
Increased Diagnostic Yield of Array Comparative Genomic Hybridization for Autism Spectrum Disorder in One Institution in Taiwan

 ,
,  ,
,  and
and
Abstract
1. Introduction
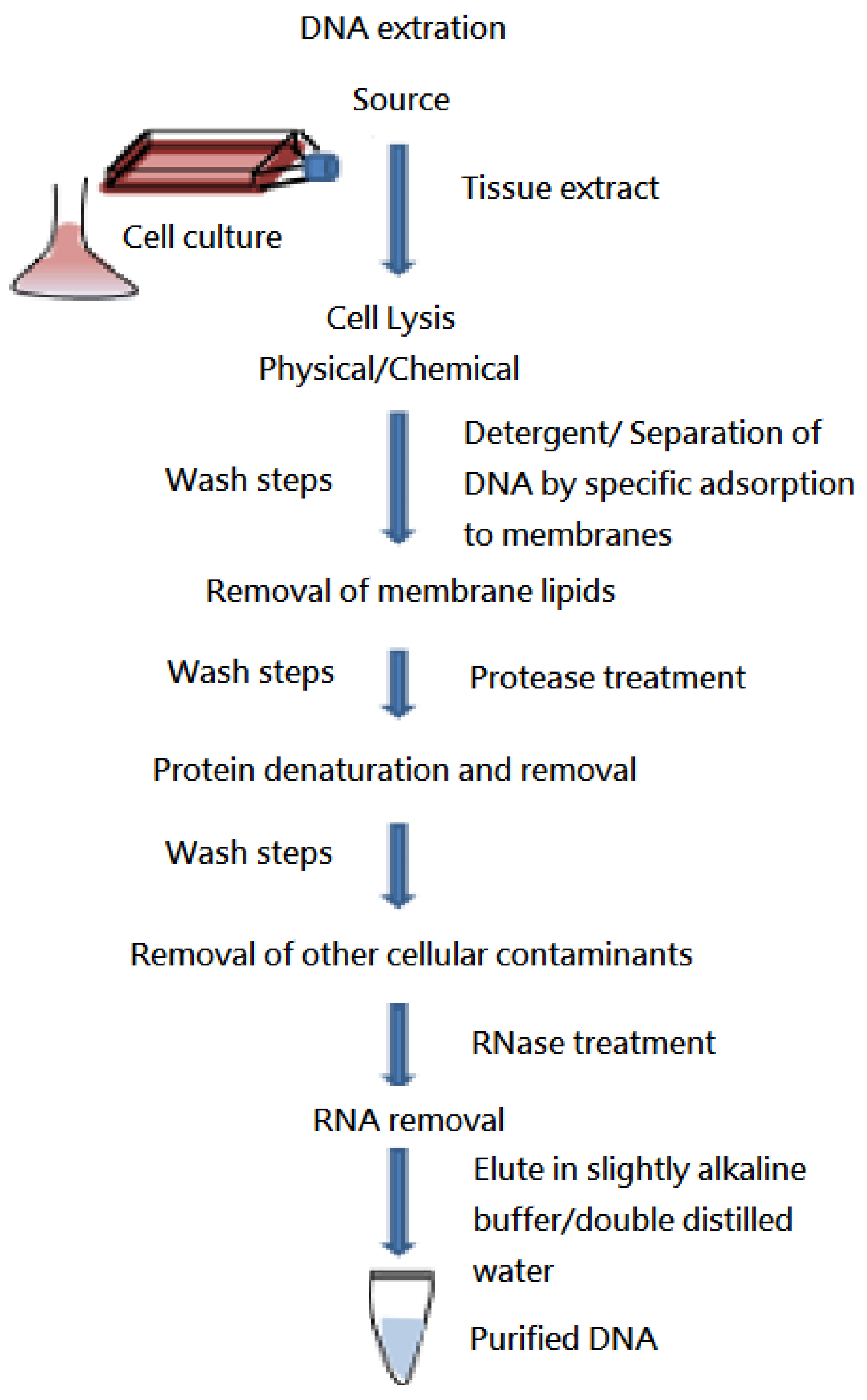
2. Materials and methods
2.1. Patients
2.2. Array-CGH and Data Interpretation
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Elsabbagh, M.; Divan, G.; Koh, Y.J.; Kim, Y.S.; Kauchali, S.; Marcín, C.; Montiel-Nava, C.; Patel, V.; Paula, C.S.; Wang, C.; et al. Global prevalence of autism and other pervasive developmental disorders. Autism Res. 2012, 5, 160–179. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Gilman, S.R.; Chiang, A.H.; Sanders, S.J.; Vitkup, D. Genotype to phenotype relationships in autism spectrum disorders. Nat. Neurosci. 2015, 18, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.L.; Lee, C.H.; Chuang, C.K.; Chiu, H.C.; Chen, Y.J.; Chou, C.L.; Wu, P.S.; Chen, C.P.; Lin, H.Y.; Lin, S.P. Array-CGH Increased the Diagnostic Rate of Developmental Delay or Intellectual Disability in Taiwan. Pediatrics Neonatol. 2019, 60, 453–460. [Google Scholar] [CrossRef]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef]
- Shevell, M.; Ashwal, S.; Donley, D.; Flint, J.; Gingold, M.; Hirtz, D.; Majnemer, A.; Noetzel, M.; Sheth, R.D. Practice parameter: Evaluation of the child with global developmental delay: Report of the Quality Standards Subcommittee of the American Academy of Neurology and The Practice Committee of the Child Neurology Society. Neurology 2003, 60, 367–380. [Google Scholar] [CrossRef]
- Ravnan, J.B.; Tepperberg, J.H.; Papenhausen, P.; Lamb, A.N.; Hedrick, J.; Eash, D.; Ledbetter, D.H.; Martin, C.L. Subtelomere FISH analysis of 11,688 cases: An evaluation of the frequency and pattern of subtelomere rearrangements in individuals with developmental disabilities. J. Med. Genet. 2006, 43, 478–489. [Google Scholar] [CrossRef]
- Rauch, A.; Hoyer, J.; Guth, S.; Zweier, C.; Kraus, C.; Becker, C.; Zenker, M.; Hüffmeier, U.; Thiel, C.; Rüschendorf, F.; et al. Diagnostic yield of various genetic approaches in patients with unexplained developmental delay or mental retardation. Am. J. Med. Genet. A 2006, 140, 2063–2074. [Google Scholar] [CrossRef]
- Moreno-De-Luca, D.; Sanders, S.J.; Willsey, A.J.; Mulle, J.G.; Lowe, J.K.; Geschwind, D.H.; State, M.W.; Martin, C.L.; Ledbetter, D.H. Using large clinical data sets to infer pathogenicity for rare copy number variants in autism cohorts. Mol. Psychiatry 2013, 18, 1090–1095. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, G.B.; Mendelsohn, N.J. Professional Practice and Guidelines Committee. Clinical genetics evaluation in identifying the etiology of autism spectrum disorders: 2013 guideline revisions. Genet. Med. 2013, 15, 399–407, Correction in Genet. Med. 2013, 15, 669. [Google Scholar] [CrossRef]
