
Impaired Folate-Mediated One-Carbon Metabolism in Type 2 Diabetes, Late-Onset Alzheimer’s Disease and Long COVID



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Impaired FOCM in T2DM
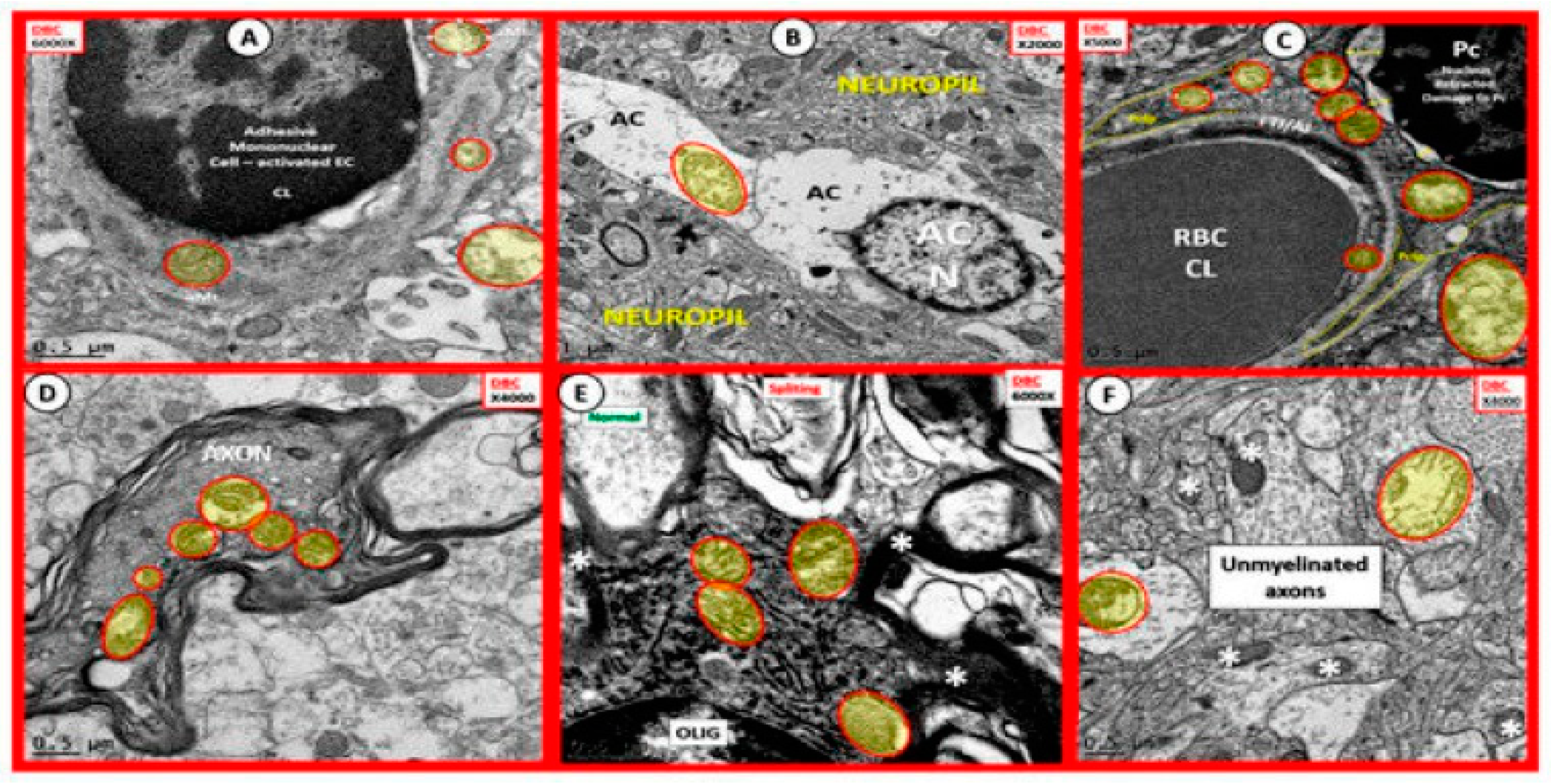
2.1. The Neurovascular Unit (NVU) in Diabetic Brain of the db/db Mouse
2.2. The Endothelial Glycocalyx (ecGCx) in T2DM
2.3. Aberrant Mitochondria and Impaired FOCM in T2DM
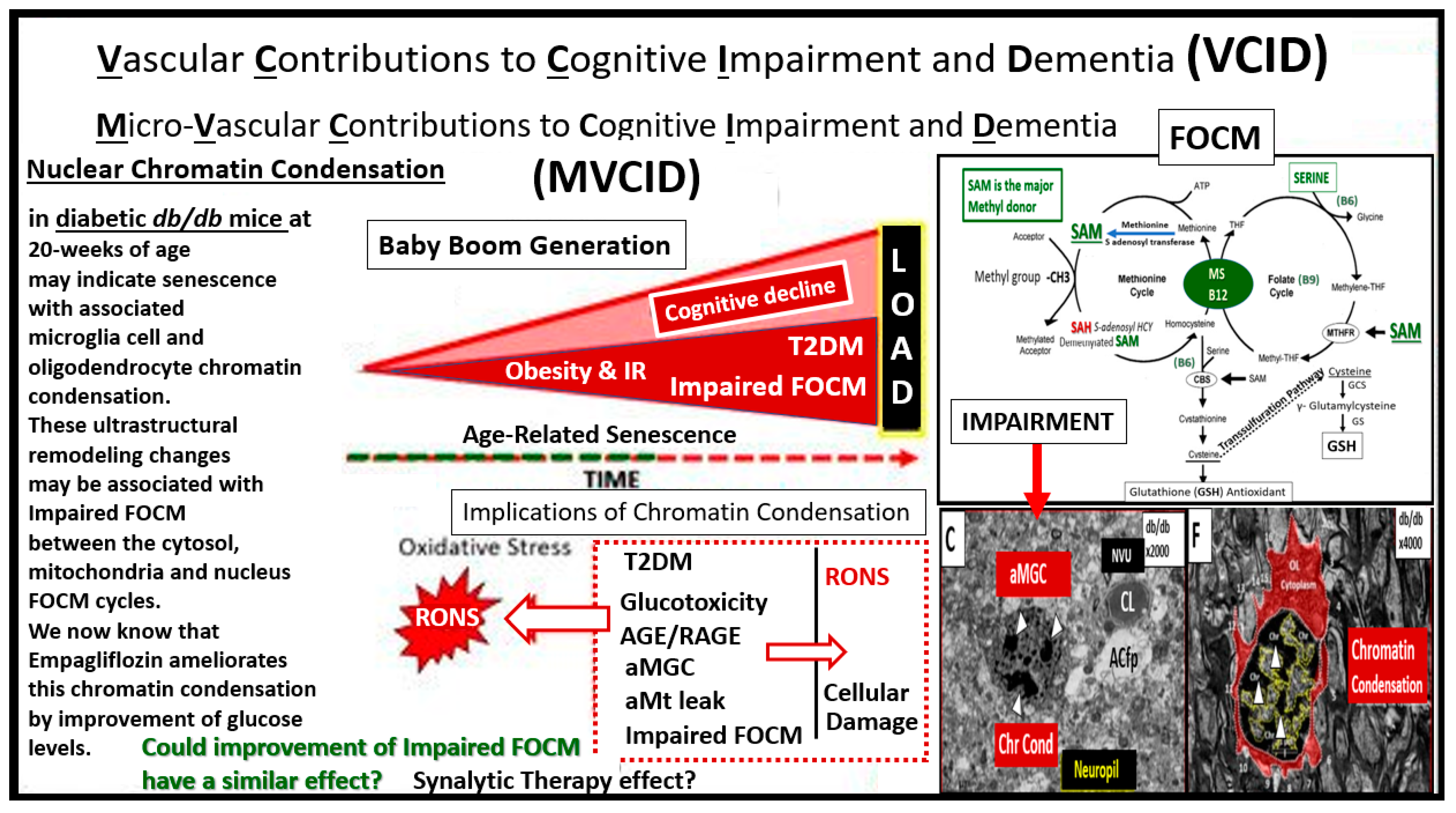
3. Impaired FOCM in Sporadic or LOAD
3.1. Vascular Contributions to Cognitive Impairment and Dementia (VCID) including LOAD and Impaired FOCM
3.2. Examining the Mitochondrial Cascade Hypothesis in LOAD and Impaired FOCM
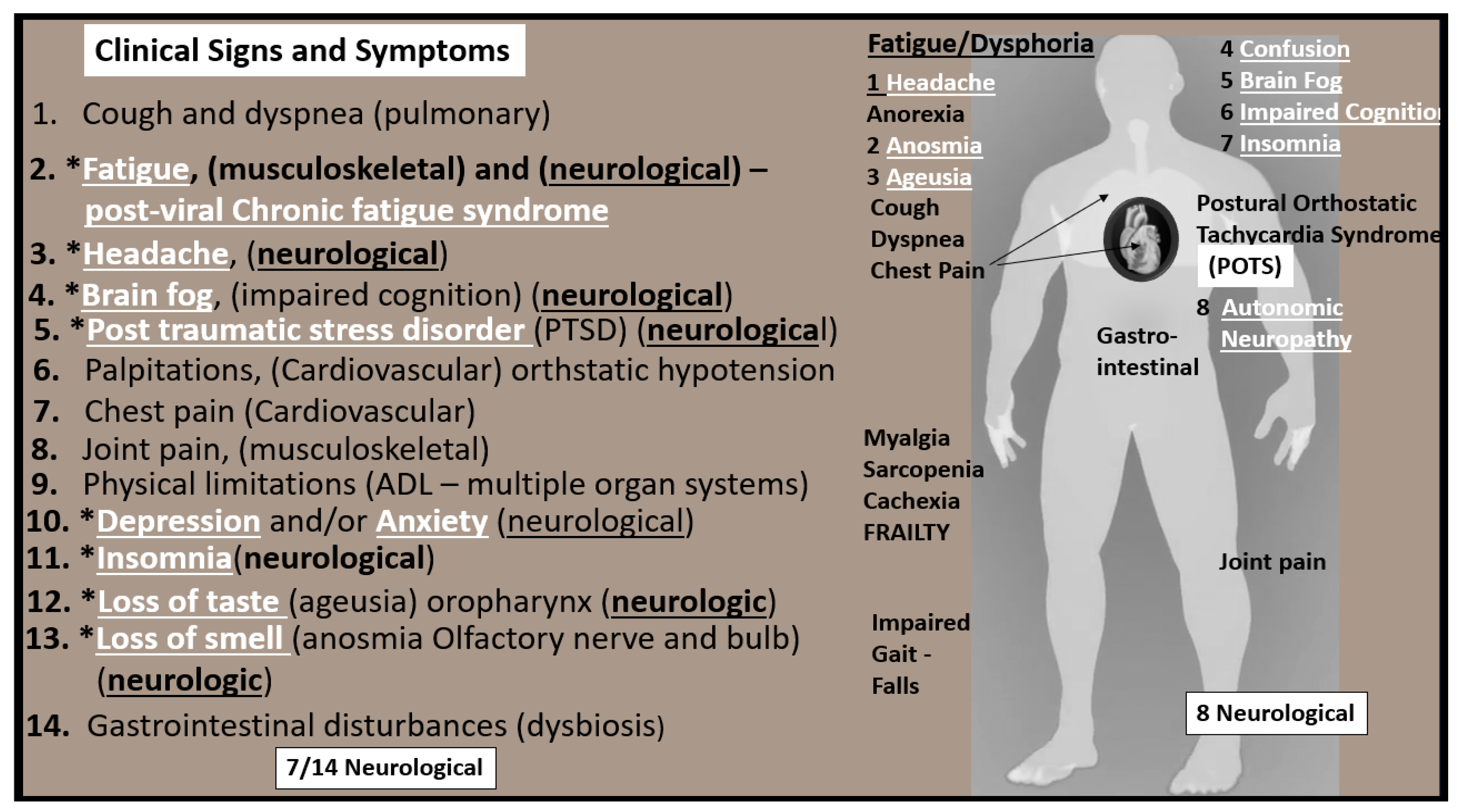
4. Impaired FOCM in LC/PASC
Compromised FOCM in LC/PASC: Importance of HHCY and Deficient Micronutrients (Vitamins B12 and B9)
5. Future Possible Treatment Options for LC/PASC
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cheng, Z.; Yang, X.; Wang, H. Hyperhomocysteinemia and Endothelial Dysfunction. Curr. Hypertens. Rev. 2009, 5, 158–165. [Google Scholar] [CrossRef] [Green Version]
- Rehman, T.; Shabbir, M.A.; Inam-Ur-Raheem, M.; Manzoor, M.F.; Ahmad, N.; Liu, Z.W.; Ahmad, M.H.; Siddeeg, A.; Abid, M.; Aadil, R.M. Cysteine and homocysteine as biomarker of various diseases. Food Sci. Nutr. 2020, 8, 4696–4707. [Google Scholar] [CrossRef]
- Abbenhardt, C.; Miller, J.W.; Song, X.; Brown, E.C.; Cheng, T.-Y.D.; Wener, M.H.; Zheng, Y.; Toriola, A.; Neuhouser, M.L.; Beresford, S.A.A.; et al. Biomarkers of One-Carbon Metabolism Are Associated with Biomarkers of Inflammation in Women. J. Nutr. 2014, 144, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Troesch, B.; Weber, P.; Mohajeri, M.H. Potential Links between Impaired One-Carbon Metabolism Due to Polymorphisms, Inadequate B-Vitamin Status, and the Development of Alzheimer’s Disease. Nutrients 2016, 8, 803. [Google Scholar] [CrossRef]
- Mason, J.B. Biomarkers of Nutrient Exposure and Status in One-Carbon (Methyl) Metabolism. J. Nutr. 2003, 133 (Suppl. 3), 941S–947S. [Google Scholar] [CrossRef]
- Hayden, M.R.; Tyagi, S.C. Homocysteine and reactive oxygen species in metabolic syndrome, type 2 diabetes mellitus, and atheroscleropathy: The pleiotropic effects of folate supplementation. Nutr. J. 2004, 3, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crapser, J.D.; Spangenberg, E.E.; Barahona, R.A.; Arreola, M.A.; Hohsfield, L.A.; Green, K.N. Microglia facilitate loss of perineuronal nets in the Alzheimer’s disease brain. EBioMedicine 2020, 58, 102919. [Google Scholar] [CrossRef]
- Barroso, M.; Handy, D.E.; Castro, R. The Link between Hyperhomocysteinemia and Hypomethylation: Implications for Cardiovascular Disease. J. Inborn Errors Metab. Screen. 2017, 5, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, W. Significance of hyperhomocysteinemia. Clin. Lab. 2006, 52, 367–374. [Google Scholar]
