
Fibroblast Growth Factor 23 Signaling Does Not Increase Inflammation from Pseudomonas aeruginosa Infection in the Cystic Fibrosis Bronchial Epithelium

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Stimulation
2.2. P. aeruginosa Strains and Infection
2.3. ELISA
2.4. RNA Purification and Quantitative Real Time PCR (qRT-PCR)
2.5. Protein Immunoblotting
2.6. Statistics
3. Results
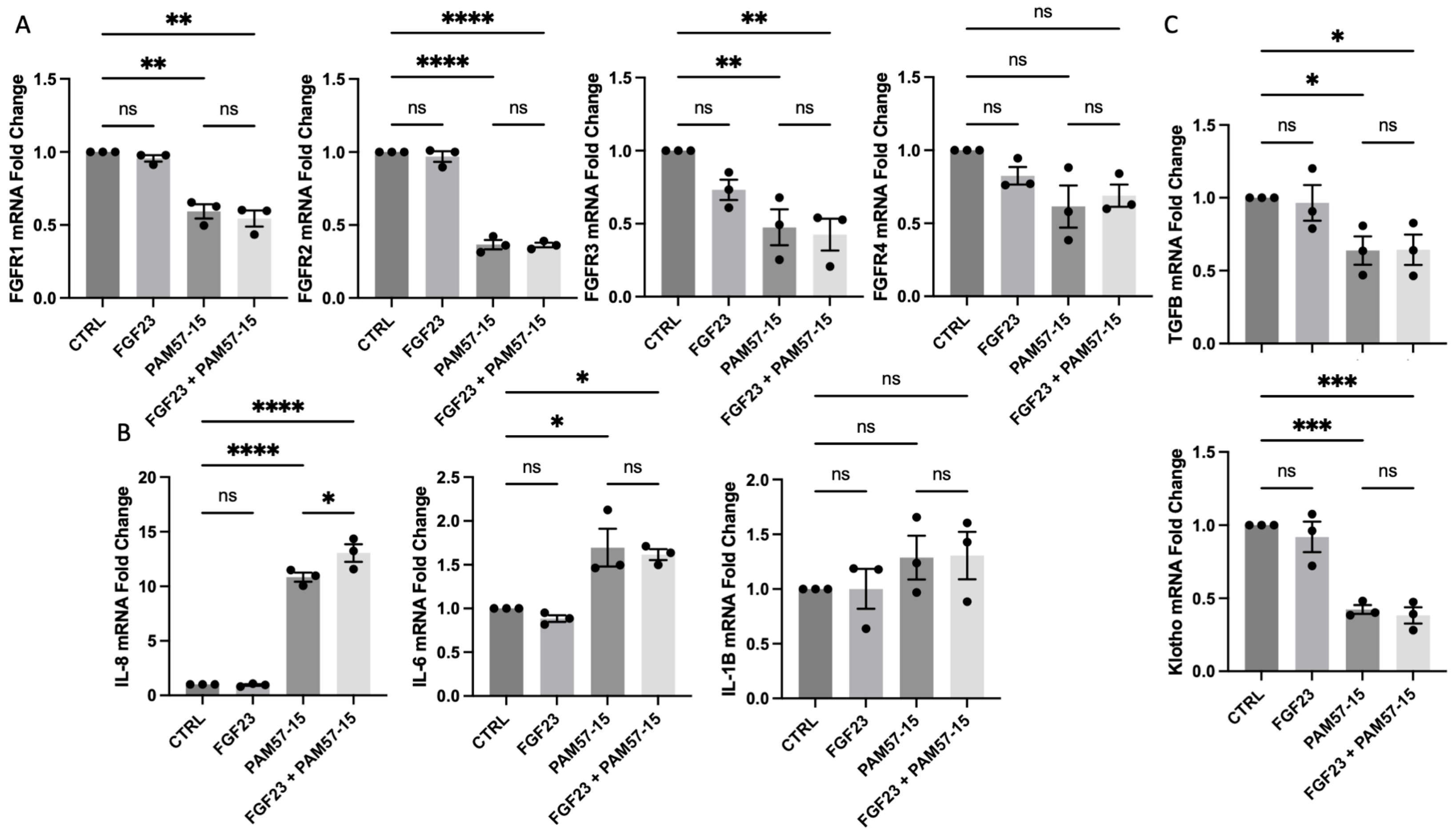