- Armengol, L.; Nevado, J.; Serra-Juhé, C.; Plaja, A.; Mediano, C.; García-Santiago, F.A.; García-Aragonés, M.; Villa, O.; Mansilla, E.; Preciado, C.; et al. Clinical utility of chromosomal microarray analysis in invasive prenatal diagnosis. Hum. Genet. 2012, 131, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Riggs, E.R.; Wain, K.E.; Riethmaier, D.; Smith-Packard, B.; Faucett, W.A.; Hoppman, N.; Thorland, E.C.; Patel, V.C.; Miller, D.T. Chromosomal microarray impacts clinical management. Clin. Genet. 2014, 85, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Wiśniowiecka-Kowalnik, B.; Kastory-Bronowska, M.; Bartnik, M.; Derwińska, K.; Dymczak-Domini, W.; Szumbarska, D.; Ziemka, E.; Szczałuba, K.; Sykulski, M.; Gambin, T.; et al. Application of custom-designed oligonucleotide array CGH in 145 patients with autistic spectrum disorders. Eur. J. Hum. Genet. 2013, 21, 620–625. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Syrmou, A.; Tzetis, M.; Fryssira, H.; Kosma, K.; Oikonomakis, V.; Giannikou, K.; Makrythanasis, P.; Kitsiou-Tzeli, S.; Kanavakis, E. Array comparative genomic hybridization as a clinical diagnostic tool in syndromic and nonsyndromic congenital heart disease. Pediatric Res. 2013, 73, 772–776. [Google Scholar] [CrossRef]
- Bartnik, M.; Szczepanik, E.; Derwińska, K.; Wiśniowiecka-Kowalnik, B.; Gambin, T.; Sykulski, M.; Ziemkiewicz, K.; Kędzior, M.; Gos, M.; Hoffman-Zacharska, D.; et al. Application of array comparative genomic hybridization in 102 patients with epilepsy and additional neurodevelopmental disorders. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2012, 159, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Sanna-Cherchi, S.; Kiryluk, K.; Burgess, K.E.; Bodria, M.; Sampson, M.G.; Hadley, D.; Nees, S.N.; Verbitsky, M.; Perry, B.J.; Sterken, R.; et al. Copy-number disorders are a common cause of congenital kidney malformations. Am. J. Hum. Genet. 2012, 91, 987–997. [Google Scholar] [CrossRef]
- Girirajan, S.; Rosenfeld, J.A.; Cooper, G.M.; Antonacci, F.; Siswara, P.; Itsara, A.; Vives, L.; Walsh, T.; McCarthy, S.E.; Baker, C.; et al. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat. Genet. 2010, 42, 203–209. [Google Scholar] [CrossRef]
- Girirajan, S.; Rosenfeld, J.A.; Coe, B.P.; Parikh, S.; Friedman, N.; Goldstein, A.; Filipink, R.A.; McConnell, J.S.; Angle, B.; Meschino, W.S.; et al. Phenotypic heterogeneity of genomic disorders and rare copy-number variants. N. Engl. J. Med. 2012, 367, 1321–1331. [Google Scholar] [CrossRef]
- Rutter, M.; Le Couteur, A.; Lord, C. Autism Diagnostic Interview-Revised (ADI–R) Manual; Western Psychological Services: Los Angeles, CA, USA, 2003. [Google Scholar]
- Wechsler, D. Wechsler Preschool and Primary Scale of Intelligence—Fourth Edition (WPPSI-IV): Technical and Interpretive Manual; NCS Pearson, Inc.: Bloomington, IN, USA, 2012. [Google Scholar]