- Graham, I.M.; O’Callaghan, P. Vitamins, homocysteine and cardiovascular risk. Cardiovasc. Drugs Ther. 2002, 16, 383–389. [Google Scholar] [CrossRef]
- Ponti, G.; Ruini, C.; Tomasi, A. Homocysteine as a potential predictor of cardiovascular risk in patients with COVID-19. Med. Hypotheses 2020, 143, 109859. [Google Scholar] [CrossRef]
- Ibrahimagić, O.Ć.; Smajlović, D.; Dostović, Z.; Vidović, M.; Tupković, E.; Kunić, S. COMMENT ON AN ARTICLE: “Homocysteine as a potential predictor of cardiovascular risk in patients with COVID-19”. Med. Hypotheses 2020, 143, 110107. [Google Scholar] [CrossRef]
- Boers, G.H. Mild hyperhomocysteinemia is an independent risk factor of arterial vascular disease. Semin. Thromb. Hemost. 2000, 26, 291–295. [Google Scholar] [CrossRef]
- Blom, H.J. Consequences of homocysteine export and oxidation in the vascular system. Semin. Thromb. Hemost. 2000, 26, 227–232. [Google Scholar] [CrossRef]
- Jakubowski, H.; Zhang, L.; Bardeguez, A.; Aviv, A. Homocysteine thiolactone and protein homocysteinylation in human endothelial cells: Implications for atherosclerosis. Circ. Res. 2000, 87, 45–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Bailey, L.B.; Stover, P.J.; McNulty, H.; Fenech, M.F.; Gregory, J.F.; Mills, J.L.; Pfeiffer, C.M.; Fazili, Z.; Zhang, M.; Ueland, P.M.; et al. Biomarkers of nutrition for development- folate review. J. Nutr. 2015, 1447, 1636S–16380S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNulty, H.; Strain, J.J.; Hughes, C.F.; Pentieva, K.; Ward, M. Evidence of a Role for One-Carbon Metabolism in Blood Pressure: Can B Vitamin Intervention Address the Genetic Risk of Hypertension Owing to a Common Folate Polymorphism? Curr. Dev. Nutr. 2019, 4, nzz102. [Google Scholar] [CrossRef]
- Oguntibeju, O.O. Type 2 diabetes mellitus, oxidative stress and inflammation: Examining the links. Int. J. Physiol. Pathophysiol. Pharmacol. 2019, 11, 45–63. [Google Scholar] [PubMed]
- Yang, Y.; Hayden, M.R.; Sowers, S.; Bagree, S.V.; Sowers, J.R. Retinal redox stress and remodeling in cardiometabolic syndrome and diabetes. Oxid. Med. Cell Longev. 2010, 3, 392–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayden, M.R.; Salam, M.; Sowers, J.R. Reactive Oxygen Species and Diabetic Peripheral Neuropathy—A Closer Look, Chapter 149; Lahey, I., Ed.; Systems Biology of Reactive Oxygen Species and Antioxidants; Springer: Berlin/Heidelberg, Germany, 2014; pp. 3375–3400. [Google Scholar]
- Hayden, M.R.; Whaley-Connell, A.; Sowers, J.R. Renal redox stress and remodeling in metabolic syndrome, type 2 diabetes mellitus, and diabetic nephropathy: Paying homage to the podocyte. Am. J. Nephrol. 2005, 25, 553–569. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R.; Tyagi, S.C. Intimal redox stress: Accelerated atherosclerosis in metabolic syndrome and type 2 diabetes mellitus. Cardiovasc. Diabetol. 2002, 1, 3. [Google Scholar] [CrossRef]
