
Congenital Absence of the Portal Vein as a Rare Cause of Portopulmonary Hypertension—A Case Study Series

 ,
,
Abstract
1. Introduction
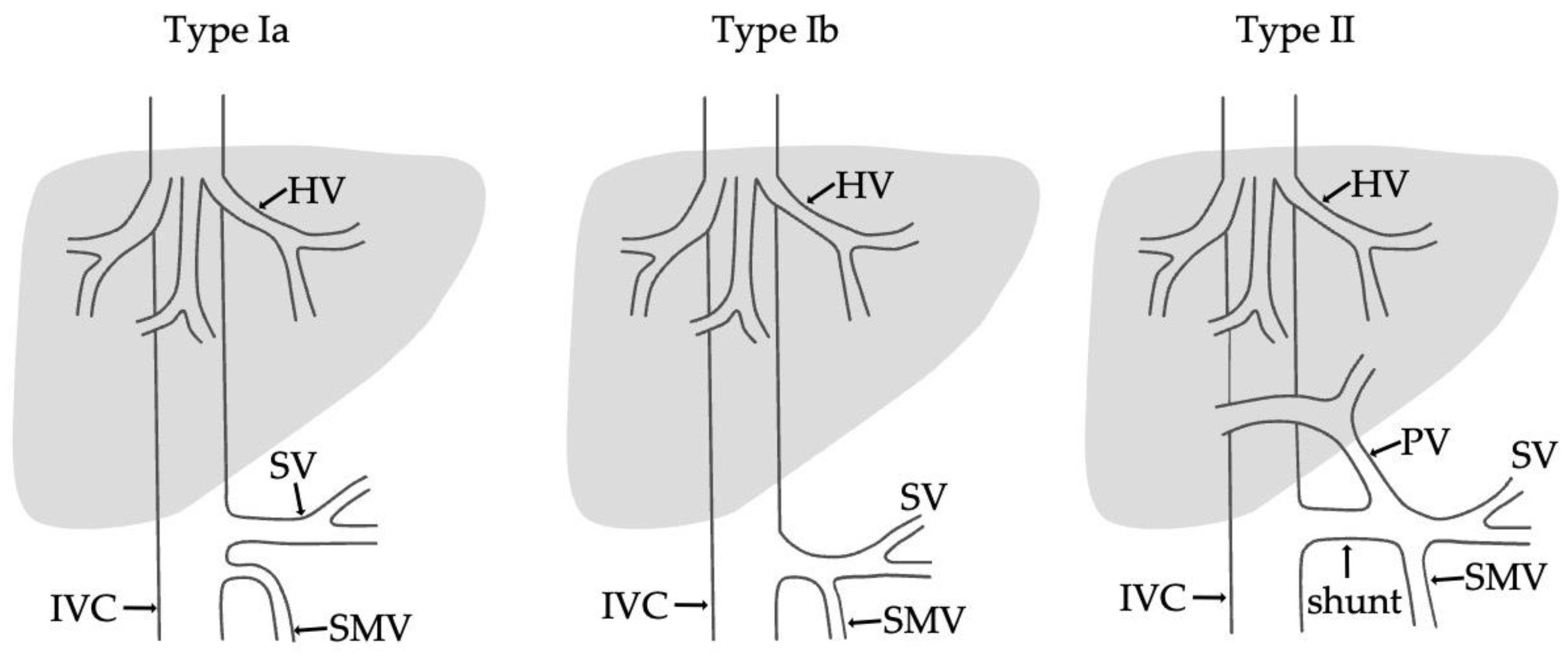
- Type I—end-to -side anastomosis with a complete portosystemic shunt exhibiting no visible portal flow in the liver due to the absence of intrahepatic portal veins. The malformation is female-predominant and associated with other congenital abnormalities, such as cardiac defects.
- Type II—side-to side-anastomosis with hypoplastic intrahepatic portal veins, and the liver is perfused with portal blood in the presence of a partial shunt (e.g., porto-hepatic venous anastomoses) [4].