- Wechsler, D. Wechsler Intelligence Scale for Children—Fifth Edition (WISC-V): Technical and Interpretive Manual; NCS Pearson Inc.: Bloomington, MN, USA, 2014. [Google Scholar]
- Sun, Y.; Yang, Y.; Luo, Y.; Chen, M.; Wang, L.; Huang, Y.; Yang, Y.; Dong, M. Lack of MECP2 gene transcription on the duplicated alleles of two related asymptomatic females with Xq28 duplications and opposite X-chromosome inactivation skewing. Hum. Mutat. 2021, 42, 1429–1442. [Google Scholar] [CrossRef]
- Zarrei, M.; MacDonald, J.R.; Merico, D.; Scherer, S.W. A copy number variation map of the human genome. Nat. Rev. Genet. 2015, 16, 172–183. [Google Scholar] [CrossRef]
- Shen, Y.; Dies, K.A.; Holm, I.A.; Bridgemohan, C.; Sobeih, M.M.; Caronna, E.B.; Miller, K.J.; Frazier, J.A.; Silverstein, I.; Picker, J.; et al. Clinical genetic testing for patients with autism spectrum disorders. Pediatrics 2010, 125, e727–e735. [Google Scholar] [CrossRef]
- Sebat, J.; Lakshmi, B.; Malhotra, D.; Troge, J.; Lese-Martin, C.; Walsh, T.; Yamrom, B.; Yoon, S.; Krasnitz, A.; Kendall, J.; et al. Strong association of de novo copy number mutations with autism. Science 2007, 316, 445–449. [Google Scholar] [CrossRef]
- Burnside, R.D.; Pasion, R.; Mikhail, F.M.; Carroll, A.J.; Robin, N.H.; Youngs, E.L.; Gadi, I.K.; Keitges, E.; Jaswaney, V.L.; Papenhausen, P.R.; et al. Microdeletion/microduplication of proximal 15q11.2 between BP1 and BP2: A susceptibility region for neurological dysfunction including developmental and language delay. Hum. Genet. 2011, 130, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Doornbos, M.; Sikkema-Raddatz, B.; Ruijvenkamp, C.A.; Dijkhuizen, T.; Bijlsma, E.K.; Gijsbers, A.C.; Hilhorst-Hofstee, Y.; Hordijk, R.; Verbruggen, K.T.; Kerstjens-Frederikse, W.S.; et al. Nine patients with a microdeletion 15q11.2 between breakpoints 1 and 2 of the Prader-Willi critical region, possibly associated with behavioural disturbances. Eur. J. Med. Genet. 2009, 52, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Siu, W.K.; Lam, C.W.; Mak, C.M.; Lau, E.T.; Tang, M.H.; Tang, W.F.; Poon-Mak, R.S.; Lee, C.C.; Hung, S.F.; Leung, P.W.; et al. Diagnostic yield of array CGH in patients with autism spectrum disorder in Hong Kong. Clin. Transl. Med. 2016, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Heard, P.L.; Carter, E.M.; Crandall, A.C.; Sebold, C.; Hale, D.E.; Cody, J.D. High resolution genomic analysis of 18q- using oligo-microarray comparative genomic hybridization (aCGH). Am. J. Med. Genet A 2009, 149, 1431–1437. [Google Scholar] [CrossRef]
- Cody, J.D.; Hasi, M.; Soileau, B.; Heard, P.; Carter, E.; Sebold, C.; O’Donnell, L.; Perry, B.; Stratton, R.F.; Hale, D.E. Establishing a reference group for distal 18q-: Clinical description and molecular basis. Hum. Genet. 2014, 133, 199–209. [Google Scholar] [CrossRef][Green Version]
