
Expression Analysis, Diagnostic Significance and Biological Functions of BAG4 in Acute Myeloid Leukemia


Abstract

1. Introduction
2. Materials and Methods
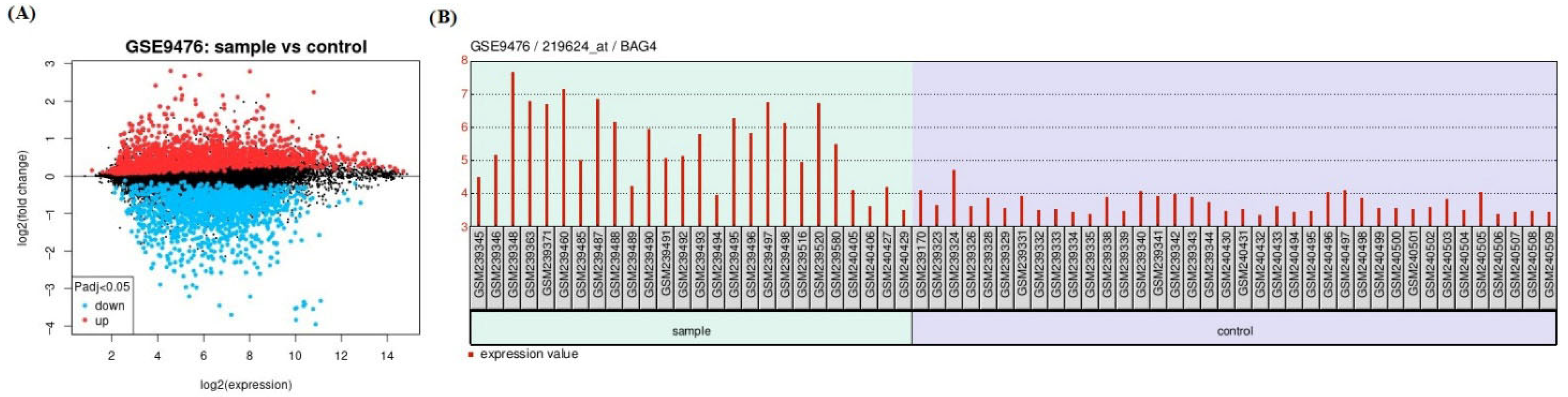
2.1. Data Collection and Identification of Differentially Expressed Genes (DEGs)
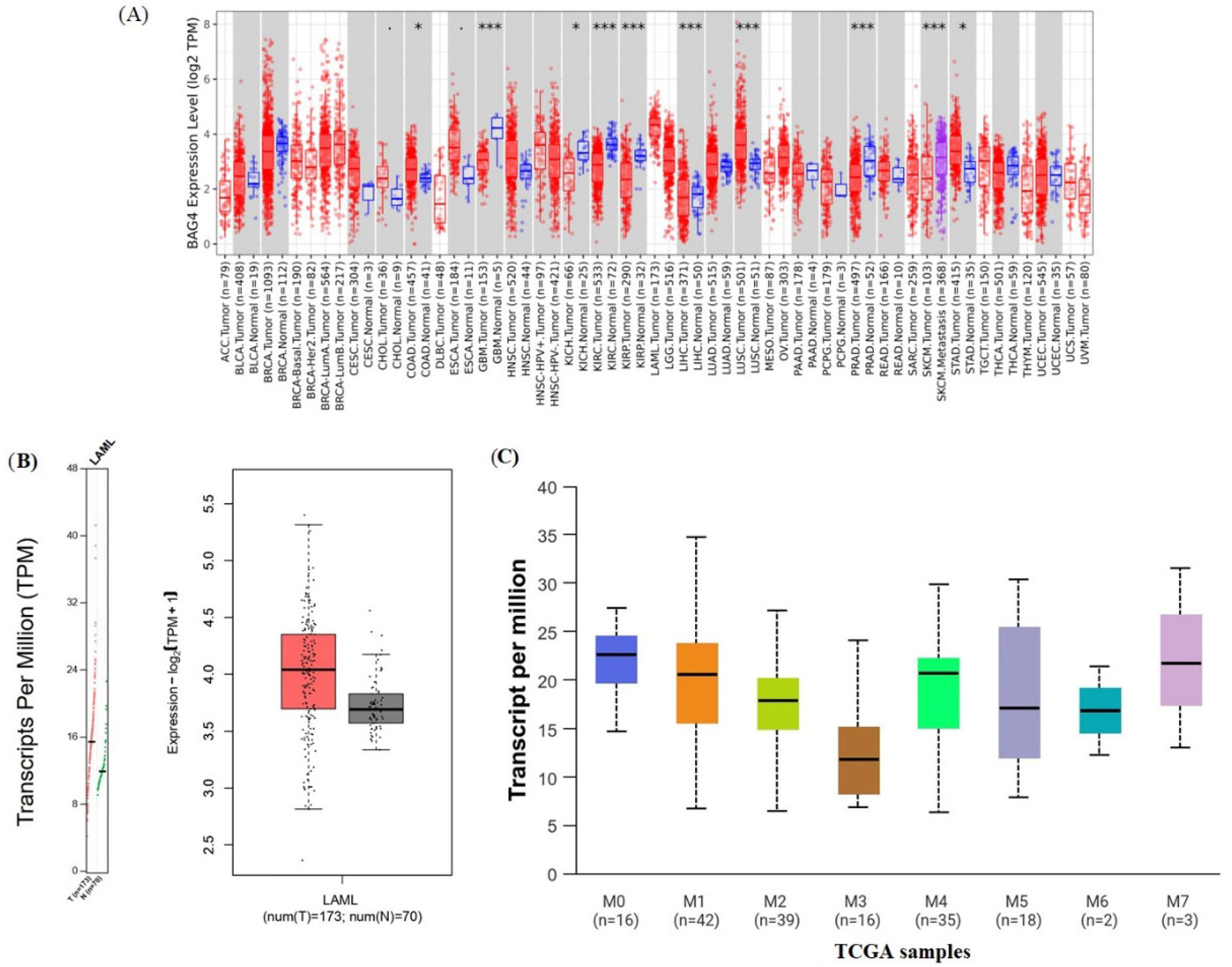
2.2. Analysis of BAG4 Expression in Tumor and Normal Tissues
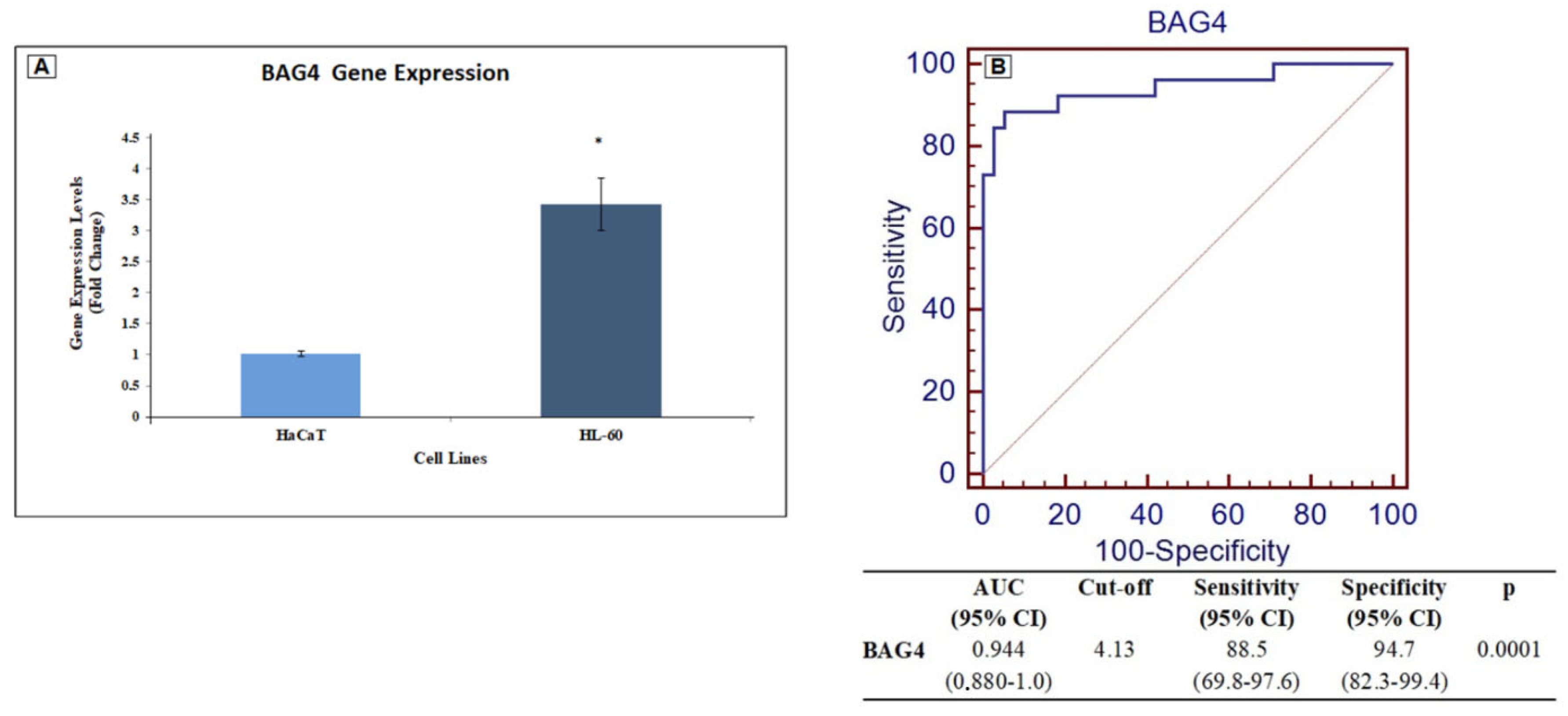
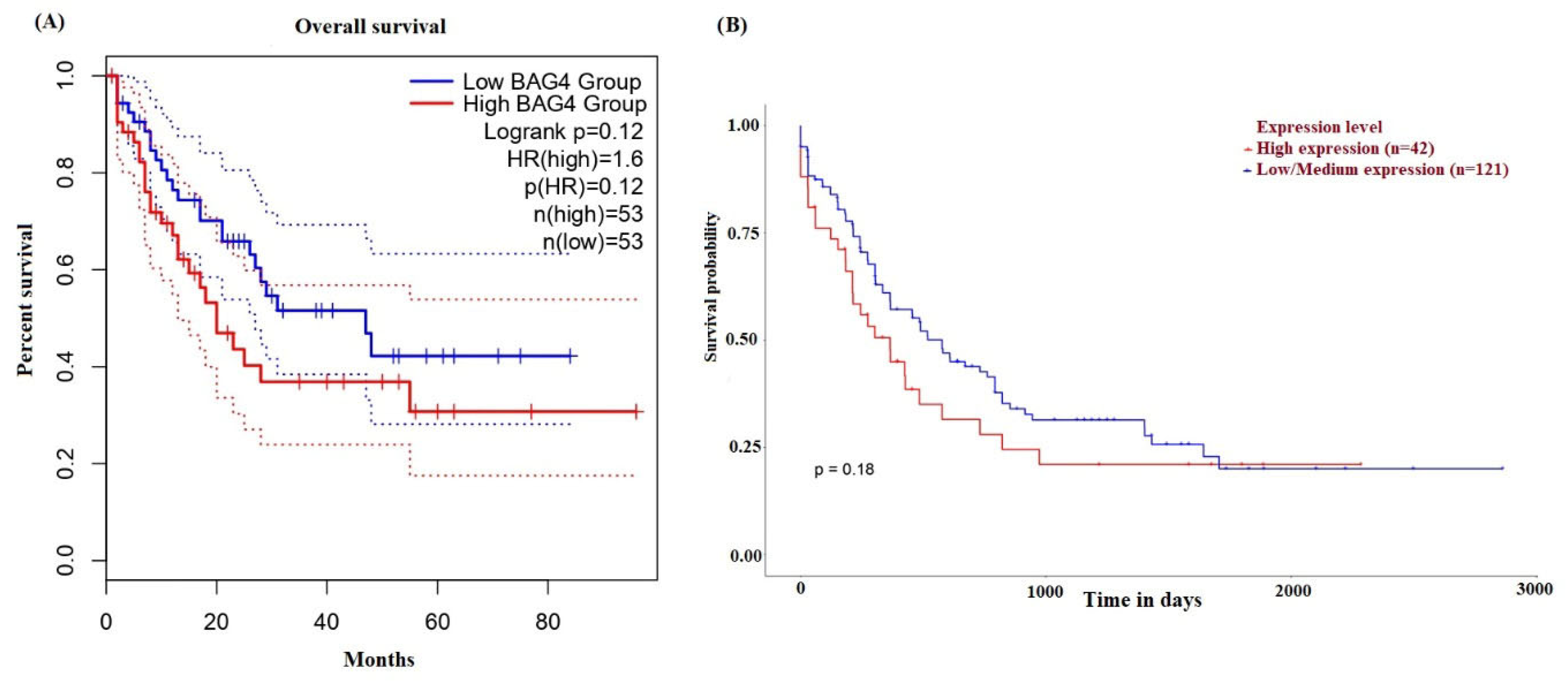
2.3. Diagnostic and Prognostic Value of BAG4
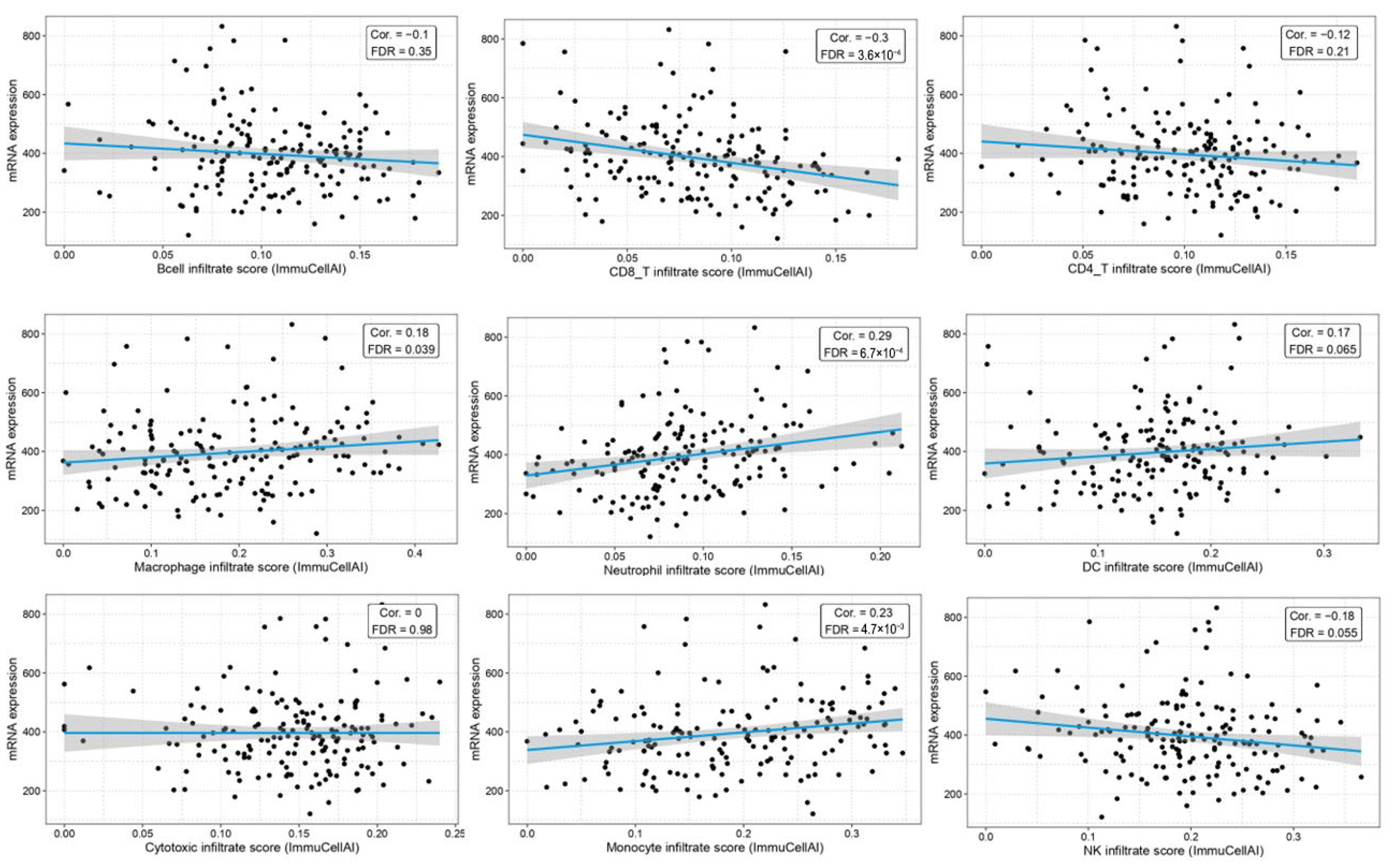
2.4. Correlation Between BAG4 Expression and Immune Infiltration
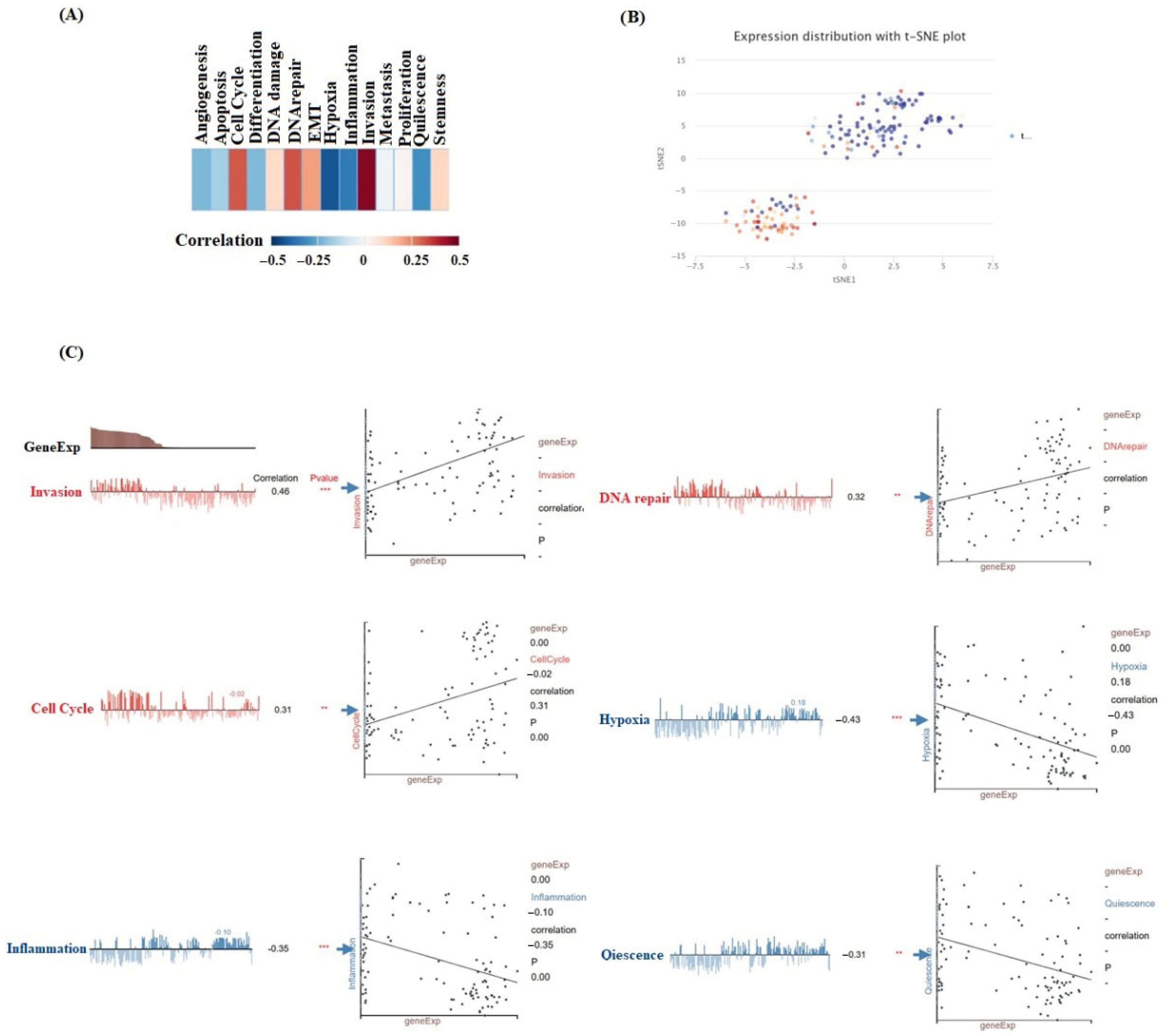
2.5. Single-Cell Function in Cancer
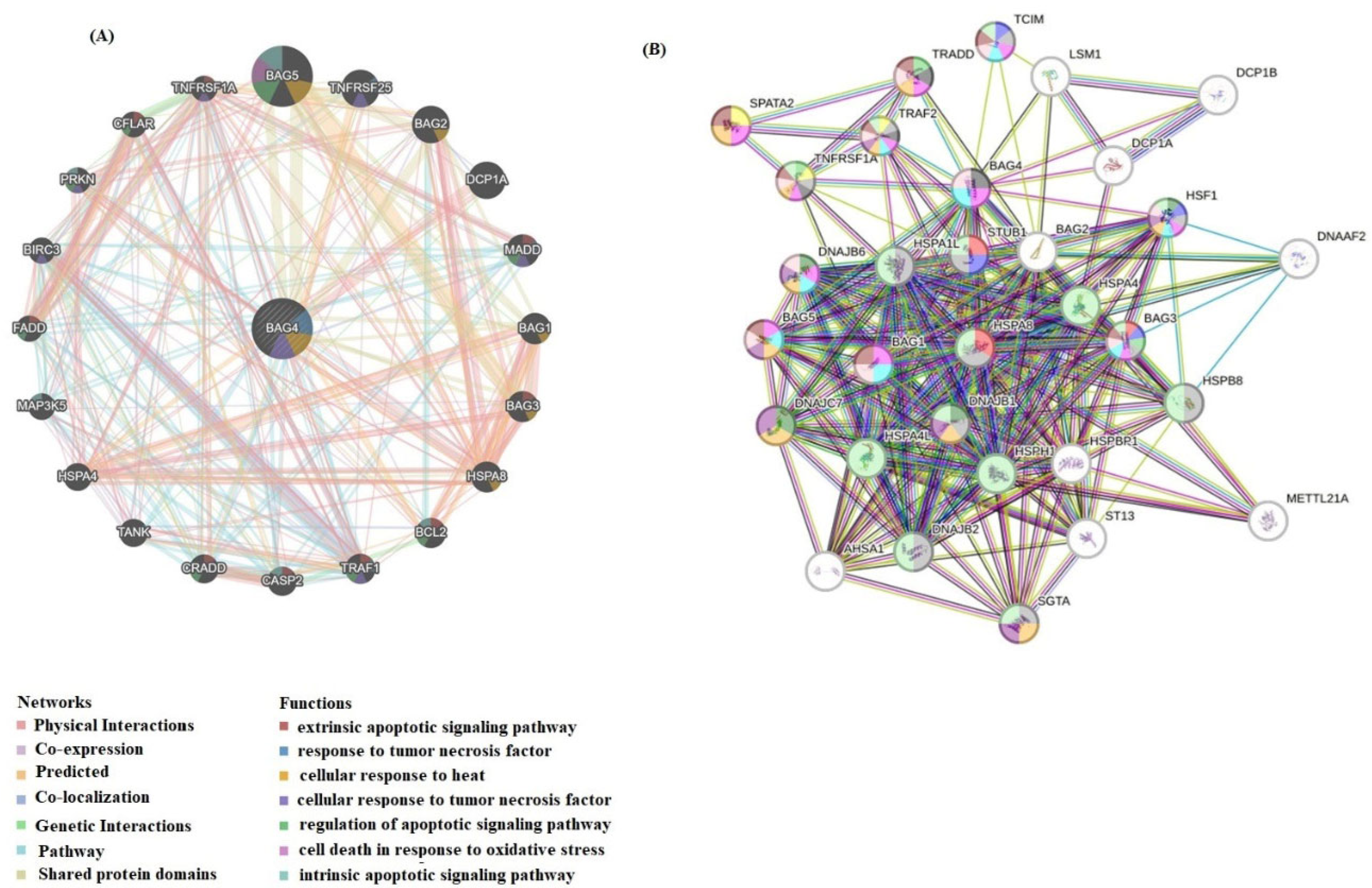
2.6. Interaction Network Analysis of BAG4 and Functional Enrichment Analysis
2.7. Cell Culture and RT–qPCR
2.8. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AML | Acute myeloid leukemia |
| BAG4 | BCL-2-associated athanogene 4 |
| ROC | Receiver operating characteristic |
| AUC | Area under the curve |
| OS | Overall survival |
| BCL2 | B-cell lymphoma 2 |
| TNFRSF1A | TNF receptor superfamily member 1A |
| HSF1 | Heat shock factor 1 |
| FADD | Fas associated via death domain |
| TME | Tumor microenvironment |
References
- Fu, D.; Zhang, B.; Wu, S.; Zhang, Y.; Xie, J.; Ning, W.; Jiang, H. Prognosis and characterization of immune microenvironment in acute myeloid leukemia through identification of an autophagy-related signature. Front. Immunol. 2021, 12, 695865. [Google Scholar] [CrossRef]
- El Omri, H.; Taha, R.Y.; Elomri, A.; Kacem, N.; Elsabah, H.; Ellahie, A.Y.; Gamil, A.; Ibrahim, F.; Soliman, D.S.A.; Akiki, S.J.L.E.; et al. Acute myeloid leukemia in qatar (2010–2016): Clinical, biological, and prognostic factors and treatment outcomes. Front. Genet. 2020, 11, 553. [Google Scholar] [CrossRef]