2. Case Presentation
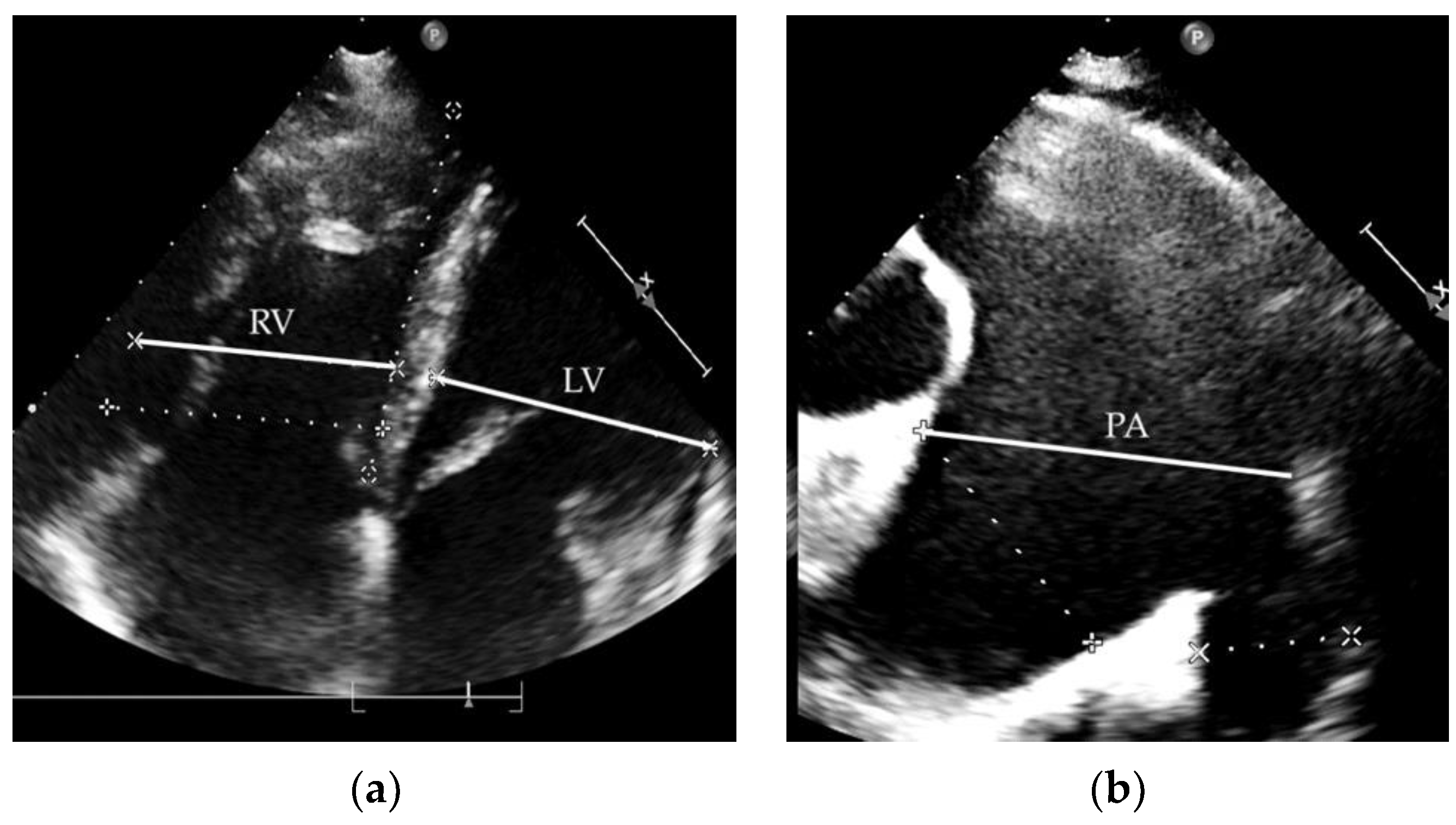
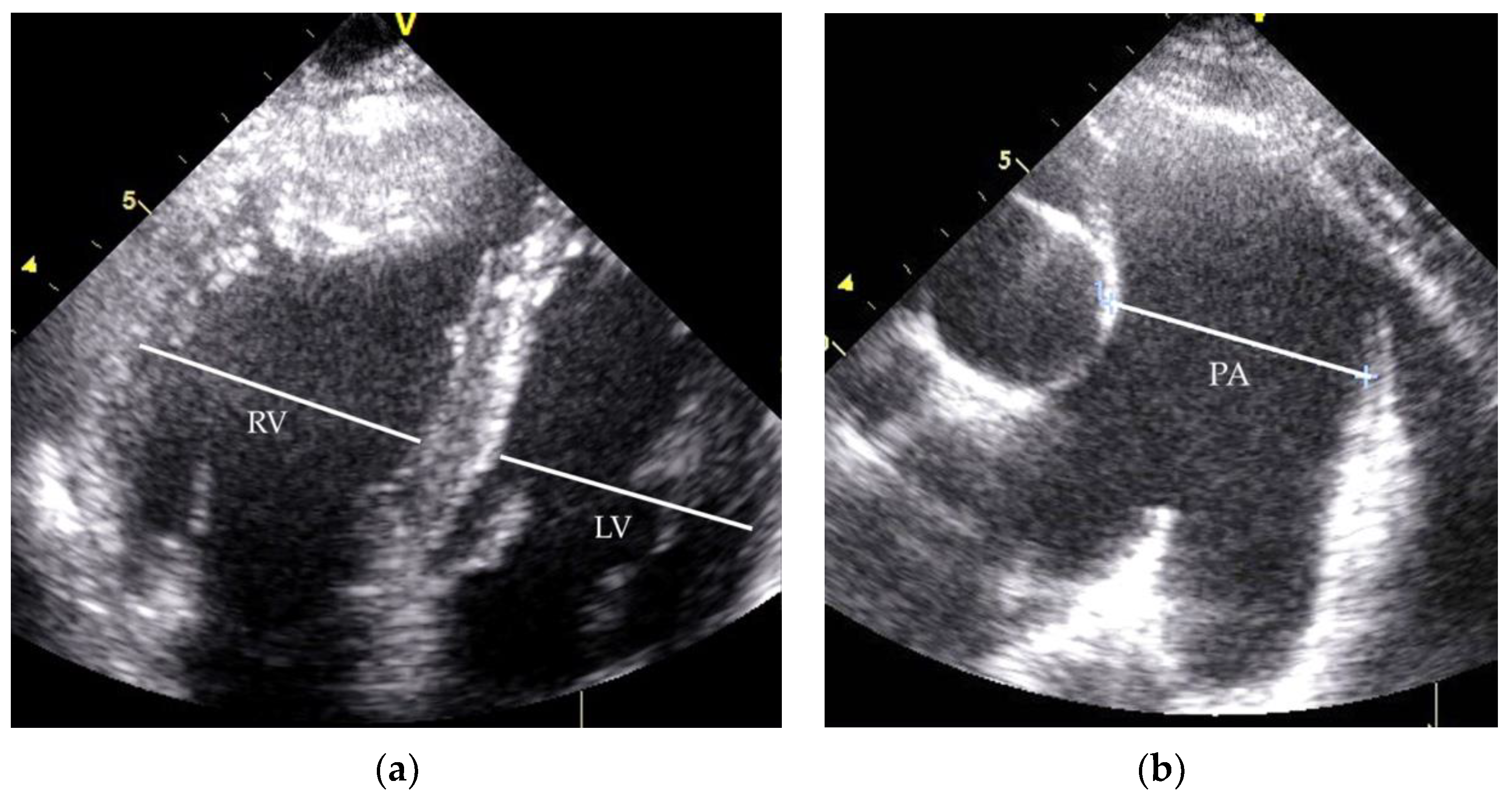
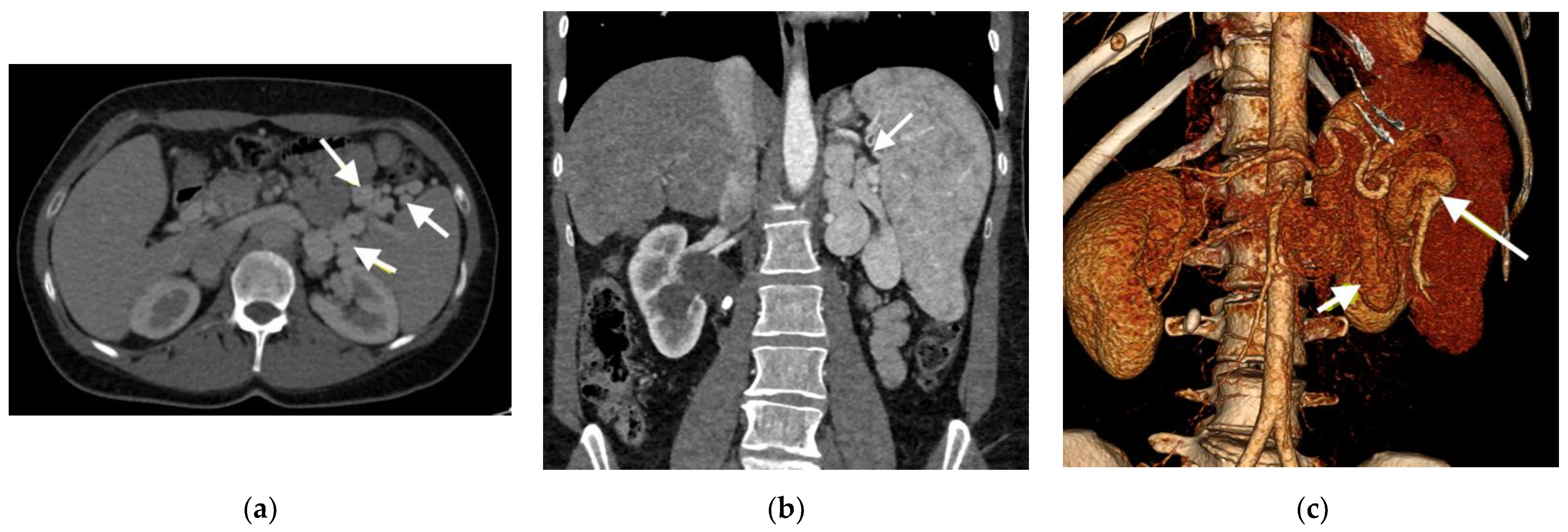
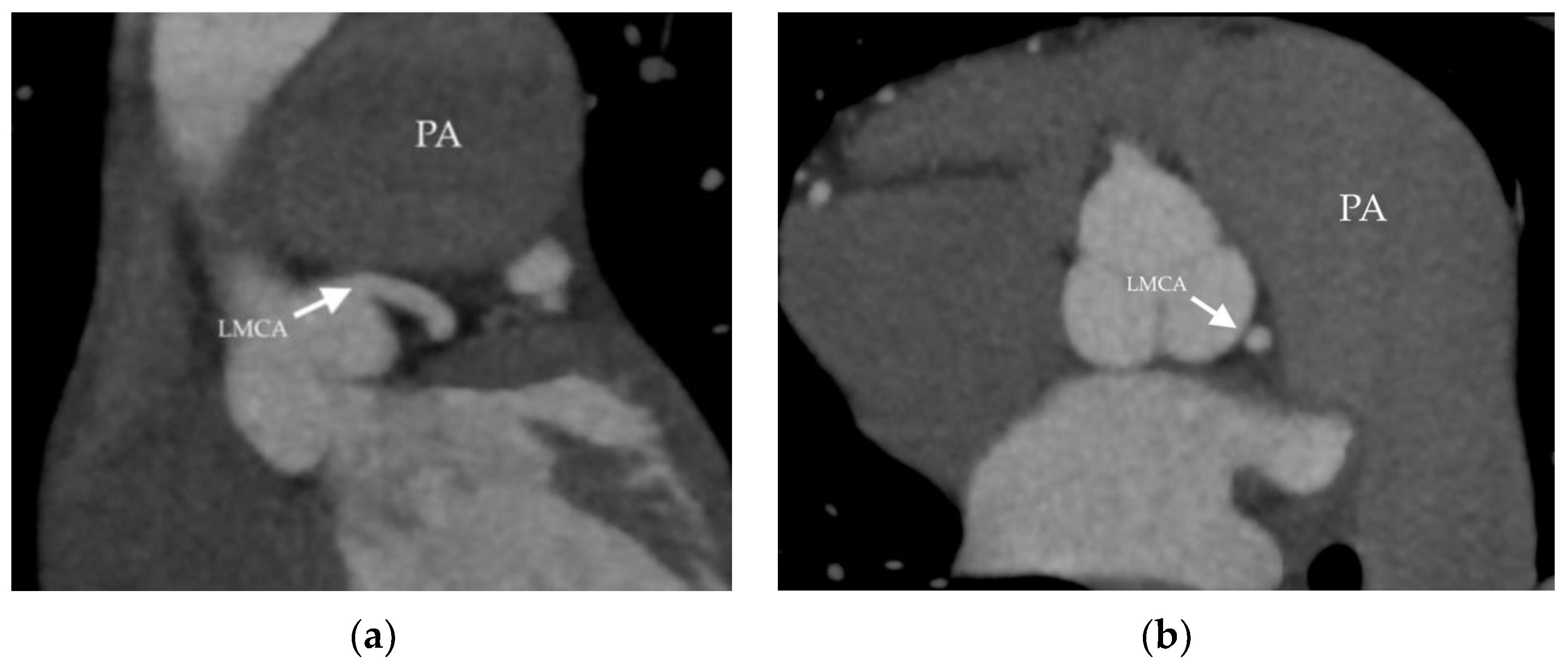
2.1. Clinical Case 1
2.2. Clinical Case 2
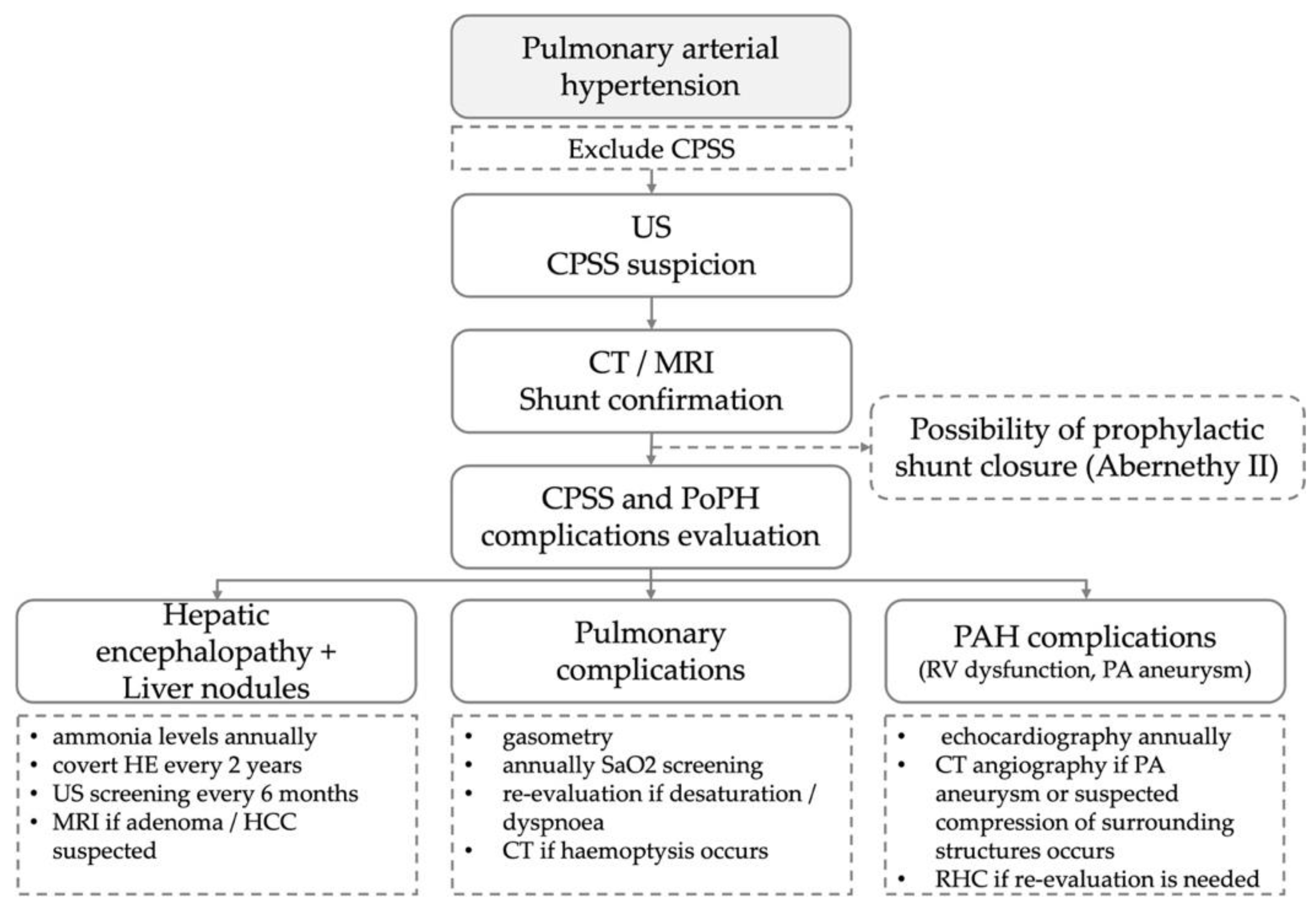
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Knirsch, W.; Benz, D.C.; Bühr, P.; Quandt, D.; Weber, R.; Kellenberger, C.; Braegger, C.P.; Kretschmar, O. Catheter Interventional Treatment of Congenital Portosystemic Venous Shunts in Childhood. Catheter. Cardiovasc. Interv. 2016, 87, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Bobhate, P.; Garg, S.; Sharma, A.; Roy, D.; Raut, A.; Pawar, R.; Karande, T.; Kulkarni, S. Congenital Extrahepatic Portocaval Malformation: Rare but Potentially Treatable Cause of Pulmonary Hypertension. Indian Heart J. 2021, 73, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Abernethy, J. Account of Two Instances of Uncommon Formation in the Viscera of the Human Body: From the Philosophical Transactions of the Royal Society of London. Med. Facts Obs. 1797, 7, 100–108. [Google Scholar] [PubMed]
- Morgan, G.; Superina, R. Congenital Absence of the Portal Vein: Two Cases and a Proposed Classification System for Portasystemic Vascular Anomalies. J. Pediatr. Surg. 1994, 29, 1239–1241. [Google Scholar] [CrossRef]