- Hayden, M.R.; Sowers, J.R. Isletopathy in Type 2 diabetes mellitus: Implications of islet RAS, islet fibrosis, islet amyloid, remodeling, and oxidative stress. Antiox Redox Signal. 2007, 9, 891–910. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R.; Tyagi, S.C. Is type 2 diabetes mellitus a vascular disease (atheroscleropathy) with hyperglycemia a late manifestation? The role of NOS, NO, and redox stress. Cardiovasc. Diabetol. 2003, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R.; Banks, W.A.; Shah, G.N.; Gu, Z.; Sowers, J.R. Cardiorenal metabolic syndrome and diabetic cognopathy. Cardiorenal. Med. 2013, 3, 265–282. [Google Scholar] [CrossRef] [Green Version]
- Hayden, M.R. Type 2 Diabetes Mellitus Increases the Risk of Late-Onset Alzheimer’s Disease: Ultrastructural Remodeling of the Neurovascular Unit and Diabetic Gliopathy. Brain Sci. 2019, 9, 262. [Google Scholar] [CrossRef] [Green Version]
- Joshi, M.B.; Baipadithaya, G.; Balakrishnan, A.; Hegde, M.; Vohra, M.; Ahamed, R.; Nagri, S.K.; Ramachandra, L.; Satyamoorthy, K. Elevated homocysteine levels in type 2 diabetes induce constitutive neutrophil extracellular traps. Sci. Rep. 2016, 6, 36362. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.; Ren, J.; Haung, J.; Li, D. Association of homocysteine with type 2 diabetes: A meta-analysis implementing Mendelian randomization approach. BMC Genom. 2013, 14, 867. [Google Scholar] [CrossRef] [Green Version]
- Finer, S.; Saravanan, P.; Hitman, G.; Yajnik, C. The role of the one-carbon cycle in the developmental origins of Type 2 diabetes and obesity. Diabet. Med. 2014, 31, 263–272. [Google Scholar] [CrossRef]
- Mursleen, M.T.; Riaz, S. Implication of homocysteine in diabetes and impact of folate and vitamin B12 in diabetic population. Diabetes Metab. Syndr. 2017, 11 (Suppl. 1), S141–S146. [Google Scholar] [CrossRef]
- Hayden, M.R.; Grant, D.G.; Aroor, A.R.; DeMarco, V.G. Ultrastructural Remodeling of the Neurovascular Unit in the Female Diabetic db/db Model—Part II: Microglia and Mitochondria. Neuroglia. Neuroglia 2018, 1, 311–326. [Google Scholar] [CrossRef] [Green Version]
- Hayden, M.R.; Grant, D.G.; Aroor, A.R.; DeMarco, V.G. Ultrastructural Remodeling of the Neurovascular Unit in the Female Diabetic db/db Model–Part III: Oligodendrocyte and Myelin. Neuroglia 2018, 1, 351–364. [Google Scholar] [CrossRef] [Green Version]