- Santoro, N.; Salutari, P.; Di Ianni, M.; Marra, A. Precision Medicine Approaches in Acute Myeloid Leukemia with Adverse Genetics. Int. J. Mol. Sci. 2024, 25, 4259. [Google Scholar] [CrossRef]
- Deng, L.; Jiang, L.; Lin, X.H.; Tseng, K.-F.; Liu, Y.; Zhang, X.; Dong, R.H.; Lu, Z.G.; Wang, X.J. The PI3K/mTOR dual inhibitor BEZ235 suppresses proliferation and migration and reverses multidrug resistance in acute myeloid leukemia. Acta Pharmacol. Sin. 2017, 38, 382–391. [Google Scholar] [CrossRef]
- Lin, M.S.; Zhong, H.Y.; Yim, R.L.H.; Chen, Q.Y.; Du, H.L.; He, H.Q.; Lin, K.; Zhao, P.; Gao, R.; Gao, F.; et al. Pan-cancer analysis of oncogenic TNFAIP2 identifying its prognostic value and immunological function in acute myeloid leukemia. BMC Cancer 2022, 22, 1068. [Google Scholar] [CrossRef]
- Abulimiti, M.; Jia, Z.Y.; Wu, Y.; Yu, J.; Gong, Y.H.; Guan, N.; Xiong, D.-Q.; Ding, N.; Uddin, N.; Wang, J. Exploring and clinical validation of prognostic significance and therapeutic implications of copper homeostasis-related gene dysregulation in acute myeloid leukemia. Ann. Hematol. 2024, 103, 2797–2826. [Google Scholar] [CrossRef]
- Wang, F. Identification of tumor antigens and immune subtypes of acute myeloid leukemia for mRNA vaccine development. Clin. Transl. Oncol. 2023, 25, 2204–2223. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Guo, D.; Kong, D.; Bian, S.; Zhao, L.; Li, Q.; Lin, L.; Hao, J.; Sun, L.; Li, Y. Expression analysis, clinical significance and potential function of PLXNB2 in acute myeloid leukaemia. Mol. Biol. Rep. 2023, 50, 8445–8457. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Borthakur, G.; Daver, N.; DiNardo, C.D.; Issa, G.; Jabbour, E.; Kadia, T.; Sasaki, K.; Short, N.J.; Yilmaz, M.; et al. Current status and research directions in acute myeloid leukemia. Blood Cancer J. 2024, 14, 163. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Chen, Y.; Min, G.; Wang, J.; Chen, W.; Wang, H.; Wang, X.; Yao, N. Bcl2-associated athanogene 4 promotes the invasion and metastasis of gastric cancer cells by activating the PI3K/AKT/NF-κB/ZEB1 axis. Cancer Lett. 2021, 520, 409–421. [Google Scholar] [CrossRef]
- Mariotto, E.; Viola, G.; Zanon, C.; Aveic, S. A BAG’s life: Every connection matters in cancer. Pharmacol. Ther. 2020, 209, 107498. [Google Scholar] [CrossRef] [PubMed]
- Bao, F.; An, S.; Yang, Y.; Xu, T.R. SODD promotes lung cancer tumorigenesis by activating the PDK1/AKT and RAF/MEK/ERK signaling. Genes 2023, 14, 829. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, F.; Friess, H.; Zimmermann, A.; Kleeff, J.; Büchler, M.W. Enhanced expression of Silencer of death domains (SODD/BAG-4) in pancreatic cancer. Biochem. Biophys. Res. Commun. 2000, 271, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, C.M.; Kleinberg, L.; Davidson, B.; Berner, A.; Gius, D.; Tchabo, N.; Steinberg, S.M.; Kohn, E.C. BAG-4/SODD and associated antiapoptotic proteins are linked to aggressiveness of epithelial ovarian cancer. Clin. Cancer Res. 2007, 13, 6585–6592. [Google Scholar] [CrossRef]
- Davidson, B.; Reinertsen, K.V.; Trinh, D.; Reed, W.; Bøhler, P.J. BAG-1/SODD, HSP70, and HSP90 are potential prognostic markers of poor survival in node-negative breast carcinoma. Hum. Pathol. 2016, 54, 64–73. [Google Scholar] [CrossRef]
- Cisterne, A.; Baraz, R.; Khan, N.I.; Welschinger, R.; Basnett, J.; Fung, C.; Rizos, H.; Bradstock, K.F.; Bendall, L.J. Silencer of death domains controls cell death through tumour necrosis factor-receptor 1 and caspase-10 in acute lymphoblastic leukemia. PLoS ONE 2014, 9, e103383. [Google Scholar] [CrossRef]
