
A Rare Case of Orbital Castleman Disease with Overlapping IgG4-Related Disease



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
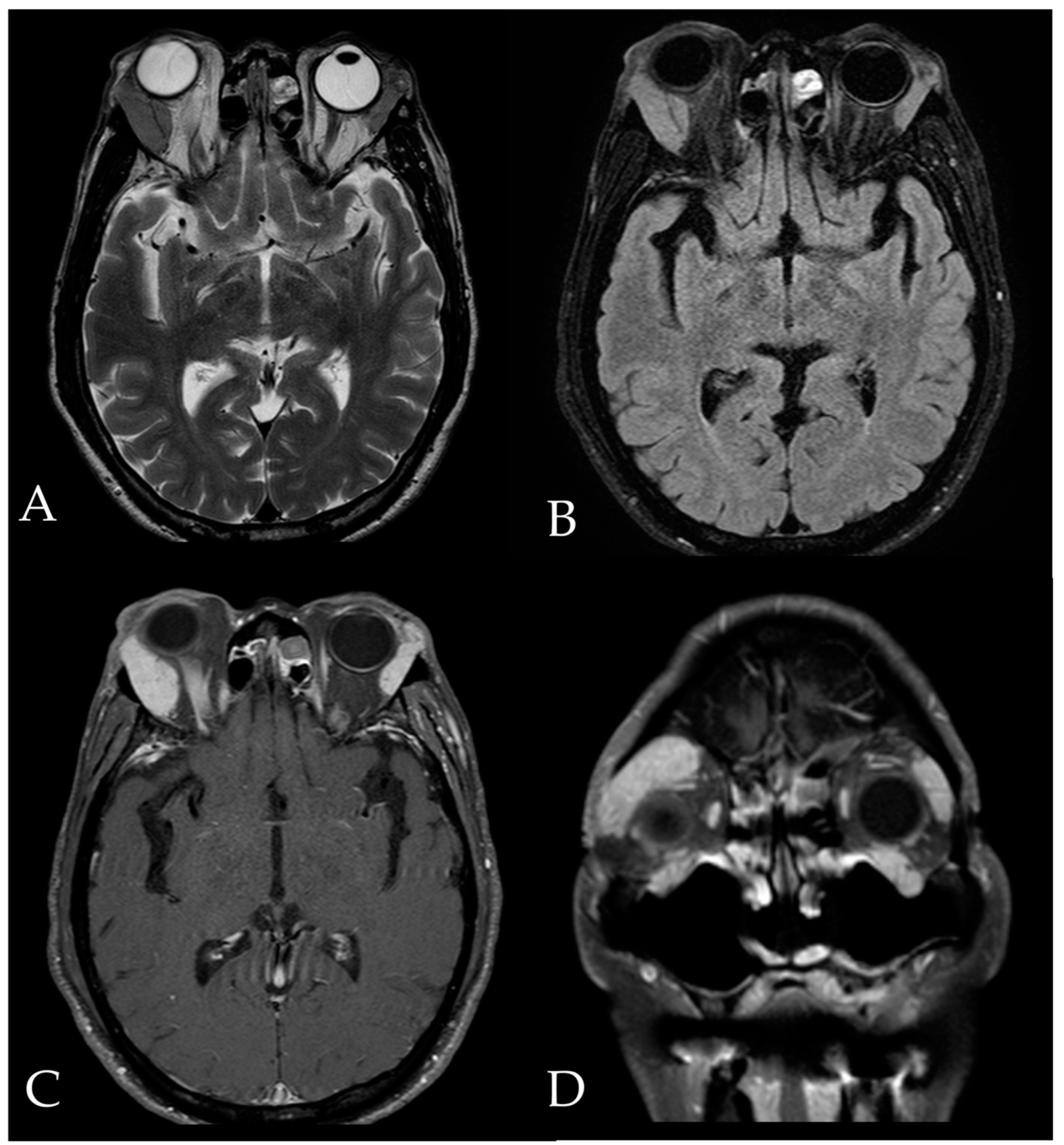
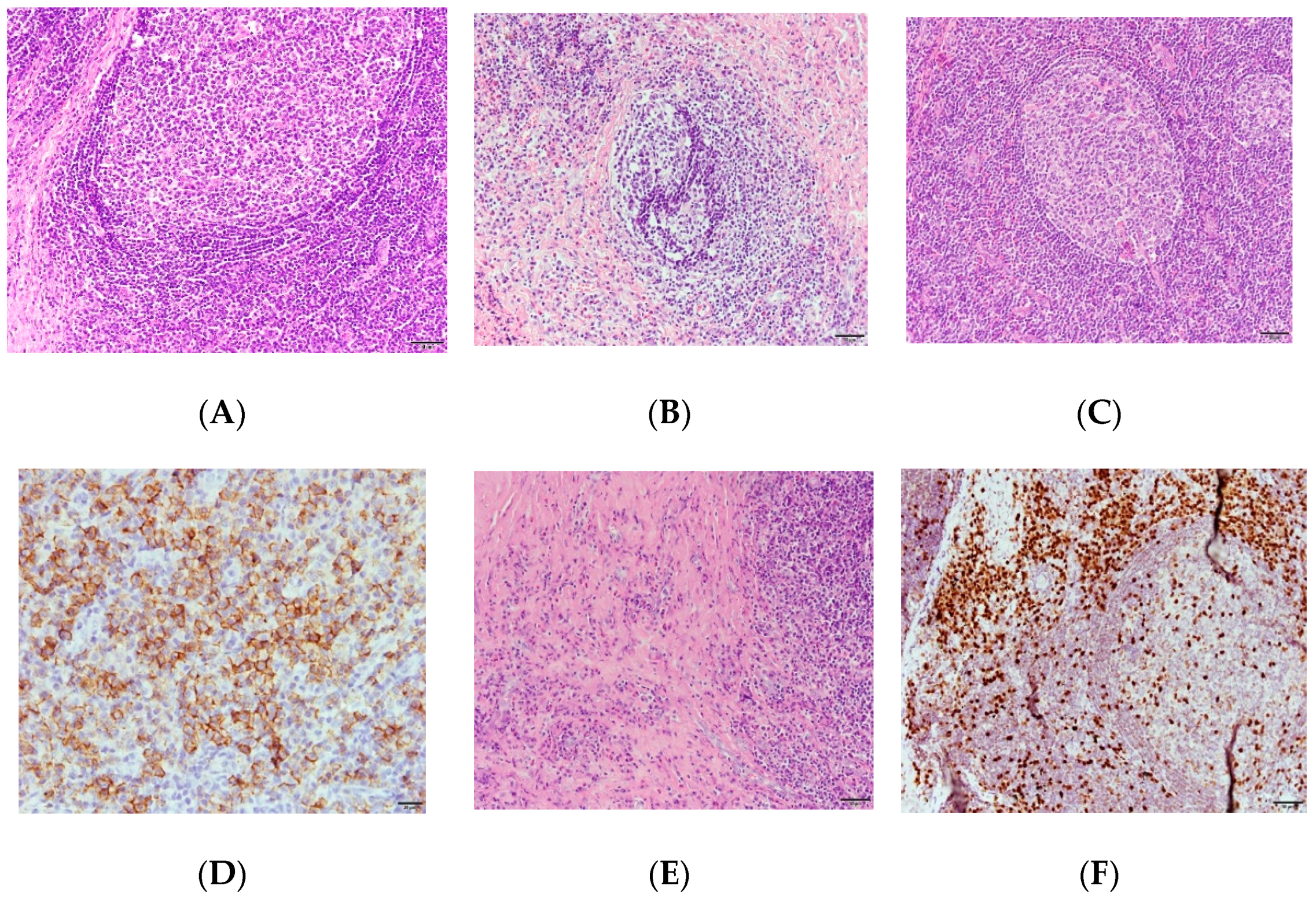
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gittinger, J.W., Jr. Ocular involvement in Castleman’s disease. Response to radiotherapy. Ophthalmology 1989, 96, 1646–1649. [Google Scholar] [CrossRef] [PubMed]
- Goel, R.; Raut, A.; Agarwal, A.; Raghav, S.; Kumar, S.; Chaudhary, S.; Golhait, P.; Kumar, S.; Saran, R. A rare presentation of orbital Castleman’s disease. Case Rep. Ophthalmol. Med. 2020, 2020, 1012759. [Google Scholar] [CrossRef] [PubMed]
- Dispenzieri, A.; Fajgenbaum, D.C. Overview of Castleman disease. Blood 2020, 135, 1353–1364. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.H.; Zen, Y.; Deshpande, V. IgG4-related disease. N. Engl. J. Med. 2012, 366, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, S.; Goyal, S.; Yadav, A.K.; Goyal, A. Multi-organ IgG4-related disease: Demystifying the diagnostic enigma. J. Postgrad. Med. 2018, 64, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Martín-Nares, E.; Hernández-Molina, G.; Baenas, D.F.; Paira, S. IgG4-related disease: Mimickers and diagnostic pitfalls. J. Clin. Rheumatol. 2022, 28, e596–e604. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Akiyama, M.; Kaneko, Y.; Takeuchi, T. Immunoglobulin G4-related disease and idiopathic multicentric Castleman’s disease: Confusable immune-mediated disorders. Rheumatology 2022, 61, 490–501. [Google Scholar] [CrossRef] [PubMed]
- Koppens, J.M.; Pon, J.A.; Allen, J.; Sloan, B.H. Castleman’s disease of the lacrimal gland. Clin. Exp. Ophthalmol. 2004, 32, 108–110. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Tang, D.; Sun, F. Clinical analysis of Castleman’s disease of the lacrimal gland. J. Ophthalmol. 2020, 2020, 3718305. [Google Scholar] [CrossRef] [PubMed]