3.1. FGF23 Did Not Further Increase the Expression of Pro-Inflammatory Markers in the PA-Infected CF Bronchial Epithelium
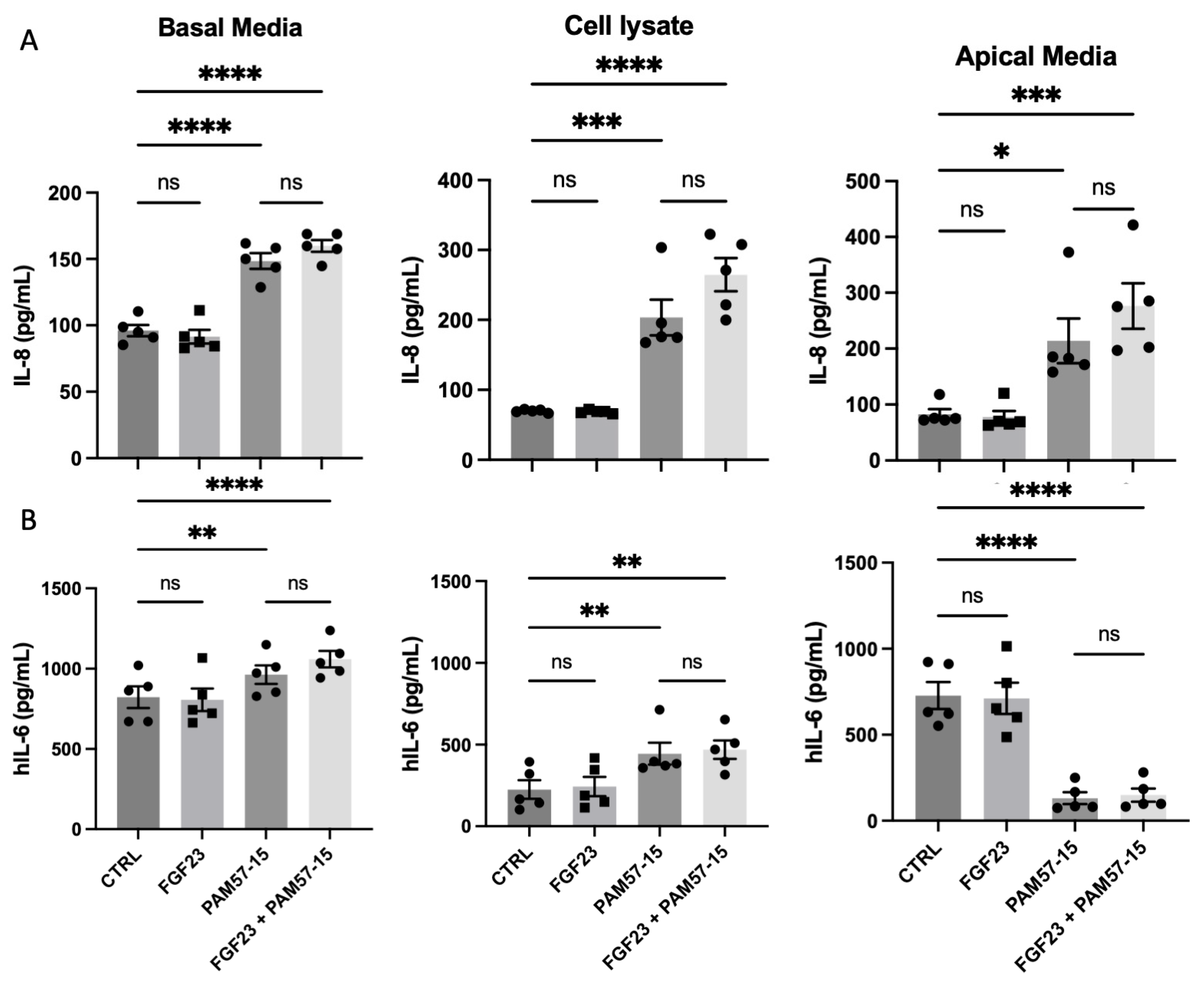
3.2. FGF23 Did Not Alter PA-Induced Pro-Inflammatory Marker Production and Secretion in the CF Bronchial Epithelium
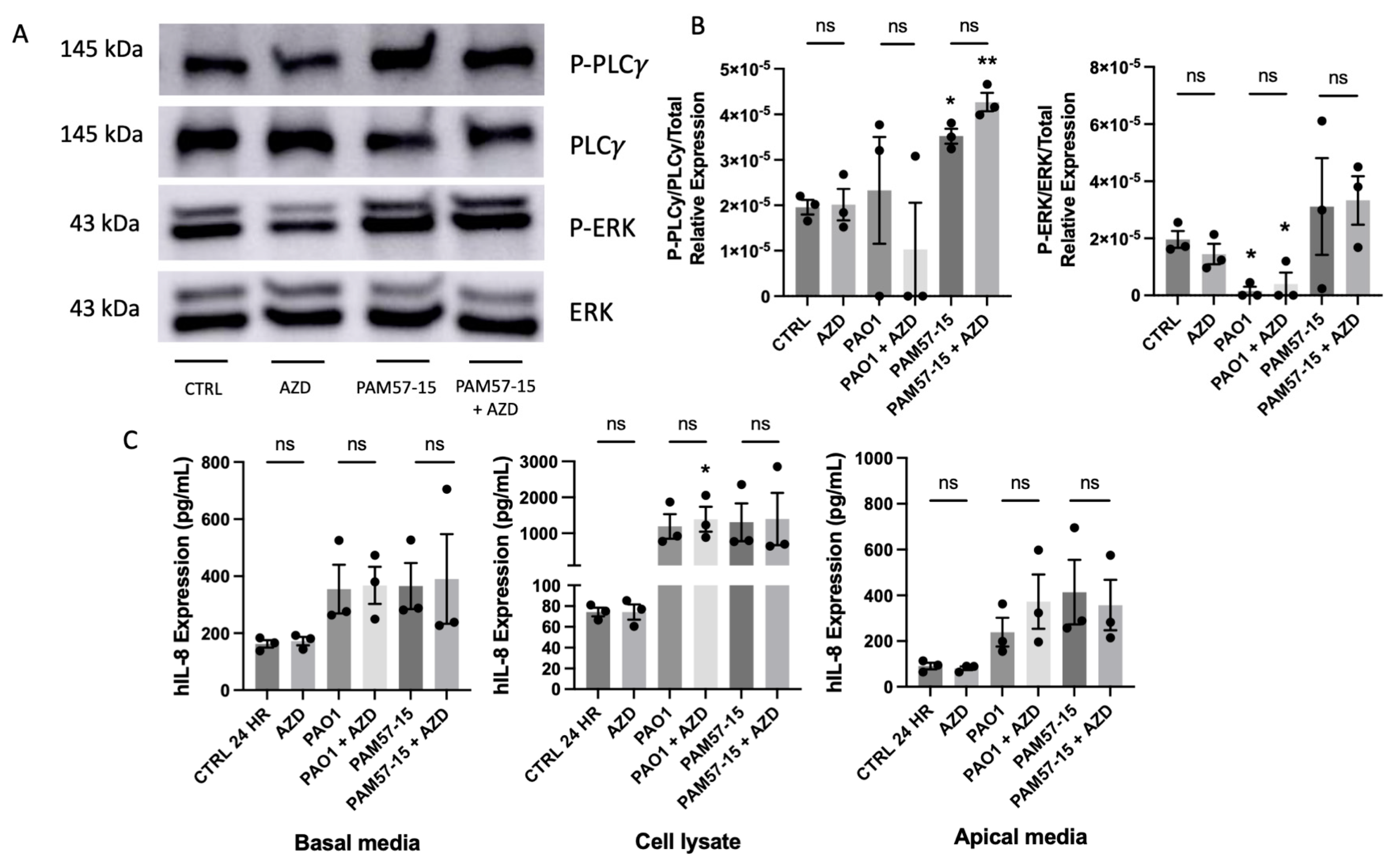
3.3. Phosphorylation of PLCγ and ERK Was Not Significantly Affected by FGF23 in PA-Infected CFBEs
3.4. Isoform-Specific FGFR Blockade Did Not Alter the Phosphorylation of PLCγ or ERK or Pro-Inflammatory Cytokine Expression in PA-Infected CFBEs