- Rho, J.H.; Ladd, J.J.; Li, C.I.; Potter, J.D.; Zhang, Y.; Shelley, D.; Shibata, D.; Coppola, D.; Yamada, H.; Toyoda, H.; et al. Protein and glycomic plasma markers for early detection of adenoma and colon cancer. Gut 2018, 67, 473–484. [Google Scholar] [CrossRef]
- Carter, B.Z.; Mak, P.Y.; Ruvolo, V.; Tao, W.; Hughes, P.; Chen, X.; Morrow, P.K.; Andreeff, M. Targeting Mcl-1 enhances the activity of tyrosine kinase inhibitor gilteritinib in FLT3 mutated AML. Blood 2020, 136, 30–31. [Google Scholar] [CrossRef]
- Huang, J.; Mao, L.; Lei, Q.; Guo, A.Y. Bioinformatics tools and resources for cancer and application. Chin. Med. J. 2024, 137, 2052–2064. [Google Scholar] [CrossRef]
- Rosati, D.; Palmieri, M.; Brunelli, G.; Morrione, A.; Iannelli, F.; Frullanti, E.; Giordano, A. Differential gene expression analysis pipelines and bioinformatic tools for the identification of specific biomarkers: A review. Comput. Struct. Biotechnol. J. 2024, 23, 1154–1168. [Google Scholar] [CrossRef]
- Stirewalt, D.L.; Meshinchi, S.; Kopecky, K.J.; Fan, W.; Pogosova-Agadjanyan, E.L.; Engel, J.H.; Cronk, M.R.; Dorcy, K.S.; McQuary, A.R.; Hockenbery, D.; et al. Identification of genes with abnormal expression changes in acute myeloid leukemia. Genes Chromosomes Cancer 2008, 47, 8–20. [Google Scholar] [CrossRef]
- Bao, M.; Jiang, G. Differential expression and functional analysis of lung cancer gene expression datasets: A systems biology perspective. Oncol. Lett. 2019, 18, 776–782. [Google Scholar] [CrossRef]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.J.; Hu, F.F.; Xia, M.X.; Han, L.; Zhang, Q.; Guo, A.Y. GSCALite: A Web Server for Gene Set Cancer Analysis. Bioinformatics 2018, 34, 3771–7372. [Google Scholar] [CrossRef]