- Kohda, E.; Saeki, M.; Nakano, M.; Masaki, H.; Ogawa, K.; Nirasawa, M.; Hiramatsu, K. Congenital Absence of the Portal Vein in a Boy. Pediatr. Radiol. 1999, 29, 235–237. [Google Scholar] [CrossRef]
- Mistinova, J.; Valacsai, F.; Varga, I. Congenital Absence of the Portal Vein--Case Report and a Review of Literature. Clin. Anat. 2010, 23, 750–758. [Google Scholar] [CrossRef]
- Baiges, A.; Turon, F.; Simón-Talero, M.; Tasayco, S.; Bueno, J.; Zekrini, K.; Plessier, A.; Franchi-Abella, S.; Guerin, F.; Mukund, A.; et al. Congenital Extrahepatic Portosystemic Shunts (Abernethy Malformation): An International Observational Study. Hepatology 2020, 71, 658–669. [Google Scholar] [CrossRef]
- Kanazawa, H.; Nosaka, S.; Miyazaki, O.; Sakamoto, S.; Fukuda, A.; Shigeta, T.; Nakazawa, A.; Kasahara, M. The Classification Based on Intrahepatic Portal System for Congenital Portosystemic Shunts. J. Pediatr. Surg. 2015, 50, 688–695. [Google Scholar] [CrossRef]
- Ponziani, F.R.; Faccia, M.; Zocco, M.A.; Giannelli, V.; Pellicelli, A.; Ettorre, G.M.; De Matthaeis, N.; Pizzolante, F.; De Gaetano, A.M.; Riccardi, L.; et al. Congenital Extrahepatic Portosystemic Shunt: Description of Four Cases and Review of the Literature. J. Ultrasound 2018, 22, 349–358. [Google Scholar] [CrossRef]
- Le Borgne, J.; Paineau, J.; Hamy, A.; Dupas, B.; Lerat, F.; Raoul, S.; Hamel, A.; Robert, R.; Armstrong, O.; Rogez, J.M. Interruption of the Inferior Vena Cava with Azygos Termination Associated with Congenital Absence of Portal Vein. Surg. Radiol. Anat. 2000, 22, 197–202. [Google Scholar] [CrossRef]