- Hayden, M.R.; Grant, D.G.; Aroor, A.R.; DeMarco, V.G. Empagliflozin Ameliorates Type 2 Diabetes-Induced Ultrastructural Remodeling of the Neurovascular Unit and Neuroglia in the Female db/db Mouse. Brain Sci. 2019, 9, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietzke, M.; Meiser, J.; Vazquez, A. Formate metabolism in health and disease. Mol. Metab. 2020, 33, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.; Pai, Y.J.; Mahmood, M.R.; Leung, K.Y.; Savery, D.; Waddington, S.N.; Copp, A.J.; Greene, N. Impaired folate 1-carbon metabolism causes formate-preventable hydrocephalus in glycine decarboxylase-deficient mice. J. Clin. Investig. 2020, 130, 1446–1452. [Google Scholar] [CrossRef]
- Zhao, M.; Yuan, M.M.; Yuan, L.; Huang, L.L.; Liao, J.H.; Yu, X.L.; Su, C.; Chen, Y.H.; Yang, Y.Y.; Yu, H.; et al. Chronic folate deficiency induces glucose and lipid metabolism disorders and subsequent cognitive dysfunction in mice. PLoS ONE 2018, 13, e0202910. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhai, J.X.; Liu, D.W. Serum folate, vitamin B12 levels and diabetic peripheral neuropathy in type 2 diabetes: A meta-analysis. Mol. Cell Endocrinol. 2017, 443, 72–79. [Google Scholar] [CrossRef]
- Wee, A.K. Serum folate predicts muscle strength: A pilot cross-sectional study of the association between serum vitamin levels and muscle strength and gait measures in patients >65 years old with diabetes mellitus in a primary care setting. Nutr. J. 2016, 15, 89. [Google Scholar] [CrossRef] [Green Version]
- Hayden, M.R.; Banks, W.A. Deficient Leptin Cellular Signaling Plays a Key Role in Brain Ultrastructural Remodeling in Obesity and Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2021, 22, 5427. [Google Scholar] [CrossRef]
- Hayden, M.R.; Grant, D.G.; Aroor, A.R.; DeMarco, V.G. Ultrastructural Remodeling of the Neurovascular Unit in the Female Diabetic db/db Model—Part I: Astrocyte. Neuroglia 2018, 1, 220–244. [Google Scholar] [CrossRef] [Green Version]
- Luft, J.H. Fine structures of capillary and endocapillary layer as revealed by ruthenium red. Fed. Proc. 1966, 25, 1773–1783. [Google Scholar]
- Reitsma, S.; Slaaf, D.W.; Vink, H.; van Zandvoort, M.A.; Oude Egbrink, M.G. The endothelial glycocalyx: Composition, functions, and visualization. Pflug. Arch. 2007, 454, 345–359. [Google Scholar] [CrossRef] [Green Version]
- Haeren, R.H.L.; Rijkers, K.; Schijns, O.E.M.G.; Dings, J.; Hoogland, G.; van Zandvoort, M.A.M.J.; Vink, H.; van Overbeeke, J.J. In vivo assessment of the human cerebral microcirculation and its glycocalyx: A technical report. J. Neurosci. Methods 2018, 303, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Jeong, Y. In vivo imaging for neurovascular disease research. Arch. Pharm. Res. 2019, 42, 263–273. [Google Scholar] [CrossRef]
- Reed, M.J.; Damodarasamy, M.; Banks, W.A. The extracellular matrix of the blood-brain barrier: Structural and functional roles in health, aging, and Alzheimer’s disease. Tissue Barriers 2019, 7, 1651157. [Google Scholar] [CrossRef]