- Liu, C.J.; Hu, F.F.; Xie, G.Y.; Miao, Y.R.; Li, X.W.; Zeng, Y.; Guo, A.Y. GSCA: An Integrated Platform for Gene Set Cancer Analysis at Genomic, Pharmacogenomic, and Immunogenomic Levels. Brief. Bioinform. 2023, 24, bbac558. [Google Scholar] [CrossRef]
- Yuan, H.; Yan, M.; Zhang, G.; Liu, W.; Deng, C.; Liao, G.; Xu, L.; Luo, T.; Yan, H.; Long, Z.; et al. CancerSEA: A cancer single-cell state atlas. Nucleic Acids Res. 2019, 47, D900–D908. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Kuhn, M.; Simonovic, M.; Roth, A.; Minguez, P.; Doerks, T.; Stark, M.; Muller, J.; Bork, P.; et al. The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2010, 39, D561–D568. [Google Scholar] [CrossRef]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The Gene MANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Kir, J.; Liu, D.; Bryant, D.; Guo, Y.; Stephens, R.; Baseler, M.W.; Lane, H.C.; et al. DAVID Bioinformatics Resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007, 35, W169–W175. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using realtime quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Hahn, S.; Choi, Y.J.; Cho, H.; Chung, H.; Jang, J.E.; Lyu, C.J.; Lee, S.T.; Choi, J.R.; Cheong, J.W.; et al. Comprehensive insights into AML relapse: Genetic mutations, clonal evolution, and clinical outcomes. Cancer Cell Int. 2024, 24, 174. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Wei, A.H.; Löwenberg, B. Towards precision medicine for AML. Nat. Rev. Clin. Oncol. 2021, 18, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhao, H. Bioinformatics analysis of the expression and clinical significance of the NUP210 Gene in acute myeloid leukaemia. Hematology 2022, 27, 456–462. [Google Scholar] [CrossRef]