- Versacci, P.; Digilio, M.C.; Sauer, U.; Dallapiccola, B.; Marino, B. Absent pulmonary valve with intact ventricular septum and patent ductus arteriosus: A specific cardiac phenotype associated with deletion 18q syndrome. Am. J. Med. Genet. A 2005, 138, 185–186. [Google Scholar] [CrossRef]
- Van Trier, D.C.; Feenstra, I.; Bot, P.; de Leeuw, N.; Draaisma, J.M. Cardiac anomalies in individuals with the 18q deletion syndrome; report of a child with Ebstein anomaly and review of the literature. Eur. J. Med. Genet. 2013, 56, 426–431. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, L.; Soileau, B.; Heard, P.; Carter, E.; Sebold, C.; Gelfond, J.; Hale, D.E.; Cody, J.D. Genetic determinants of autism in individuals with deletions of 18q. Hum. Genet. 2010, 128, 155–164. [Google Scholar] [CrossRef]
- Dauber, A.; Lafranchi, S.H.; Maliga, Z.; Lui, J.C.; Moon, J.E.; McDeed, C.; Henke, K.; Zonana, J.; Kingman, G.A.; Pers, T.H.; et al. Novel microcephalic primordial dwarfism disorder associated with variants in the centrosomal protein ninein. J. Clin. Endocrinol. Metab. 2012, 97, E2140–E2151. [Google Scholar] [CrossRef]
- Wang, X.; Tsai, J.W.; Imai, J.H.; Lian, W.N.; Vallee, R.B.; Shi, S.H. Asymmetric centrosome inheritance maintains neural progenitors in the neocortex. Nature 2009, 461, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Srivatsa, S.; Parthasarathy, S.; Molnár, Z.; Tarabykin, V. Sip1 downstream Effector ninein controls neocortical axonal growth, ipsilateral branching, and microtubule growth and stability. Neuron 2015, 85, 998–1012. [Google Scholar] [CrossRef] [PubMed]
- Blockus, H.; Chédotal, A. The multifaceted roles of Slits and Robos in cortical circuits: From proliferation to axon guidance and neurological diseases. Curr. Opin. Neurobiol. 2014, 27, 82–88. [Google Scholar] [CrossRef]
- Chu, J.; Anderson, S.A. Development of cortical interneurons. Neuropsychopharmacology 2015, 40, 16–23. [Google Scholar] [CrossRef]
- Marín, O. Interneuron dysfunction in psychiatric disorders. Nat. Rev. Neurosci. 2012, 13, 107–120. [Google Scholar] [CrossRef]
- Robichaux, M.A.; Cowan, C.W. Signaling mechanisms of axon guidance and early synaptogenesis. Curr. Top. Behav. Neurosci. 2014, 16, 19–48. [Google Scholar]
- Pinto, D.; Pagnamenta, A.T.; Klei, L.; Anney, R.; Merico, D.; Regan, R.; Conroy, J.; Magalhaes, T.R.; Correia, C.; Abrahams, B.S.; et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010, 466, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Tammimies, K.; Marshall, C.R.; Walker, S.; Kaur, G.; Thiruvahindrapuram, B.; Lionel, A.C.; Yuen, R.K.; Uddin, M.; Roberts, W.; Weksberg, R.; et al. Molecular Diagnostic Yield of Chromosomal Microarray Analysis and Whole-Exome Sequencing in Children with Autism Spectrum Disorder. JAMA 2015, 314, 895–903. [Google Scholar] [CrossRef]