- Hayden, M.R. Endothelial activation and dysfunction in metabolic syndrome, type 2 diabetes and coronavirus disease 2019. J. Int. Med. Res. 2020, 48, 300060520939746. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R. Hypothesis: Neuroglia Activation Due to Increased Peripheral and CNS Proinflammatory Cytokines/Chemokines with Neuroinflammation May Result in Long COVID. Neuroglia 2021, 2, 7–35. [Google Scholar] [CrossRef]
- Pahakis, M.Y.; Kosky, J.R.; Dull, R.O.; Tarbell, J.M. The role of endothelial glycocalyx components in mechanotransduction of fluid shear stress. Biochem. Biophys. Res. Commun. 2007, 355, 228–233. [Google Scholar] [CrossRef] [Green Version]
- Tarbell, J.M.; Pahakis, M.Y. Mechanotransduction and the glycocalyx. J. Int. Med. 2006, 259, 339–350. [Google Scholar] [CrossRef] [PubMed]
- du Preez, H.N.; Aldous, C.; Hayden, M.R.; Kruger, H.G.; Lin, J. Pathogenesis of COVID-19 described through the lens of an undersulfated and degraded epithelial and endothelial glycocalyx. FASEB J. 2022, 36, e22052. [Google Scholar] [CrossRef]
- Fuso, A.; Scarpa, S. One-carbon metabolism and Alzheimer’s disease: Is it all a methylation matter? Neurobiol. Aging 2011, 32, 1192–1195. [Google Scholar] [CrossRef] [PubMed]
- Coppedè, F. One-Carbon Metabolism and Alzheimer’s Disease: Focus on Epigenetics. Curr. Genom. 2010, 11, 246–260. [Google Scholar] [CrossRef] [PubMed]
- Dayon, L.; Guiraud, S.P.; Corthésy, J.; Da Silva, L.; Migliavacca, E.; Tautvydaitė, D.; Oikonomidi, A.; Moullet, B.; Henry, H.; Métairon, S.; et al. One-carbon metabolism, cognitive impairment and CSF measures of Alzheimer pathology: Homocysteine and beyond. Alzheimers Res. Ther. 2017, 9, 43. [Google Scholar] [CrossRef] [PubMed]
- Stover, P.J. Polymorphisms in 1-Carbon Metabolism, Epigenetics and Folate-Related Pathologies. J. Nutrigenet. Nutr. 2011, 4, 293–305. [Google Scholar] [CrossRef] [Green Version]
- Coppedè, F.; Tannorella, P.; Pezzini, I.; Migheli, F.; Ricci, G.; Ienco, E.C.; Piaceri, I.; Polini, A.; Nacmias, B.; Monzani, F.; et al. Folate, homocysteine, vitamin B12, and polymorphisms of genes participating in one-carbon metabolism in late-onset Alzheimer’s disease patients and healthy controls. Antioxid Redox Signal. 2012, 17, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Grossi, E.; Stoccoro, A.; Tannorella, P.; Migliore, L.; Coppedè, F. Artificial Neural Networks Link One-Carbon Metabolism to Gene-Promoter Methylation in Alzheimer’s Disease. J. Alzheimers Dis. 2016, 53, 1517–1522. [Google Scholar] [CrossRef] [Green Version]
- Coppedè, F. One-carbon epigenetics and redox biology of neurodegeneration. One-Carbon Epigenetics Redox Biol. Neurodegener. Free Radic Biol Med. 2021, 170, 19–33. [Google Scholar] [CrossRef]