- Wang, N. Analysis of prognostic biomarker models and immune microenvironment in acute myeloid leukemia by integrative bioinformatics. J. Cancer Res. Clin. Oncol. 2023, 149, 9609–9619. [Google Scholar] [CrossRef]
- Doong, H.; Vrailas, A.; Kohn, E.C. What’s in the ‘BAG’?—A functional domain analysis of the BAG-family proteins. Cancer Lett. 2002, 188, 25–32. [Google Scholar] [CrossRef]
- Tao, H.; Hu, Q.; Fang, J.; Liu, A.; Liu, S.; Zhang, L.; Hu, Y. Expression of SODD and P65 in ALL of children and its relationship with chemotherapeutic drugs. J. Huazhong Univ. Sci. Technol. Med. Sci. 2007, 27, 326–329. [Google Scholar] [CrossRef]
- Guo, X.; Cao, H.; Hu, M.; Wu, Y.; Li, J. KIF22 in the Prognosis and Immune Biomarking of Pan-Cancer. J. Immunol. Res. 2023, 1, 9542311. [Google Scholar] [CrossRef]
- Lei, X.; Lei, Y.; Li, J.K.; Du, W.X.; Li, R.G.; Yang, J.; Li, J.; Li, F.; Tan, H.B. Immune cells within the tumor microenvironment: Biological functions and roles in cancer immunotherapy. Cancer Lett. 2020, 470, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Zhang, Q.; Zhai, J.; Chen, P.; Deng, X.; Miao, L.; Zhang, X. Systematic pan-cancer analysis identifies ZBTB11 as a potential pan-cancer biomarker and immunotherapy target in multiple tumor types. Discov. Oncol. 2024, 15, 830. [Google Scholar] [CrossRef]
- Madeleine, M.M.; Johnson, L.G.; Malkki, M.; Resler, A.J.; Petersdorf, E.W.; McKnight, B.; Malone, K.E. Genetic variation in proinflammatory cytokines IL6, IL6R, TNF-region, and TNFRSF1A and risk of breast cancer. Breast Cancer Res. Treat. 2011, 29, 887–899. [Google Scholar] [CrossRef]
- Chin, Y.; Gumilar, K.E.; Li, X.G.; Tjokroprawiro, B.A.; Lu, C.H.; Lu, J.; Zhou, M.; Sobol, R.W.; Tan, M. Targeting HSF1 for cancer treatment: Mechanisms and inhibitor development. Theranostics 2023, 13, 2281–2300. [Google Scholar] [CrossRef]