- De Gaetano, A.M.; Gui, B.; Macis, G.; Manfredi, R.; Di Stasi, C. Congenital Absence of the Portal Vein Associated with Focal Nodular Hyperplasia in the Liver in an Adult Woman: Imaging and Review of the Literature. Abdom. Imaging 2004, 29, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.; Lee, C.; Wong, C.; Chan, A. Congenital Absence of Portal Vein Presenting as Hepatopulmonary Syndrome. J. Paediatr. Child Health 2005, 41, 72–75. [Google Scholar] [CrossRef] [PubMed]
- Namgoong, J.-M.; Hwang, S.; Park, G.-C.; Kwon, H.; Kim, K.M.; Oh, S.H. Living Donor Liver Transplantation in a Pediatric Patient with Congenital Absence of the Portal Vein. Ann. Hepatobiliary Pancreat. Surg. 2021, 25, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Lambert, V.; Ladarre, D.; Fortas, F.; Durand, P.; Hervé, P.; Gonzales, E.; Guérin, F.; Savale, L.; McLin, V.A.; Ackermann, O.; et al. Cardiovascular Disorders in Patients with Congenital Portosystemic Shunts: 23 Years of Experience in a Tertiary Referral Centre. Arch. Cardiovasc. Dis. 2021, 114, 221–231. [Google Scholar] [CrossRef]
- Ringle, M.L.; Loomba, R.; Dykes, J.C.; Khan, D.; Schidlow, D.; Wernovsky, G. The Multisystem Nature of Isomerism: Left Isomerism Complicated by Abernethy Malformation and Portopulmonary Hypertension. Cardiol. Young 2021, 31, 532–540. [Google Scholar] [CrossRef]
