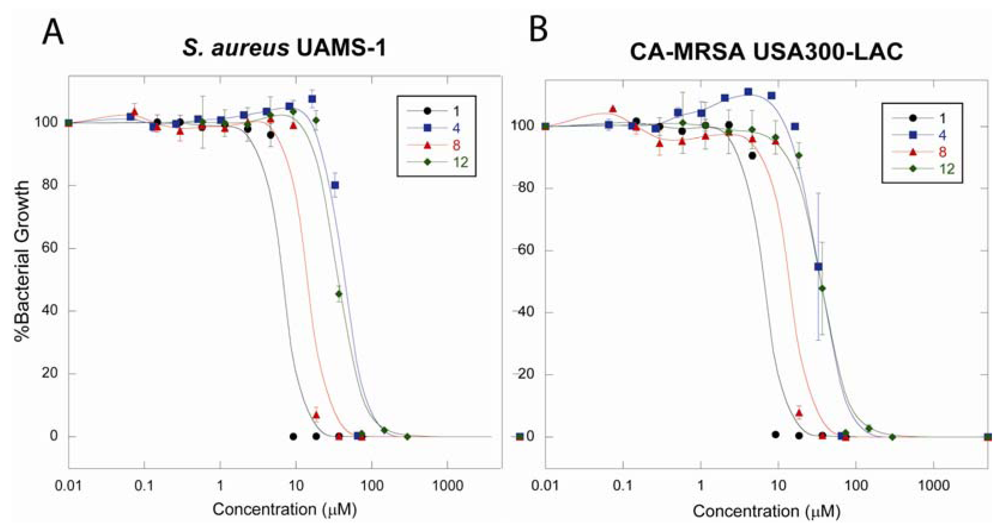
2.4.3. Antibacterial Activity against S. aureus UAMS-1 and CA-MRSA USA300-LAC
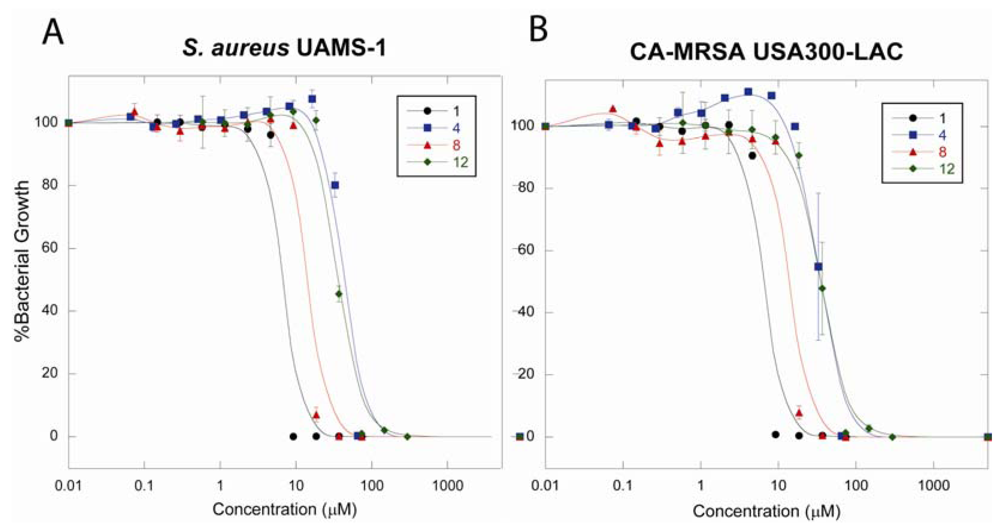
2.5. Discussion
3. Experimental Section
3.1. Biological Material
3.2. LC-MS
3.3. Mass Spectrometry and MS/MS Fragmentation
3.4. General Synthesis
3.5. Anti-Staphylococcal Activity
4. Conclusions
Acknowledgments
References
- Diekema, D.J.; Pfaller, M.A.; Schmitz, F.J.; Smayevsky, J.; Bell, J.; Jones, R.N.; Beach, M. Survey of infections due to Staphylococcus species: Frequency of occurrence and antimicrobial susceptibility of isolates collected in the United States, Canada, Latin America, Europe, and the Western Pacific region for the SENTRY Antimicrobial Surveillance Program, 1997–1999. Clin. Infect. Dis. 2001, 32 (Suppl. 2), S114–S132. [Google Scholar] [CrossRef]
- Kirby, W.M. Extraction of a highly potent penicillin inactivator from penicillin resistant Staphylococci. Science 1944, 99, 452–453. [Google Scholar]
- Barber, M.; Rozwadowska-Dowzenko, M. Infection by penicillin-resistant staphylococci. Lancet 1948, 2, 641–644. [Google Scholar]
- Jevons, M.P. “Celbenin”-resistant Staphylococci. Br. Med. J. 1961, 1, 124–125. [Google Scholar] [CrossRef]
- Stewart, G.T.; Holt, R.J. Evolution of natural resistance to the newer penicillins. Br. Med. J. 1963, 1, 308–311. [Google Scholar] [CrossRef]
- Mato, R.; Sanches, S.I.; Venditti, M.; Platt, D.J.; Brown, A.; Chung, M.; de Lencastre, H. Spread of the multiresistant Iberian clone of methicillin-resistant Staphylococcus aureus (MRSA) to Italy and Scotland. Microb. Drug Resist. 1998, 4, 107–112. [Google Scholar] [CrossRef]
- Klevens, R.M.; Morrison, M.A.; Nadle, J.; Petit, S.; Gershman, K.; Ray, S.; Harrison, L.H.; Lynfield, R.; Dumyati, G.; Townes, J.M.; et al. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. J. Am. Med. Assoc. 2007, 298, 1763–1771. [Google Scholar]
- Bordon, J.; Master, R.N.; Clark, R.B.; Duvvuri, P.; Karlowsky, J.A.; Ayesu, K.; Klotchko, A.; Kapoor, R.; Ramirez, J. Methicillin-resistant Staphylococcus aureus resistance to non-beta-lactam antimicrobials in the United States from 1996 to 2008. Diagn. Microbiol. Infect. Dis. 2010, 67, 395–398. [Google Scholar] [CrossRef]
- Moran, G.J.; Krishnadasan, A.; Gorwitz, R.J.; Fosheim, G.E.; McDougal, L.K.; Carey, R.B.; Talan, D.A. Methicillin-resistant S. aureus infections among patients in the emergency department. N. Engl. J. Med. 2006, 355, 666–674. [Google Scholar] [CrossRef]