- Carpenter, R.L.; Gökmen-Polar, Y. HSF1 as a cancer biomarker and therapeutic target. Curr. Cancer Drug Targets 2019, 19, 515–524. [Google Scholar] [CrossRef]
- Kaloni, D.; Diepstraten, S.T.; Strasser, A.; Kelly, G.L. BCL-2 protein family: Attractive targets for cancer therapy. Apoptosis 2023, 28, 20–38. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Term | p-Value |
|---|---|---|
| GOTERM_BP_DIRECT | extrinsic apoptotic signaling pathway via death domain receptors | 4.5 × 10−5 |
| protein folding | 7.8 × 10−4 | |
| negative regulation of protein targeting to mitochondrion | 1.5 × 10−3 | |
| protein stabilization | 1.6 × 10−3 | |
| cellular response to unfolded protein | 4.6 × 10−3 | |
| negative regulation of apoptotic process | 6.4 × 10−3 | |
| extrinsic apoptotic signaling pathway in absence of ligand | 8.7 × 10−3 | |
| intrinsic apoptotic signaling pathway in response to DNA damage | 1.3 × 10−2 | |
| cellular response to heat | 1.5 × 10−2 | |
| positive regulation of tyrosine phosphorylation of STAT protein | 1.6 × 10−2 | |
| positive regulation of peptidyl-serine phosphorylation | 1.8 × 10−2 | |
| cellular response to mechanical stimulus | 1.9 × 10−2 | |
| protein localization to plasma membrane | 4 × 10−2 | |
| GOTERM_CC_DIRECT | protein folding chaperone complex | 7.5 × 10−3 |
| membrane | 1.9 × 10−2 | |
| cytosol | 2.1 × 10−2 | |
| cytoplasm | 2.5 × 10−2 | |
| nucleus | 3 × 10−2 | |
| GOTERM_MF_DIRECT | adenyl-nucleotide exchange factor activity | 5.7 × 10−6 |
| protein-folding chaperone binding | 3 × 10−4 | |
| ubiquitin protein ligase binding | 2.5 × 10−3 | |
| sequence-specific DNA binding | 8.3 × 10−2 | |
| protein-containing complex binding | 8.7 × 10−2 | |