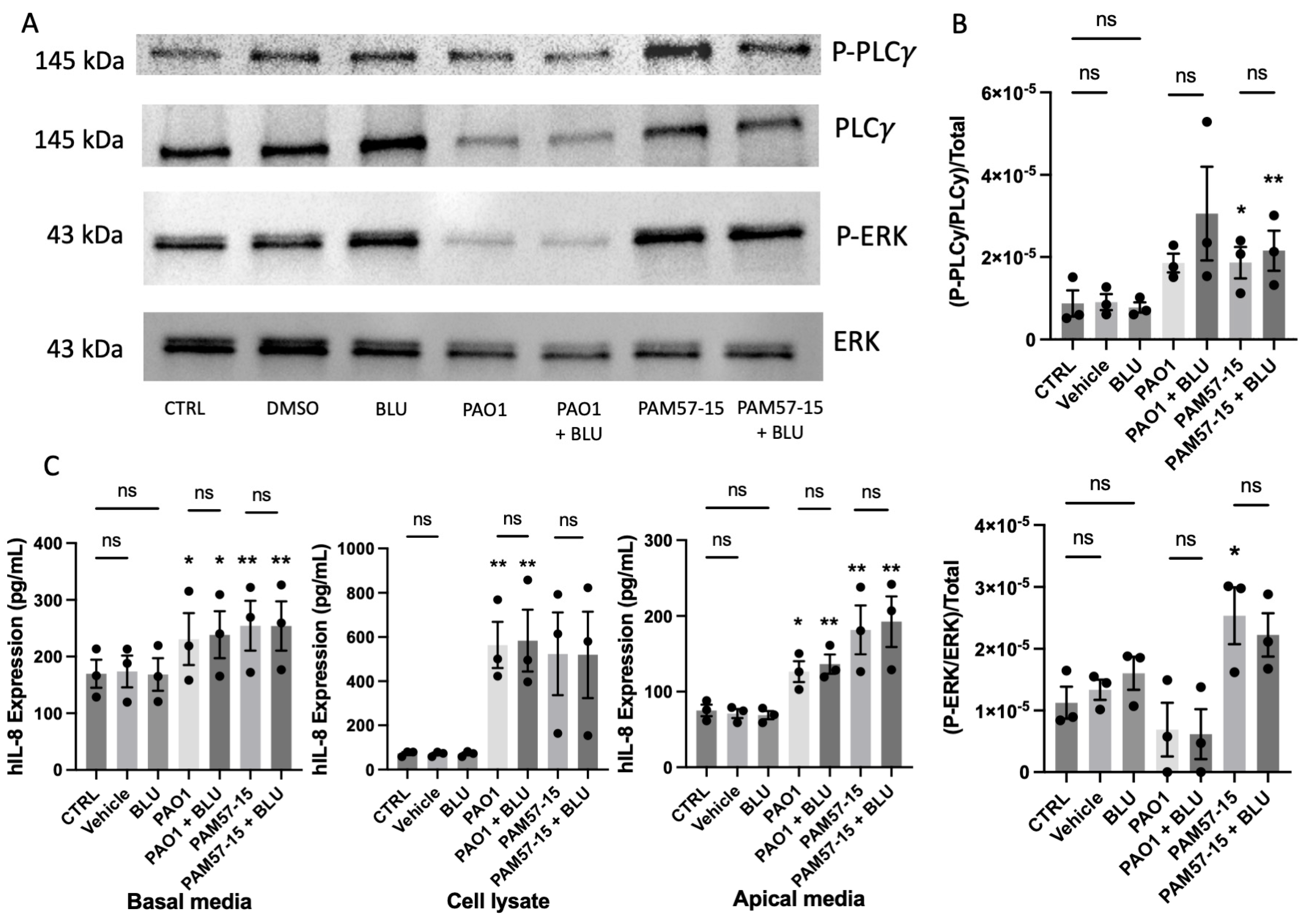
3.5. Inhibition of FGFR4 Did Not Decrease the Phosphorylation of PLCγ or ERK or Pro-Inflammatory Cytokine Expression in PA-Infected CFBEs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ratjen, F.; Doring, G. Cystic fibrosis. Lancet 2003, 361, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef] [PubMed]
- Nichols, D.; Chmiel, J.; Berger, M. Chronic inflammation in the cystic fibrosis lung: Alterations in inter- and intracellular signaling. Clin. Rev. Allergy Immunol. 2008, 34, 146–162. [Google Scholar] [CrossRef]
- Malhotra, S.; Hayes, D., Jr.; Wozniak, D.J. Cystic Fibrosis and Pseudomonas aeruginosa: The Host-Microbe Interface. Clin. Microbiol. Rev. 2019, 32, e00138-18. [Google Scholar] [CrossRef] [PubMed]
- Cystic Fibrosis Foundation Patient Registry: 2019 Annual Data Report 2020; Cystic Fibrosis Foundation: Bethesda, MD, USA, 2019.