- Pinto, D.; Delaby, E.; Merico, D.; Barbosa, M.; Merikangas, A.; Klei, L.; Thiruvahindrapuram, B.; Xu, X.; Ziman, R.; Wang, Z.; et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 2014, 94, 677–694. [Google Scholar] [CrossRef]
- Girirajan, S.; Dennis, M.Y.; Baker, C.; Malig, M.; Coe, B.P.; Campbell, C.D.; Mark, K.; Vu, T.H.; Alkan, C.; Cheng, Z.; et al. Refinement and discovery of new hotspots of copy-number variation associated with autism spectrum disorder. Am. J. Hum. Genet. 2013, 92, 221–237. [Google Scholar] [CrossRef]
- Marshall, C.R.; Noor, A.; Vincent, J.B.; Lionel, A.C.; Feuk, L.; Skaug, J.; Shago, M.; Moessner, R.; Pinto, D.; Ren, Y.; et al. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 2008, 82, 477–488. [Google Scholar] [CrossRef]
- Newschaer, C.J.; Croen, L.A.; Daniels, J.; Giarelli, E.; Grether, J.K.; Levy, S.E.; Mandell, D.S.; Miller, L.A.; Pinto-Martin, J.; Reaven, J.; et al. The Epidemiology of Autism Spectrum Disorders. Annu. Rev. Public Health 2007, 28, 235–258. [Google Scholar] [CrossRef]
- Elsabbagh, M.; Mercure, E.; Hudry, K.; Chandler, S.; Pasco, G.; Charman, T.; Pickles, A.; Baron-Cohen, S.; Bolton, P.; Johnson, M.H. Infant Neural Sensitivity to Dynamic Eye Gaze Is Associated with Later Emerging Autism. Curr. Biol. 2012, 22, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Benvenuto, A.; Moavero, R.; Alessandrelli, R.; Manzi, B.; Curatolo, P. Syndromic autism: Causes and pathogenetic pathways. World J. Pediatr. 2009, 5, 169–176. [Google Scholar] [CrossRef]
- Masini, E.; Loi, E.; Vega-Benedetti, A.F.; Carta, M.; Doneddu, G.; Fadda, R.; Zavattari, P. An Overview of the Main Genetic, Epigenetic and Environmental Factors Involved in Autism Spectrum Disorder Focusing on Synaptic Activity. Int. J. Mol. Sci. 2020, 21, 8290. [Google Scholar] [CrossRef]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.-Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584. [Google Scholar] [CrossRef] [PubMed]
- Herrero, M.J.; Velmeshev, D.; Hernandez-Pineda, D.; Sethi, S.; Sorrells, S.; Banerjee, P.; Sullivan, C.; Gupta, A.R.; Kriegstein, A.R.; Corbin, J.G. Identification of amygdala-expressed genes associated with autism spectrum disorder. Mol. Autism 2020, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Cappuccio, G.; Vitiello, F.; Casertano, A.; Fontana, P.; Genesio, R.; Bruzzese, D.; Ginocchio, V.M.; Mormile, A.; Nitsch, L.; Andria, G.; et al. New insights in the interpretation of array-CGH: Autism spectrum disorder and positive family history for intellectual disability predict the detection of pathogenic variants. Ital. J. Pediatrics 2016, 42, 39. [Google Scholar] [CrossRef]