- Miller, L.G.; Diep, B.A. Clinical practice: Colonization, fomites, and virulence: Rethinking the pathogenesis of community-associated methicillin-resistant Staphylococcus aureus infection. Clin. Infect. Dis. 2008, 46, 752–760. [Google Scholar] [CrossRef]
- Baba, T.; Takeuchi, F.; Kuroda, M.; Yuzawa, H.; Aoki, K.; Oguchi, A.; Nagai, Y.; Iwama, N.; Asano, K.; Naimi, T.; et al. Genome and virulence determinants of high virulence community-acquired MRSA. Lancet 2002, 359, 1819–1827. [Google Scholar]
- Daum, R.S.; Ito, T.; Hiramatsu, K.; Hussain, F.; Mongkolrattanothai, K.; Jamklang, M.; Boyle-Vavra, S. A novel methicillin-resistance cassette in community-acquired methicillin-resistant Staphylococcus aureus isolates of diverse genetic backgrounds. J. Infect. Dis. 2002, 186, 1344–1347. [Google Scholar] [CrossRef]
- Okuma, K.; Iwakawa, K.; Turnidge, J.D.; Grubb, W.B.; Bell, J.M.; O’Brien, F.G.; Coombs, G.W.; Pearman, J.W.; Tenover, F.C.; Kapi, M.; et al. Dissemination of new methicillin-resistant Staphylococcus aureus clones in the community. J. Clin. Microbiol. 2002, 40, 4289–4294. [Google Scholar]
- Vandenesch, F.; Naimi, T.; Enright, M.C.; Lina, G.; Nimmo, G.R.; Heffernan, H.; Liassine, N.; Bes, M.; Greenland, T.; Reverdy, M.E.; et al. Community-acquired methicillin-resistant Staphylococcus aureus carrying Panton-Valentine leukocidin genes: Worldwide emergence. Emerg. Infect. Dis. 2003, 9, 978–984. [Google Scholar]
- Lina, G.; Piemont, Y.; Godail-Gamot, F.; Bes, M.; Peter, M.O.; Gauduchon, V.; Vandenesch, F.; Etienne, J. Involvement of Panton-Valentine leukocidin-producing Staphylococcus aureus in primary skin infections and pneumonia. Clin. Infect. Dis. 1999, 29, 1128–1132. [Google Scholar] [CrossRef]
- Moellering, R.C., Jr.; Abbott, G.F.; Ferraro, M.J. Case records of the Massachusetts General Hospital. Case 2–2011. A 30-year-old woman with shock after treatment of a furuncle. N. Engl. J. Med. 2011, 364, 266–275. [Google Scholar] [CrossRef]
- Deleo, F.R.; Otto, M.; Kreiswirth, B.N.; Chambers, H.F. Community-associated meticillin-resistant Staphylococcus aureus. Lancet 2010, 375, 1557–1568. [Google Scholar]
- Abi-Hanna, P.; Frank, A.L.; Quinn, J.P.; Kelkar, S.; Schreckenberger, P.C.; Hayden, M.K.; Marcinak, J.F. Clonal features of community-acquired methicillin-resistant Staphylococcus aureus in children. Clin. Infect. Dis. 2000, 30, 630–631. [Google Scholar]