- Garcia-Clemente, M.; de la Rosa, D.; Maiz, L.; Giron, R.; Blanco, M.; Olveira, C.; Canton, R.; Martinez-García, M.A. Impact of Pseudomonas aeruginosa Infection on Patients with Chronic Inflammatory Airway Diseases. J. Clin. Med. 2020, 9, 3800. [Google Scholar] [CrossRef]
- Cabrini, G.; Rimessi, A.; Borgatti, M.; Pinton, P.; Gambari, R. Overview of CF lung pathophysiology. Curr. Opin. Pharmacol. 2022, 64, 102214. [Google Scholar] [CrossRef]
- Lyczak, J.B.; Cannon, C.L.; Pier, G.B. Lung infections associated with cystic fibrosis. Clin. Microbiol. Rev. 2002, 15, 194–222. [Google Scholar] [CrossRef]
- Davies, J.C.; Martin, I. New anti-pseudomonal agents for cystic fibrosis- still needed in the era of small molecule CFTR modulators? Expert Opin. Pharmacother. 2018, 19, 1327–1336. [Google Scholar] [CrossRef]
- Zemanick, E.T.; Bell, S.C. Prevention of chronic infection with Pseudomonas aeruginosa infection in cystic fibrosis. Curr. Opin. Pulm. Med. 2019, 25, 636–645. [Google Scholar] [CrossRef]
- King, S.J.; Tierney, A.C.; Edgeworth, D.; Keating, D.; Williams, E.; Kotsimbos, T.; Button, B.M.; Wilson, J.W. Body composition and weight changes after ivacaftor treatment in adults with cystic fibrosis carrying the G551 D cystic fibrosis transmembrane conductance regulator mutation: A double-blind, placebo-controlled, randomized, crossover study with open-label extension. Nutrition 2021, 85, 111124. [Google Scholar]
- Middleton, P.G.; Taylor-Cousar, J.L. Development of elexacaftor—tezacaftor—ivacaftor: Highly effective CFTR modulation for the majority of people with Cystic Fibrosis. Expert Rev. Respir. Med. 2021, 15, 723–735. [Google Scholar] [CrossRef]
- Hisert, K.B.; Heltshe, S.L.; Pope, C.; Jorth, P.; Wu, X.; Edwards, R.M.; Radey, M.; Accurso, F.J.; Wolter, D.J.; Cooke, G.; et al. Restoring Cystic Fibrosis Transmembrane Conductance Regulator Function Reduces Airway Bacteria and Inflammation in People with Cystic Fibrosis and Chronic Lung Infections. Am. J. Respir. Crit. Care Med. 2017, 195, 1617–1628. [Google Scholar] [CrossRef]
- Jurado-Martin, I.; Sainz-Mejias, M.; McClean, S. Pseudomonas aeruginosa: An Audacious Pathogen with an Adaptable Arsenal of Virulence Factors. Int. J. Mol. Sci. 2021, 22, 3128. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, S.; Hayes, D., Jr.; Wozniak, D.J. Mucoid Pseudomonas aeruginosa and regional inflammation in the cystic fibrosis lung. J. Cyst. Fibros. 2019, 18, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef]
- Czaya, B.; Faul, C. The Role of Fibroblast Growth Factor 23 in Inflammation and Anemia. Int. J. Mol. Sci. 2019, 20, 4195. [Google Scholar] [CrossRef]
- Grabner, A.; Mazzaferro, S.; Cianciolo, G.; Krick, S.; Capelli, I.; Rotondi, S.; Ronco, C.; La Manna, G.; Faul, C. Fibroblast Growth Factor 23: Mineral Metabolism and Beyond. Contrib. Nephrol. 2017, 190, 83–95. [Google Scholar] [PubMed]
- Musgrove, J.; Wolf, M. Regulation and Effects of FGF23 in Chronic Kidney Disease. Annu. Rev. Physiol. 2020, 82, 365–390. [Google Scholar] [CrossRef]
- Grabner, A.; Amaral, A.P.; Schramm, K.; Singh, S.; Sloan, A.; Yanucil, C.; Li, J.; Shehadeh, L.A.; Hare, J.M.; David, V.; et al. Activation of Cardiac Fibroblast Growth Factor Receptor 4 Causes Left Ventricular Hypertrophy. Cell Metab. 2015, 22, 1020–1032. [Google Scholar] [CrossRef]