| protein heterodimerization activity | 9.7 × 10−2 |
| Term | Count | p-Value |
|---|---|---|
| Apoptosis multiple species | 2 | 1.1 × 10−2 |
| NF-kappa B signaling pathway | 2 | 3.5 × 10−2 |
| Toxoplasmosis | 2 | 3.7 × 10−2 |
| TNF signaling pathway | 2 | 4 × 10−2 |
| Sphingolipid signaling pathway | 2 | 4.1 × 10−2 |
| Apoptosis | 2 | 4.5 × 10−2 |
| Fluid shear stress and atherosclerosis | 2 | 4.7 × 10−2 |
| Necroptosis | 2 | 5.3 × 10−2 |
| Tuberculosis | 2 | 6 × 10−2 |
| Human immunodeficiency virus 1 infection | 2 | 7 × 10−2 |
| Lipid and atherosclerosis | 2 | 7.1 × 10−2 |
| Shigellosis | 2 | 8.2 × 10−2 |
| Salmonella infection | 2 | 8.3 × 10−2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Published by MDPI on behalf of the Lithuanian University of Health Sciences. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akidan, O.; Yaman, S.; Ozer Yaman, S.; Misir, S. Expression Analysis, Diagnostic Significance and Biological Functions of BAG4 in Acute Myeloid Leukemia. Medicina 2025, 61, 1333. https://doi.org/10.3390/medicina61081333
Akidan O, Yaman S, Ozer Yaman S, Misir S. Expression Analysis, Diagnostic Significance and Biological Functions of BAG4 in Acute Myeloid Leukemia. Medicina. 2025; 61(8):1333. https://doi.org/10.3390/medicina61081333
Chicago/Turabian StyleAkidan, Osman, Selçuk Yaman, Serap Ozer Yaman, and Sema Misir. 2025. "Expression Analysis, Diagnostic Significance and Biological Functions of BAG4 in Acute Myeloid Leukemia" Medicina 61, no. 8: 1333. https://doi.org/10.3390/medicina61081333
APA StyleAkidan, O., Yaman, S., Ozer Yaman, S., & Misir, S. (2025). Expression Analysis, Diagnostic Significance and Biological Functions of BAG4 in Acute Myeloid Leukemia. Medicina, 61(8), 1333. https://doi.org/10.3390/medicina61081333