- Hino, T.; Hayashida, A.; Okahashi, N.; Wada, N.; Watanabe, N.; Obase, K.; Neishi, Y.; Kawamoto, T.; Okura, H.; Yoshida, K. Portopulmonary Hypertension Associated with Congenital Absence of the Portal Vein Treated with Bosentan. Intern. Med. 2009, 48, 597–600. [Google Scholar] [CrossRef][Green Version]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic Definitions and Updated Clinical Classification of Pulmonary Hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef]
- Savale, L.; Guimas, M.; Ebstein, N.; Fertin, M.; Jevnikar, M.; Renard, S.; Horeau-Langlard, D.; Tromeur, C.; Chabanne, C.; Prevot, G.; et al. Portopulmonary Hypertension in the Current Era of Pulmonary Hypertension Management. J. Hepatol. 2020, 73, 130–139. [Google Scholar] [CrossRef]
- Lebrec, D.; Capron, J.P.; Dhumeaux, D.; Benhamou, J.P. Pulmonary Hypertension Complicating Portal Hypertension. Am. Rev. Respir Dis 1979, 120, 849–856. [Google Scholar] [CrossRef]
- Huertas, A.; Guignabert, C.; Barberà, J.A.; Bärtsch, P.; Bhattacharya, J.; Bhattacharya, S.; Bonsignore, M.R.; Dewachter, L.; Dinh-Xuan, A.T.; Dorfmüller, P.; et al. Pulmonary Vascular Endothelium: The Orchestra Conductor in Respiratory Diseases: Highlights from Basic Research to Therapy. Eur. Respir. J. 2018, 51, 1700745. [Google Scholar] [CrossRef]