- Tenover, F.C.; McDougal, L.K.; Goering, R.V.; Killgore, G.; Projan, S.J.; Patel, J.B.; Dunman, P.M. Characterization of a strain of community-associated methicillin-resistant Staphylococcus aureus widely disseminated in the United States. J. Clin. Microbiol. 2006, 44, 108–118. [Google Scholar]
- Diep, B.A.; Stone, G.G.; Basuino, L.; Graber, C.J.; Miller, A.; des Etages, S.A.; Jones, A.; Palazzolo-Ballance, A.M.; Perdreau-Remington, F.; Sensabaugh, G.F.; et al. The arginine catabolic mobile element and staphylococcal chromosomal cassette mec linkage: Convergence of virulence and resistance in the USA300 clone of methicillin-resistant Staphylococcus aureus. J. Infect. Dis. 2008, 197, 1523–1530. [Google Scholar]
- Gorwitz, R.J. Understanding the success of methicillin-resistant Staphylococcus aureus strains causing epidemic disease in the community. J. Infect. Dis. 2008, 197, 179–182. [Google Scholar] [CrossRef]
- Montgomery, C.P.; Boyle-Vavra, S.; Adem, P.V.; Lee, J.C.; Husain, A.N.; Clasen, J.; Daum, R.S. Comparison of virulence in community-associated methicillin-resistant Staphylococcus aureus pulsotypes USA300 and USA400 in a rat model of pneumonia. J. Infect. Dis. 2008, 198, 561–570. [Google Scholar] [CrossRef]
- Payne, D.J.; Gwynn, M.N.; Holmes, D.J.; Pompliano, D.L. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2007, 6, 29–40. [Google Scholar]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef]
- Appelbaum, P.C. Reduced glycopeptide susceptibility in methicillin-resistant Staphylococcus aureus (MRSA). Int. J. Antimicrob. Agents 2007, 30, 398–408. [Google Scholar] [CrossRef]
- Zetola, N.; Francis, J.S.; Nuermberger, E.L.; Bishai, W.R. Community-acquired meticillin-resistant Staphylococcus aureus: An emerging threat. Lancet Infect. Dis. 2005, 5, 275–286. [Google Scholar] [CrossRef]
- Plaza, A.; Keffer, J.L.; Bifulco, G.; Lloyd, J.R.; Bewley, C.A. Chrysophaentins A–H, antibacterial bisdiarylbutene macrocycles that inhibit the bacterial cell division protein FtsZ. J. Am. Chem. Soc. 2010, 132, 9069–9077. [Google Scholar]
- Google Earth. Available online: http://www.google.com/earth/index.html (accessed on 16 February 2012). Image: U.S. Geological Survey, Image copyright: 2012 Digital Globe, GeoEye, Google..