- Ravaglia, G.; Forti, P.; Maioli, F.; Chiappelli, M.; Montesi, F.; Bianchin, M.; Federico Licastro, F.; Patterson, C. Apolipoprotein E e4 allele affects risk of hyperhomocysteinemia in the elderly. Am. J. Clin. Nutr. 2006, 84, 1473–1480. [Google Scholar] [CrossRef]
- Kloske, C.M.; Wilcock, D.M. The Important Interface between Apolipoprotein E and Neuroinflammation in Alzheimer’s Disease. Front. Immunol. 2020, 11, 754. [Google Scholar] [CrossRef]
- Snyder, H.M.; Corriveau, R.A.; Craft, S.; Faber, J.E.; Greenberg, S.M.; Knopman, D.; Lamb, B.T.; Montine, T.J.; Nedergaard, M.; Schaffer, C.B.; et al. Vascular contributions to cognitive impairment and dementia including Alzheimer’s disease. Alzheimer’s Dement. 2015, 11, 710–717. [Google Scholar] [CrossRef] [Green Version]
- Sudduth, T.L.; Powell, D.K.; Smith, C.D.; Greenstein, A.; Wilcock, D.M. Induction of hyperhomocysteinemia models vascular dementia by induction of cerebral microhemorrhages and neuroinflammation. J. Cereb. Blood Flow Metab. 2013, 33, 708–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kammoun, S.; Gold, G.; Bouras, C.; Giannakopoulos, P.; McGee, W.; Herrmann, F.; Michel, J.P. Immediate causes of death of demented and non-demented elderly. Acta Neurol. Scand. Suppl. 2000, 176, 96–99. [Google Scholar] [CrossRef]
- Corriveau, R.A.; Bosetti, F.; Emr, M.; Gladman, J.T.; Koenig, J.I.; Moy, C.S.; Pahigiannis, K.; Waddy, S.P.; Koroshetz, W. The Science of Vascular Contributions to Cognitive Impairment and Dementia (VCID): A Framework for Advancing Research Priorities in the Cerebrovascular Biology of Cognitive Decline. Cell Mol. Neurobiol. 2016, 36, 281–288. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P.; Corriveau, R.A.; Wilcock, D.M. Vascular contributions to cognitive impairment and dementia (VCID). Biochim. Biophys. Acta 2016, 1862, 857–859. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Montagne, A.; Sagare, A.P.; Nation, D.A.; Schneider, L.S.; Chui, H.C.; Harrington, M.G.; Pa, J.; Law, M.; Wang, D.J.J.; et al. The disregarded partner of Alzheimer’s disease. Alzheimers Dement. 2019, 15, 158–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Cerebrospinal fluid biomarkers of neurovascular dysfunction in mild dementia and Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2015, 35, 1055–1068. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, R.H.; Khan, S.M. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med. Hypotheses 2004, 63, 8–20. [Google Scholar] [CrossRef]
- Cabrera Martimbianco, A.L.; Pacheco, R.L.; Bagattini, Â.M.; Riera, R. Frequency, signs and symptoms, and criteria adopted for long COVID-19: A systematic review. Int. J. Clin. Pract. 2021, 75, e14357. [Google Scholar] [CrossRef]
- Huang, C.; Huang, L.; Wang, Y.; Li, X.; Ren, L.; Gu, X.; Kang, L.; Guo, L.; Liu, M.; Zhou, X.; et al. 6-month consequences of COVID-19 in patients discharged from hospital: A cohort study. Lancet 2021, 397, 220–232. [Google Scholar] [CrossRef]