- Thomas, C.; Glinskii, V.; de Jesus Perez, V.; Sahay, S. Portopulmonary Hypertension: From Bench to Bedside. Front. Med. 2020, 7, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Laverdure, N.; Lallier, M.; Dubois, J.; Paganelli, M. Congenital Absence of the Portal Vein: Define the Portosystemic Shunt, Avoid Liver Transplantation. CanLivJ 2021, 4, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Puri, A.; McGoon, M.D.; Kushwaha, S.S. Pulmonary Arterial Hypertension: Current Therapeutic Strategies. Nat. Clin. Pract. Cardiovasc. Med. 2007, 4, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Kreibich, M.; Siepe, M.; Kroll, J.; Höhn, R.; Grohmann, J.; Beyersdorf, F. Aneurysms of the Pulmonary Artery. Circulation 2015, 131, 310–316. [Google Scholar] [CrossRef] [PubMed]
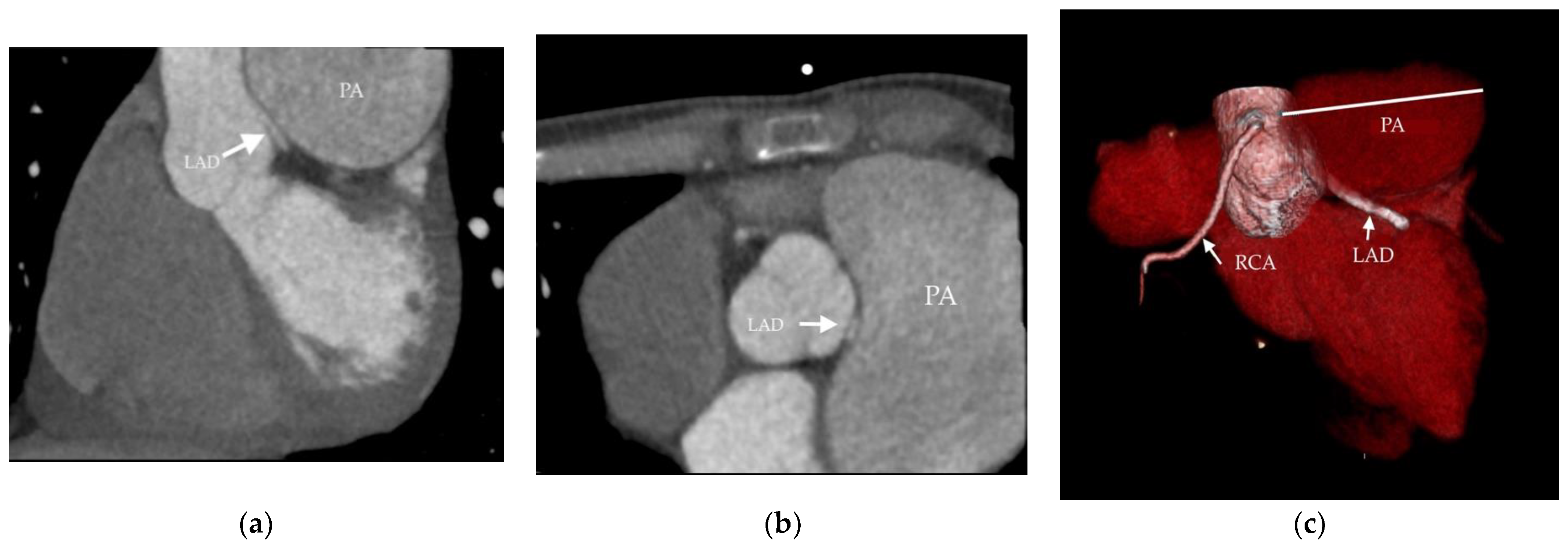
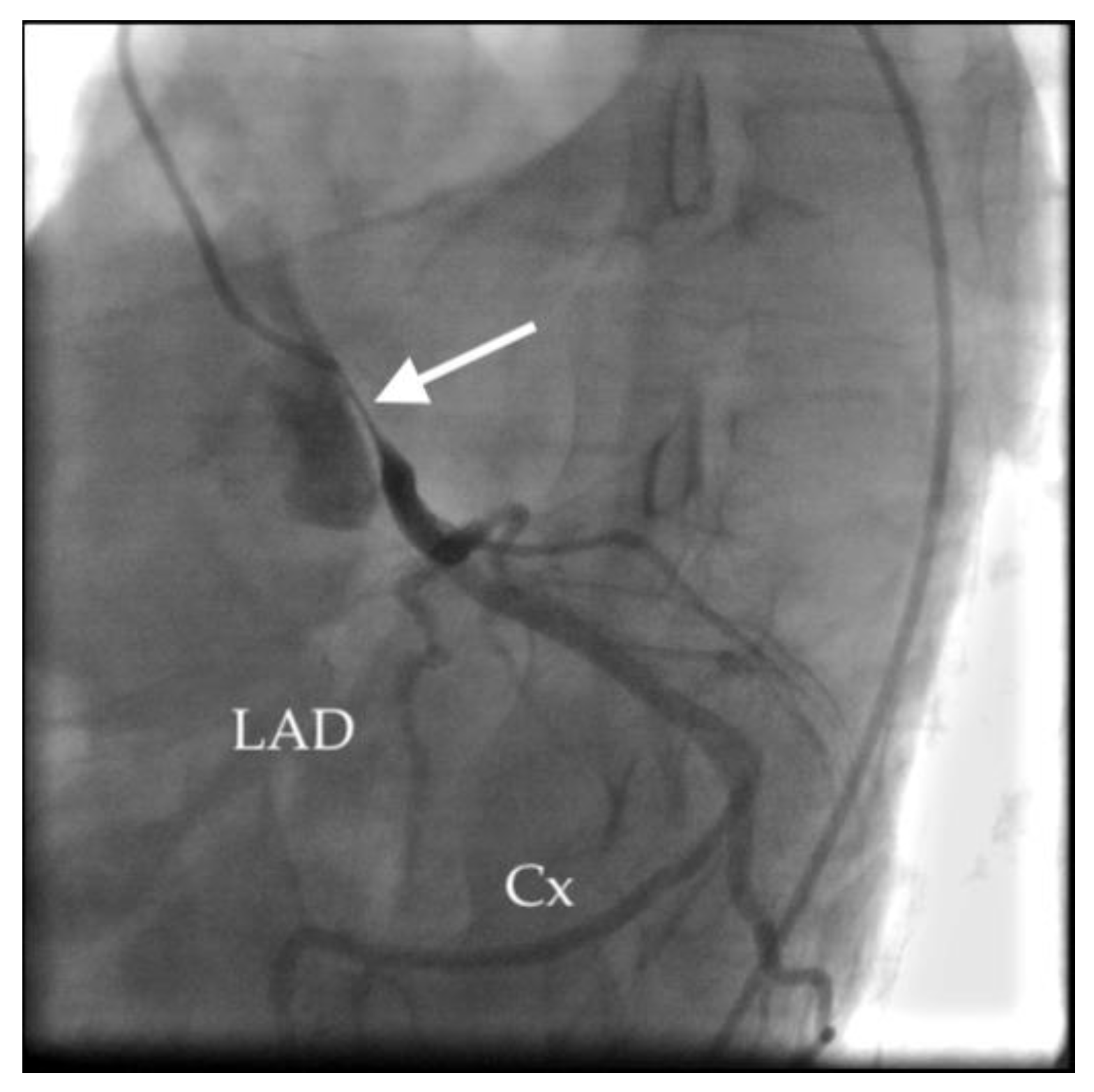
- Akbal, O.Y.; Kaymaz, C.; Tanboga, I.H.; Hakgor, A.; Yilmaz, F.; Turkday, S.; Dogan, C.; Tanyeri, S.; Demir, D.; Bayram, Z.; et al. Extrinsic Compression of Left Main Coronary Artery by Aneurysmal Pulmonary Artery in Severe Pulmonary Hypertension: Its Correlates, Clinical Impact, and Management Strategies. Eur. Heart J. Cardiovasc. Imaging 2018, 19, 1302–1308. [Google Scholar] [CrossRef]
- Hernández, V.; Ruiz-Cano, M.J.; Escribano, P.; Sánchez, M.A. Complications of Proximal Pulmonary Artery Aneurysm in Patients with Severe Pulmonary Arterial Hypertension. Rev. Esp. Cardiol. 2010, 63, 617–618. [Google Scholar] [CrossRef]