- Kozlowski, M.C.; Dugan, E.C.; DiVirgilio, E.S.; Maksimenka, K.; Bringmann, G. Asymmetric total synthesis of nigerone and ent-nigerone: Enantioselective oxidative biaryl coupling of highly hindered naphthols. Adv. Synth. Catal. 2007, 349, 583–594. [Google Scholar] [CrossRef]
- Gopalsamuthiram, V.; Wulff, W.D. A new convergent strategy for the synthesis of calixarenes via a triple annulation of Fischer carbene complexes. J. Am. Chem. Soc. 2004, 126, 13936–13937. [Google Scholar] [CrossRef]
- Bellina, F.; Colzi, F.; Mannina, L.; Rossi, R.; Viel, S. Reaction of alkynes with iodine monochloride revisited. J. Org. Chem. 2003, 68, 10175–10177. [Google Scholar]
- Uemura, S.; Okazaki, H.; Onoe, A.; Okano, M. Chlorination and chloroiodination of acetylenes with copper(II) chloride. J. Chem. Soc. Perkin Trans. 1 1977, 6, 676–680. [Google Scholar]
- Wright, B.J.D.; Hartung, J.; Peng, F.; van de Water, R.; Liu, H.B.; Tan, Q.H.; Chou, T.C.; Danishefsky, S.J. Synthesis of pluraflavin A “aglycone”. J. Am. Chem. Soc. 2008, 130, 16786–16790. [Google Scholar]
- Bloomer, J.L.; Stagliano, K.W.; Gazzillo, J.A. Preparation of functionalized juglone acetates and juglones via 1,4-dimethoxynaphthalene derivatives: Synthesis of anthraquinones related to rhein and aloe emodin. J. Org. Chem. 1993, 58, 7906–7912. [Google Scholar]
- Oswald, C.L.; Carrillo-Marquez, T.; Caggiano, L.; Jackson, R.F.W. Negishi cross-coupling reactions of alpha-amino acid-derived organozinc reagents and aromatic bromides. Tetrahedron 2008, 64, 681–687. [Google Scholar]
- Sanches, I.S.; Ramirez, M.; Troni, H.; Abecassis, M.; Padua, M.; Tomasz, A.; de Lencastre, H. Evidence for the geographic spread of a methicillin-resistant Staphylococcus aureus clone between Portugal and Spain. J. Clin. Microbiol. 1995, 33, 1243–1246. [Google Scholar]
- Gillaspy, A.F.; Hickmon, S.G.; Skinner, R.A.; Thomas, J.R.; Nelson, C.L.; Smeltzer, M.S. Role of the accessory gene regulator (agr) in pathogenesis of staphylococcal osteomyelitis. Infect. Immun. 1995, 63, 3373–3380. [Google Scholar]
- McDougal, L.K.; Steward, C.D.; Killgore, G.E.; Chaitram, J.M.; McAllister, S.K.; Tenover, F.C. Pulsed-field gel electrophoresis typing of oxacillin-resistant Staphylococcus aureus isolates from the United States: Establishing a national database. J. Clin. Microbiol. 2003, 41, 5113–5120. [Google Scholar]
- Burlak, C.; Hammer, C.H.; Robinson, M.A.; Whitney, A.R.; McGavin, M.J.; Kreiswirth, B.N.; Deleo, F.R. Global analysis of community-associated methicillin-resistant Staphylococcus aureus exoproteins reveals molecules produced in vitro and during infection. Cell Microbiol. 2007, 9, 1172–1190. [Google Scholar]
- Bryan, H.F.; Lewis, I.F. A new protophyte from the Dry Tortugas. Am. J. Bot. 1941, 28, 343–348. [Google Scholar] [CrossRef]
- Gerwick, W.H.; Lopez, A.; van Duyne, G.D.; Clardy, J.; Ortiz, W.; Baez, A. Hormothamnione, a novel cytotoxic styrylchromone from the marine cyanophyte Hormothamnion enteromorphoides grunow. Tetrahedron Lett. 1986, 27, 1979–1982. [Google Scholar]