- Palka Bayard de Volo, C.; Alfonsi, M.; Morizio, E.; Guaciali-Franchi, P.; Mohn, A.; Chiarelli, F. A 343 Italian cohort of patients analysed with array-comparative genomic hybridization: Unsolved problems and genetic counselling difficulties. J. Intellect. Disabil. Res. 2021, 65, 863–869. [Google Scholar] [CrossRef]
- Monteiro, S.; Pinto, J.; Mira Coelho, A.; Leão, M.; Dória, S. Identification of Copy Number Variation by Array-CGH in Portuguese Children and Adolescents Diagnosed with Autism Spectrum Disorders. Neuropediatrics 2019, 50, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Napoli, E.; Russo, S.; Casula, L.; Alesi, V.; Amendola, F.A.; Angioni, A.; Novelli, A.; Valeri, G.; Menghini, D.; Vicari, S. Array-CGH Analysis in a Cohort of Phenotypically Well-Characterized Individuals with “Essential” Autism Spectrum Disorders. J. Autism Dev. Disord. 2018, 48, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Gümüşlü, K.E.; Savli, H.; Sünnetçi, D.; Çine, N.; Kara, B.; Eren Keskin, S.; Akkoyunlu, R.U. A CGH array study in nonsyndromic (primary) autism patients: Deletions on 16p13.11, 16p11.2, 1q21.1, 2q21.1q21.2, and 8p23.1. Turk. J. Med. Sci. 2015, 45, 313–319. [Google Scholar] [CrossRef]
- Kousoulidou, L.; Moutafi, M.; Nicolaides, P.; Hadjiloizou, S.; Christofi, C.; Paradesiotou, A.; Anastasiadou, V.; Sismani, C.; Patsalis, P.C. Screening of 50 cypriot patients with autism spectrum disorders or autistic features using 400K custom array-CGH. Biomed Res. Int. 2013, 2013, 843027. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Hou, Y.; Bickhart, D.; Song, J.; Liu, G. Comparative analysis of CNV calling algorithms: Literature survey and a case study using bovine high-density SNP data. Microarrays 2013, 2, 171–185. [Google Scholar] [CrossRef]
- Hollenbeck, D.; Williams, C.L.; Drazba, K.; Descartes, M.; Korf, B.R.; Rutledge, S.L.; Lose, E.J.; Robin, N.H.; Carroll, A.J.; Mikhail, F.M. Clinical relevance of small copy-number variants in chromosomal microarray clinical testing. Genet. Med. 2017, 19, 377–385. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Number of Patients | 80 |
| Male | 48 |
| Female | 32 |
| Age range (median) (years) | 2–16 (6) |
| Total number of CNV | 47 |
| (Detection rate %) | (58.8) |
| Detected in male | 30 |
| (Detection rate %) | (62.5) |
| Detected in female | 17 |
| (Detection rate %) | (53.1) |
| Clinically significant CNV | 31 |
| (Diagnostic yield %) | (38.8) |
| Detected in male | 18 |
| (Diagnostic yield %) | (37.5) |
| Detected in female | 13 |
| (Diagnostic yield %) | (40.6) |
| Patient Number | Gender | Array CGH Result (hg18) | Chromosome Region (Genes Associated with ASD Phenotype) | Aberration Type | Size (Mb) | Clinical Significance | IQ | Additional Clinical Features |
|---|---|---|---|---|---|---|---|---|