- Maltezou, H.; Pavli, A.; Tsakris, A. Post-COVID syndrome: An insight on its pathogenesis. Post-COVID Syndrome: An Insight on Its Pathogenesis. Vaccines 2021, 9, 497. [Google Scholar] [CrossRef]
- Greenhalgh, T.; Knight, M.; A’Court, M.; Buxton, M.; Husain, L. Management of post-acute COVID-19 in primary care. BMJ 2020, 370, m3026. [Google Scholar] [CrossRef] [PubMed]
- Rubin, R. As Their Numbers Grow, COVID-19 “Long Haulers” Stump Experts. JAMA 2020, 324, 1381–1383. [Google Scholar] [CrossRef] [PubMed]
- Komaroff, A.L.; Bateman, L. Will COVID-19 Lead to Myalgic Encephalomyelitis/Chronic Fatigue Syndrome? Front. Med. 2021, 7, 606824. [Google Scholar] [CrossRef]
- Becker, R.C. COVID-19 and its sequelae: A platform for optimal patient care, discovery and training. J. Thromb. Thrombolysis 2021, 51, 587–594. [Google Scholar] [CrossRef] [PubMed]
- McCaddon, A.; Regland, B. COVID-19: A methyl-group assault? Med. Hypotheses 2021, 149, 110543. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, R.; Kim, S.H.; Shah, H.; Zhang, S.; Liang, J.H.; Fang, Y.; Gentili, M.; Leary, C.N.O.; Elledge, S.J.; et al. SARS-CoV-2 hijacks folate and one-carbon metabolism for viral replication. Nat. Commun. 2021, 12, 1676. [Google Scholar] [CrossRef] [PubMed]
- Regland, B.; Forsmark, S.; Halaouate, L.; Matousek, M.; Peilot, B.; Zachrisson, O. Response to vitamin B12 and folic acid in myalgic encephalomyelitis and fibromyalgia. PLoS ONE 2015, 10, e0124648. [Google Scholar] [CrossRef]
- Froese, S.; Fowler, B.; Baumgartner, M.R. Vitamin B12, folate, and the methionine remethylation cycle-biochemistry, pathways, and regulation. J. Inherit. Metab. Dis. 2019, 42, 673–685. [Google Scholar] [CrossRef] [Green Version]
- Paul, B.D.; Lemle, M.D.; Komaroff, A.L.; Snyder, S.H. Redox imbalance links COVID-19 and myalgic encephalomyelitis/chronic fatigue syndrome. Proc. Natl. Acad. Sci. USA 2021, 118, e2024358118. [Google Scholar] [CrossRef]
- Ponti, G.; Maccaferri, M.; Ruini, C.; Tomasi, A.; Ozben, T. Biomarkers associated with COVID-19 disease progression. Crit. Rev. Clin. Lab. Sci. 2020, 57, 389–399. [Google Scholar] [CrossRef]
- Ponti, G.; Roli, L.; Oliva, G.; Manfredini, M.; Trenti, T.; Kaleci, S.; Iannella, R.; Balzano, B.; Coppola, A.; Fiorentino, G.; et al. Homocysteine (Hcy) assessment to predict outcomes of hospitalized COVID-19 patients: A multicenter study on 313 COVID-19 patients. Clin. Chem. Lab. Med. 2021, 59, e354–e357. [Google Scholar] [CrossRef]
- Ponti, G.; Pastorino, L.; Manfredini, M.; Ozben, T.; Oliva, G.; Kaleci, S.; Iannella, R.; Tomasi, A. COVID-19 spreading across world correlates with C677T allele of the methylenetetrahydrofolate reductase (MTHFR) gene prevalence. J. Clin. Lab. Anal. 2021, 35, e23798. [Google Scholar] [CrossRef] [PubMed]