- Caldera, A.E.; Cruz-Gonzalez, I.; Bezerra, H.G.; Cury, R.C.; Palacios, I.F.; Cockrill, B.A.; Inglessis-Azuaje, I. Endovascular Therapy for Left Main Compression Syndrome. Case Report and Literature Review. Chest 2009, 135, 1648–1650. [Google Scholar] [CrossRef]
- Galiè, N.; Saia, F.; Palazzini, M.; Manes, A.; Russo, V.; Bacchi Reggiani, M.L.; Dall’Ara, G.; Monti, E.; Dardi, F.; Albini, A.; et al. Left Main Coronary Artery Compression in Patients With Pulmonary Arterial Hypertension and Angina. J. Am. Coll. Cardiol. 2017, 69, 2808–2817. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RHC Parameters | Time of Diagnosis | Before Initiation of the Specific Therapy | 8 Years of Specific Therapy |
|---|---|---|---|
| mPAP (mmHg) | 59 | 90 | 49 |
| PVR (WU) | 8.1 | 12.6 | 6.9 |
| PCWP (mmHg) | 8 | 10 | 13 |
| CO (L/min) | 6.1 | 5.5 | 5.2 |
| Before ST | 1 Year of ST | 5 Years of ST | 10 Years of ST | |
|---|---|---|---|---|
| 6MWT (m) | 575 | 690 | 670 | 630 |
| NYHA class | III | I-II | II | II |
| TR grad. (mmHg) | 80 | 70 | 66 | 50 |
| TAPSE (mm) | 15 | 21 | 29 | 21 |
| PA diameter (mm) | 52 | 56 | 58 | 66 |
| RHC Parameters | Time of Diagnosis | Before Initiation of the Specific Therapy |
|---|---|---|
| mPAP (mmHg) | 58 | 65 |
| PVR (WU) | 8.5 | 7.2 |
| PCWP (mmHg) | 9 | 2 |
| CO (L/min) | 5.8 | 8.7 |