- Bollenbecker, S.; Heitman, K.; Czaya, B.; Easter, M.; Hirsch, M.J.; Vang, S.; Harris, E.; Helton, E.S.; Barnes, J.W.; Faul, C.; et al. Phosphate induces inflammation and exacerbates injury from cigarette smoke in the bronchial epithelium. Sci. Rep. 2023, 13, 4898. [Google Scholar] [CrossRef]
- Barnes, J.W.; Duncan, D.; Helton, S.; Hutcheson, S.; Kurundkar, D.; Logsdon, N.J.; Locy, M.; Garth, J.; Denson, R.; Farver, C.; et al. Role of fibroblast growth factor 23 and klotho cross talk in idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 317, L141–L154. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, M.J.; Hughes, E.M.; Easter, M.M.; Bollenbecker, S.E.; Howze Iv, P.H.; Birket, S.E.; Iv, P.H.H.; Birket, S.E.; Barnes, J.W.; Kiedrowski, M.R.; et al. A novel in vitro model to study prolonged Pseudomonas aeruginosa infection in the cystic fibrosis bronchial epithelium. PLoS ONE 2023, 18, e0288002. [Google Scholar] [CrossRef]
- Anderson, G.G.; Moreau-Marquis, S.; Stanton, B.A.; O’Toole, G.A. In vitro analysis of tobramycin-treated Pseudomonas aeruginosa biofilms on cystic fibrosis-derived airway epithelial cells. Infect. Immun. 2008, 76, 1423–1433. [Google Scholar] [CrossRef] [PubMed]
- Bonfield, T.L.; Panuska, J.R.; Konstan, M.W.; Hilliard, K.A.; Hilliard, J.B.; Ghnaim, H.; Berger, M. Inflammatory cytokines in cystic fibrosis lungs. Am. J. Respir. Crit. Care Med. 1995, 152 Pt 1, 2111–2118. [Google Scholar] [CrossRef] [PubMed]
- Coates, M.S.; Alton, E.; Rapeport, G.W.; Davies, J.C.; Ito, K. Pseudomonas aeruginosa induces p38MAP kinase-dependent IL-6 and CXCL8 release from bronchial epithelial cells via a Syk kinase pathway. PLoS ONE 2021, 16, e0246050. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirsch, M.J.; Matthews, E.L.; Bollenbecker, S.; Easter, M.; Kiedrowski, M.R.; Barnes, J.W.; Krick, S. Fibroblast Growth Factor 23 Signaling Does Not Increase Inflammation from Pseudomonas aeruginosa Infection in the Cystic Fibrosis Bronchial Epithelium. Medicina 2023, 59, 1635. https://doi.org/10.3390/medicina59091635
Hirsch MJ, Matthews EL, Bollenbecker S, Easter M, Kiedrowski MR, Barnes JW, Krick S. Fibroblast Growth Factor 23 Signaling Does Not Increase Inflammation from Pseudomonas aeruginosa Infection in the Cystic Fibrosis Bronchial Epithelium. Medicina. 2023; 59(9):1635. https://doi.org/10.3390/medicina59091635
Chicago/Turabian StyleHirsch, Meghan June, Emma Lea Matthews, Seth Bollenbecker, Molly Easter, Megan R. Kiedrowski, Jarrod W. Barnes, and Stefanie Krick. 2023. "Fibroblast Growth Factor 23 Signaling Does Not Increase Inflammation from Pseudomonas aeruginosa Infection in the Cystic Fibrosis Bronchial Epithelium" Medicina 59, no. 9: 1635. https://doi.org/10.3390/medicina59091635
APA StyleHirsch, M. J., Matthews, E. L., Bollenbecker, S., Easter, M., Kiedrowski, M. R., Barnes, J. W., & Krick, S. (2023). Fibroblast Growth Factor 23 Signaling Does Not Increase Inflammation from Pseudomonas aeruginosa Infection in the Cystic Fibrosis Bronchial Epithelium. Medicina, 59(9), 1635. https://doi.org/10.3390/medicina59091635