- Collins, F.S. NIH Launches New Initiative to Study “Long COVID”. National Institute of Health; 23 February 2021. Available online: https://www.nih.gov/about-nih/who-we-are/nih-director/statements/nih-launches-new-initiative-study-long-covid (accessed on 10 November 2021).
- Patterson, B.K.; Guevara-Coto, J.; Yogendra, R.; Francisco, E.B.; Long, E.; Pise, A.; Rodrigues, H.; Parikh, P.; Mora, J.; Mora-Rodríguez, R.A. Immune-Based Prediction of COVID-19 Severity and Chronicity Decoded Using Machine Learning. Front. Immunol. 2021, 12, 700782. [Google Scholar] [CrossRef] [PubMed]
- Patterson, B.K.; Francisco, E.B.; Yogendra, R.; Long, E.; Pise, A.; Rodrigues, H.; Hall, E.; Herrara, M.; Parikh, P.; Guevara-Coto, J.; et al. Persistence of SARS CoV-2 S1 Protein in CD16+ Monocytes in Post-Acute Sequelae of COVID-19 (PASC) Up to 15 Months Post-Infection. bioRxiv 2021. [Google Scholar] [CrossRef]
- Au-Yeung, K.K.; Woo, C.W.; Sung, F.L.; Yip, J.C.; Siow, Y.L.; Karmin, O. Hyperhomocysteinemia activates nuclear factor-kappaB in endothelial cells via oxidative stress. Circ. Res. 2004, 94, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Umehara, H.; Bloom, E.T.; Okazaki, T.; Nagano, Y.; Yoshie, O.; Imai, T. Fractalkine in vascular biology: From basic research to clinical disease. Arter. Thromb. Vasc. Biol. 2004, 24, 34–40. [Google Scholar] [CrossRef]
- Corley, M.J.; Pang, A.P.; Dody, K.; Mudd, P.A.; Patterson, B.K.; Seethamraju, H.; Bram, Y.; Peluso, M.J.; Torres, L.; Iyer, N.S.; et al. Genome-wide DNA methylation profiling of peripheral blood reveals an epigenetic signature associated with severe COVID-19. J. Leukoc. Biol. 2021, 110, 21–26. [Google Scholar] [CrossRef]
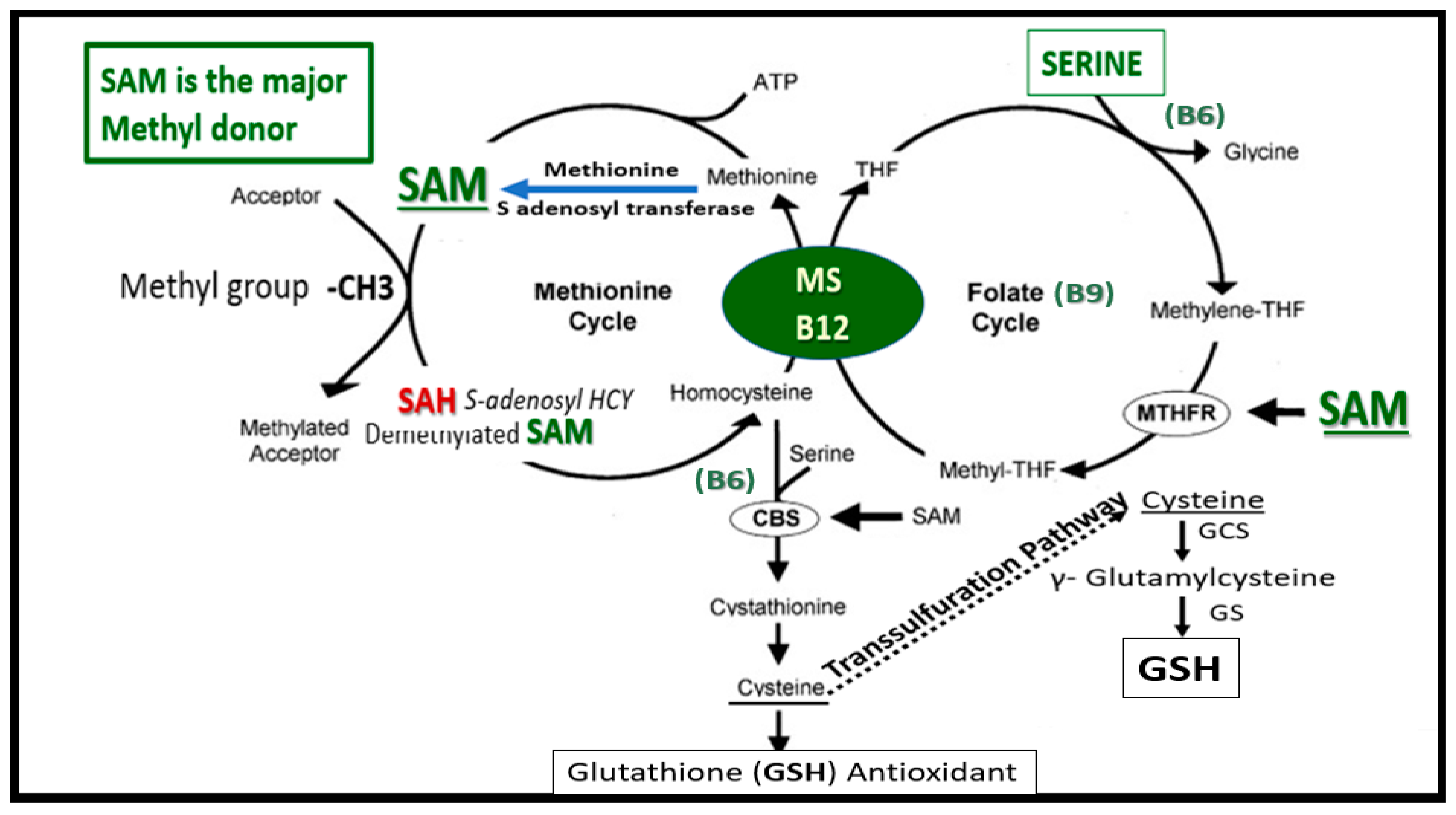
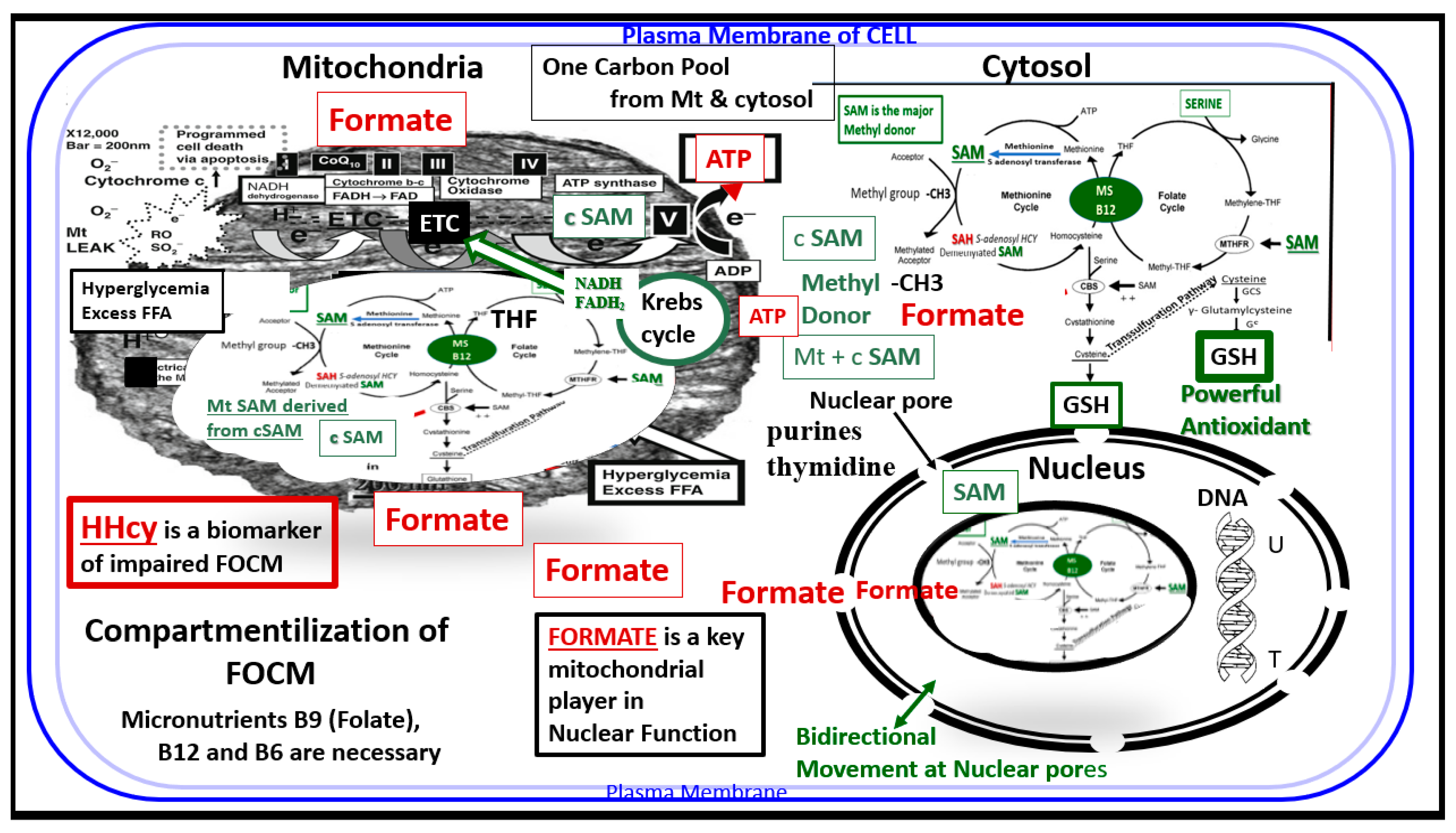
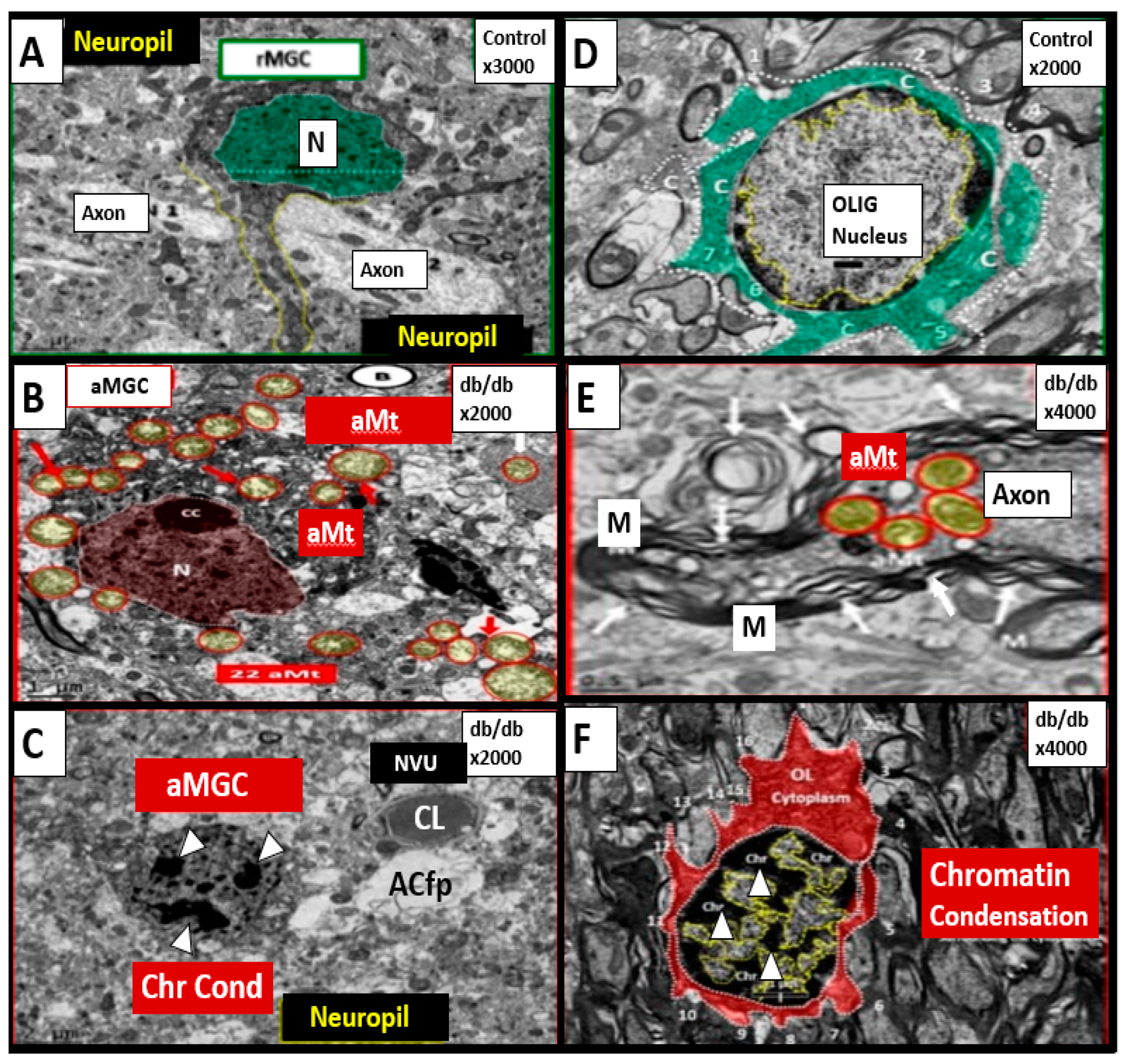

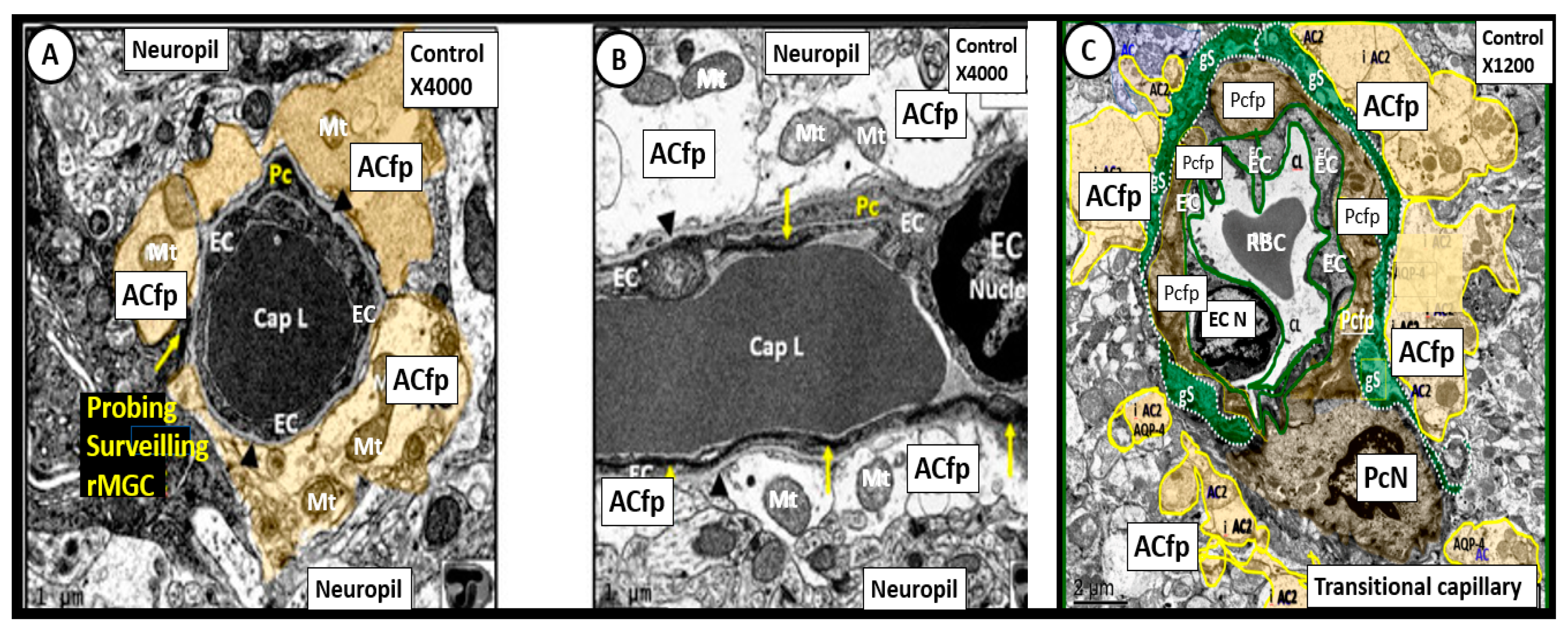





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayden, M.R.; Tyagi, S.C. Impaired Folate-Mediated One-Carbon Metabolism in Type 2 Diabetes, Late-Onset Alzheimer’s Disease and Long COVID. Medicina 2022, 58, 16. https://doi.org/10.3390/medicina58010016
Hayden MR, Tyagi SC. Impaired Folate-Mediated One-Carbon Metabolism in Type 2 Diabetes, Late-Onset Alzheimer’s Disease and Long COVID. Medicina. 2022; 58(1):16. https://doi.org/10.3390/medicina58010016
Chicago/Turabian StyleHayden, Melvin R., and Suresh C. Tyagi. 2022. "Impaired Folate-Mediated One-Carbon Metabolism in Type 2 Diabetes, Late-Onset Alzheimer’s Disease and Long COVID" Medicina 58, no. 1: 16. https://doi.org/10.3390/medicina58010016
APA StyleHayden, M. R., & Tyagi, S. C. (2022). Impaired Folate-Mediated One-Carbon Metabolism in Type 2 Diabetes, Late-Onset Alzheimer’s Disease and Long COVID. Medicina, 58(1), 16. https://doi.org/10.3390/medicina58010016