| Before ST | 1 Year of ST | 5 Years of ST | 10 Years of ST | |
|---|---|---|---|---|
| 6MWT (m) | 560 | 710 | 708 | 640 |
| NYHA class | II-III | I | I | II |
| TR grad. (mmHg) | 65 | 55 | 50 | 67 |
| TAPSE (mm) | 27 | 29 | 25 | 28 |
| PA diameter (mm) | 42 | 45 | 52 | 53 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hlavata, T.; Kaldararova, M.; Klauco, F.; Drangova, E.; Reptova, A.; Simkova, I. Congenital Absence of the Portal Vein as a Rare Cause of Portopulmonary Hypertension—A Case Study Series. Medicina 2022, 58, 1484. https://doi.org/10.3390/medicina58101484
Hlavata T, Kaldararova M, Klauco F, Drangova E, Reptova A, Simkova I. Congenital Absence of the Portal Vein as a Rare Cause of Portopulmonary Hypertension—A Case Study Series. Medicina. 2022; 58(10):1484. https://doi.org/10.3390/medicina58101484
Chicago/Turabian StyleHlavata, Tereza, Monika Kaldararova, Filip Klauco, Erika Drangova, Adriana Reptova, and Iveta Simkova. 2022. "Congenital Absence of the Portal Vein as a Rare Cause of Portopulmonary Hypertension—A Case Study Series" Medicina 58, no. 10: 1484. https://doi.org/10.3390/medicina58101484
APA StyleHlavata, T., Kaldararova, M., Klauco, F., Drangova, E., Reptova, A., & Simkova, I. (2022). Congenital Absence of the Portal Vein as a Rare Cause of Portopulmonary Hypertension—A Case Study Series. Medicina, 58(10), 1484. https://doi.org/10.3390/medicina58101484