- Gerwick, W.H. 6-Desmethoxyhormothamnione, a new cytotoxic styrylchromone from the marine cryptophyte Chrysophaeum taylori. J. Nat. Prod. 1989, 52, 252–256. [Google Scholar] [CrossRef]
- Harrowven, D.C.; Kostiuk, S.L. Macrocylic bisbibenzyl natural products and their chemical synthesis. Nat. Prod. Rep. 2012, 29, 223–242. [Google Scholar] [CrossRef]
- Asakawa, Y.; Toyota, M.; Matsuda, R.; Takikawa, K.; Takemoto, T. Distribution of novel cyclic bisbibenzyls in Marchantia and Riccardia species. Phytochemistry 1983, 22, 1413–1415. [Google Scholar]
- Scher, J.M.; Zapp, J.; Schmidt, A.; Becker, H. Bazzanins L-R, chlorinated macrocyclic bisbibenzyls from the liverwort Lepidozia incurvata. Phytochemistry 2003, 64, 791–796. [Google Scholar] [CrossRef]
- Martini, U.; Zapp, J.; Becker, H. Lignans from the liverwort Bazzania trilobata. Phytochemistry 1998, 49, 1139–1146. [Google Scholar]
- Hashimoto, T.; Irita, H.; Takaoka, S.; Tanaka, M.; Asakawa, Y. New chlorinated cyclic bis(bibenzyls) from the liverworts Herbertus sakuraii and Mastigophora diclados. Tetrahedron 2000, 56, 3153–3159. [Google Scholar]
- Friederich, S.; Maier, U.H.; Deus-Neumann, B.; Asakawa, Y.; Zenk, M.H. Biosynthesis of cyclic bis(bibenzyls) in Marchantia polymorpha. Phytochemistry 1999, 50, 589–598. [Google Scholar]
- Ren, D.; Pipes, G.D.; Hambly, D.; Bondarenko, P.V.; Treuheit, M.J.; Gadgil, H.S. Top-down N-terminal sequencing of immunoglobulin subunits with electrospray ionization time of flight mass spectrometry. Anal.Biochem. 2009, 384, 42–48. [Google Scholar]
- Bloomer, J.L.; Gazzillo, J.A. An efficient route to 3-chlorojuglones. Tetrahedron Lett. 1989, 30, 1201–1204. [Google Scholar] [CrossRef]
- Samples Availability: Available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Keffer, J.L.; Hammill, J.T.; Lloyd, J.R.; Plaza, A.; Wipf, P.; Bewley, C.A. Geographic Variability and Anti-Staphylococcal Activity of the Chrysophaentins and Their Synthetic Fragments. Mar. Drugs 2012, 10, 1103-1125. https://doi.org/10.3390/md10051103
Keffer JL, Hammill JT, Lloyd JR, Plaza A, Wipf P, Bewley CA. Geographic Variability and Anti-Staphylococcal Activity of the Chrysophaentins and Their Synthetic Fragments. Marine Drugs. 2012; 10(5):1103-1125. https://doi.org/10.3390/md10051103
Chicago/Turabian StyleKeffer, Jessica L., Jared T. Hammill, John R. Lloyd, Alberto Plaza, Peter Wipf, and Carole A. Bewley. 2012. "Geographic Variability and Anti-Staphylococcal Activity of the Chrysophaentins and Their Synthetic Fragments" Marine Drugs 10, no. 5: 1103-1125. https://doi.org/10.3390/md10051103
APA StyleKeffer, J. L., Hammill, J. T., Lloyd, J. R., Plaza, A., Wipf, P., & Bewley, C. A. (2012). Geographic Variability and Anti-Staphylococcal Activity of the Chrysophaentins and Their Synthetic Fragments. Marine Drugs, 10(5), 1103-1125. https://doi.org/10.3390/md10051103