- Venizelos, I.; Papathomas, T.G.; Papathanasiou, M.; Cheva, A.; Garypidou, V.; Coupland, S. Orbital involvement in Castleman disease. Surv. Ophthalmol. 2010, 55, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Khosroshahi, A.; Wallace, Z.S.; Crowe, J.L.; Akamizu, T.; Azumi, A.; Carruthers, M.N.; Chari, S.T.; Della-Torre, E.; Frulloni, L.; Goto, H.; et al. International consensus guidance statement on the management and treatment of IgG4-related disease. Arthritis Rheumatol. 2015, 67, 1688–1699. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.K.; Kao, S.C.; Yang, C.F.; Lee, F.L.; Tsai, C.C. Ocular adnexal IgG4-related disease: Clinical features, outcome, and factors associated with response to systemic steroids. Jpn. J. Ophthalmol. 2015, 59, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Otani, K.; Inoue, D.; Fujikura, K.; Komori, T.; Abe-Suzuki, S.; Tajiri, T.; Itoh, T.; Zen, Y. Idiopathic multicentric Castleman’s disease: A clinicopathologic study in comparison with IgG4-related disease. Oncotarget 2018, 9, 6691–6706. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Wang, H.W.; Pittaluga, S.; Jaffe, E.S. Multicentric Castleman disease and the evolution of the concept. Pathologica 2021, 113, 339–353. [Google Scholar] [CrossRef] [PubMed]
- González García, A.; Fernández-Martín, J.; Robles Marhuenda, Á. Idiopathic multicentric Castleman disease and associated autoimmune and autoinflammatory conditions: Practical guidance for diagnosis. Rheumatology 2023, 62, 1426–1435. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J. T helper 2 (Th2) cell differentiation, type 2 innate lymphoid cell (ILC2) development and regulation of interleukin-4 (IL-4) and IL-13 production. Cytokine 2015, 75, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, A.; Wali, S.; Nurieva, R. T helper 2 and T follicular helper cells: Regulation and function of interleukin-4. Cytokine Growth Factor Rev. 2016, 30, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Diehl, S.; Rincón, M. The two faces of IL-6 on Th1/Th2 differentiation. Mol. Immunol. 2002, 39, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Jain, A.; Syed, S.N.; Snodgrass, R.G.; Pflüger-Müller, B.; Leisegang, M.S.; Weigert, A.; Brandes, R.P.; Ebersberger, I.; Brüne, B.; et al. IL-6 augments IL-4-induced polarization of primary human macrophages through synergy of STAT3, STAT6 and BATF transcription factors. Oncoimmunology 2018, 7, e1494110. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, H.; Kato, M.; Higuchi, T.; Koyamada, R.; Arai, S.; Okada, S.; Eto, H. Overlap of IgG4-related disease and multicentric Castleman’s disease in a patient with skin lesions. Intern. Med. 2017, 56, 1095–1099. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, L.-C.; Chen, Y.-G.; Liu, N.-T.; Chen, Y.-H.; Chien, K.-H. A Rare Case of Orbital Castleman Disease with Overlapping IgG4-Related Disease. Medicina 2023, 59, 1381. https://doi.org/10.3390/medicina59081381
Liu L-C, Chen Y-G, Liu N-T, Chen Y-H, Chien K-H. A Rare Case of Orbital Castleman Disease with Overlapping IgG4-Related Disease. Medicina. 2023; 59(8):1381. https://doi.org/10.3390/medicina59081381
Chicago/Turabian StyleLiu, Li-Ching, Yann-Guang Chen, Nien-Tzu Liu, Yi-Hao Chen, and Ke-Hung Chien. 2023. "A Rare Case of Orbital Castleman Disease with Overlapping IgG4-Related Disease" Medicina 59, no. 8: 1381. https://doi.org/10.3390/medicina59081381
APA StyleLiu, L.-C., Chen, Y.-G., Liu, N.-T., Chen, Y.-H., & Chien, K.-H. (2023). A Rare Case of Orbital Castleman Disease with Overlapping IgG4-Related Disease. Medicina, 59(8), 1381. https://doi.org/10.3390/medicina59081381