| 1 | Male | arr15q11.2(22,842,145 − 25,235,046) × 3 | 15q11.2 (UBE3A, SNRPN, CHRNA7) | Duplication | 2.393 | Susceptibility to ASD | N/A | Developmental delay |
| arr15q11.2q13.1(25,236,676 − 28,559,402) × 4 | 15q11.2q13.1 (UBE3A, SNRPN, CHRNA7) | Duplication | 3.323 | |||||
| 3 | Male | arr17p11.2(16,782,546 − 20,219,464) × 1 | 17p11.2 (RAI1) | Deletion | 3.437 | Smith-Magenis syndrome | N/A | Developmental delay and facial dysmorphism |
| 4 | Male | arr7q11.23(72,776,313 − 74,133,332) × 1 | 7q11.23 (AUTS2) | Deletion | 1.367 | Williams syndrome | N/A | Developmental delay |
| 5 | Female | arr15q11.2q13.2(22,765,628 − 30,653,876) × 4 | 15q11.2q13.2 (UBE3A, SNRPN, CHRNA7) | Duplication | 7.888 | Susceptibility to ASD | N/A | Developmental delay |
| arr15q13.2q13.3(30,653,877 − 32,509,926) × 3 | 15q13.2q13.3 (CHRNA7) | Duplication | 1.856 | |||||
| 7 | Female | arr22q11.21(18,706,001 − 21,505,417) × 3 | 22q11.21 (CRKL, FGF8, TBX1) | Duplication | 2.799 | Susceptibility to ASD | N/A | Developmental delay and facial dysmorphism |
| 8 | Male | arr4p15.1p12(28,451,191 − 47,062,229) × 4 | 4p15.1p12 (UGDH) | Duplication | 18.611 | Susceptibility to ASD | N/A | Developmental delay |
| 9 | Female | arr22q11.23q12.1(25,695,469 − 25,903,543) × 0 | 22q11.23q12.1 (CRKL, FGF8, TBX1) | Deletion | 0.208 | Susceptibility to ASD | N/A | Developmental delay |
| 11 | Male | arr4p16.3(72,447 − 3,848,881) × 1 | 4p16.3 (WHS) | Deletion | 3.776 | Wolf-Hirschhorn syndrome | 33 | Developmental delay and facial dysmorphism |
| 12 | Female | arr15q11.2q13.3(22,770,421 − 32,915,593) × 1 | 15q11.2q13.3 (UBE3A, SNRPN, CHRNA7) | Deletion | 10.145 | Angelman syndrome | N/A | Developmental delay and facial dysmorphism |
| 13 | Female | arr4p16.3(68,345 − 4,044,985) × 1.0 | 4p16.3 (WHS) | Deletion | 3.977 | Wolf-Hirschhorn syndrome | 55 | Developmental delay and facial dysmorphism |
| 14 | Female | arr22q13.33(50,967,018 − 51,197,725) × 1 | 22q13.3 (SHANK3) | Deletion | 0.231 | Susceptibility to ASD | N/A | Developmental delay and facial dysmorphism |
| arr4p16.3p14 (68,345 − 40,111,547) × 3 | 4p16.3p14 (WHS) | Duplication | 40.000 | |||||
| 15 | Female | arr18p11.32p11.21(136,227 − 15,181,207) × 4 | 18p11.32 18p11.21 (SMCHD1) | Duplication | 15.045 | Susceptibility to ASD | 57 | Developmental delay and facial dysmorphism |
| 16 | Male | arr11q13.4q14.3(71,567,724 − 89,547,851) × 4 | 11q13.4q14.3 (SHANK2) | Duplication | 17.980 | Susceptibility to ASD | 34 | Developmental delay and facial dysmorphism |
| 18 | Female | arr2q22.1q22.3(141,332,947 − 145,948,739) × 1 | 2q22.1q22.3 (TBR1) | Deletion | 4.161 | Susceptibility to ASD | 55 | Developmental delay and facial dysmorphism |
| 21 | Male | arr1p31.3p31.1(61,947,700 − 73,030,143) × 1 | 1p31.3p31.1 (NEGR1) | Deletion | 11.080 | Susceptibility to ASD | N/A | Developmental delay and facial dysmorphism |
| 22 | Female | arr3q22.3q23(138,681,193 − 139,438,715) × 3 | 3q22.3q23 (ZBTB20) | Duplication | 0.758 | Susceptibility to ASD | N/A | Developmental delay |
| 23 | Male | arr10p15.3(162,270 − 468,133) × 3 | 10p15.3 (DIP2C) | Duplication | 0.306 | Susceptibility to ASD | 78 | Developmental delay |
| 25 | Male | arr14q21.2q22.1(45,863,061 − 50,360,747) × 0 | 14q21.2q22.1 (NIN) | Deletion | 4.500 | Deletion of the NIN gene | N/A | Developmental delay |
| 27 | Female | arr2q23.3q24.1(150,619,633 − 157,576,339) × 1.3 | 2q23.3q24.1 (MBD5) | Deletion | 6.957 | Susceptibility to ASD | N/A | Developmental delay |
| 28 | Male | arr18q21.33q23(60,414,497 − 78,003,508) × 1 | 18q21.33q23 (NETO1, FBXO15) | Deletion | 17.590 | Susceptibility to ASD | N/A | Developmental delay |
| 29 | Male | arr22q11.21(18,657,470 − 21,843,336) × 1 | 22q11.21 (CRKL, FGF8, TBX1) | Deletion | 3.190 | CATCH22 | N/A | Developmental delay |
| 30 | Male | arrXp22.31(6,450,627 − 8,141,242) × 0 | Xp22.31 (NLGN4) | Deletion | 1.690 | Susceptibility to ASD | 80 | Developmental delay |
| arrXp22.31(8,429,167 − 8,435,863) × 0.5 | Xp22.31 (NLGN4) | Deletion | 1.310 | |||||
| 31 | Female | arr15q11.2(20,760,484 − 23,601,857) × 1.1 | 15q11.2 (UBE3A, SNRPN, CHRNA7) | Deletion | 2.840 | Susceptibility to ASD | 41 | Developmental delay |
| 32 | Male | arr15q11.2(22,748,697 − 23,188,522) × 1 | 15q11.2 (UBE3A, SNRPN, CHRNA7) | Deletion | 0.440 | Susceptibility to ASD | 35 | Developmental delay |
| 33 | Male | arr15q11.2q13.1(23,614,732 − 28,536,497) × 1 | 15q11.2q13.1 (UBE3A, SNRPN, CHRNA7) | Deletion | 4.920 | Angelman syndrome | 17 | Developmental delay |
| 34 | Male | arr9q34.3 (140,687,823 − 140,695,906) × 1 | 9q34.3 (TSC1, EHMT1) | Deletion | 0.008 | Kleefstra syndrome | 59 | Developmental delay and facial dysmorphism |
| 36 | Male | arrXq28(152,956,854 − 155,270,560) × 2 | Xq28 (MECP2) | Duplication | 2.310 | Susceptibility to ASD | N/A | Developmental delay |
| Patient Number | Gender | Array CGH Result (hg18) | Chromosome Region (Genes Associated with ASD Phenotype) | Aberration Type | Size (Mb) | IQ | Additional Clinical Features |
|---|---|---|---|---|---|---|---|
| 2 | Male | arr22q11.22(22,336,268 − 22,556,733) × 1 | 22q11.22 (CRKL, FGF8, TBX1) | Deletion | 0.220 | 72 | Developmental delay |
| 6 | Male | arr17p13.3(1693 − 2,393,788) × 1 | 17p13.3 (MDLS) | Deletion | 2.392 | 85 | Developmental delay |
| 10 | Female | arr9p24.39p23(204,193 − 10,972,824) × 1 | 9p24.39p23 (KANK1) | Deletion | 10.768 | N/A | Developmental delay |
| 17 | Male | arr16q22.1q22.2(69,098,865 − 72,591,930) × 1 | 16q22.1q22.2 (SCA4) | Deletion | 3.493 | 69 | Developmental delay |
| 19 | Male | arr12p13.33p13.32(173,786 − 4,424,837) × 1 | 12p13.33p13.32 (EMG1) | Deletion | 4.250 | 69 | Developmental delay |
| 23 | Male | arr20p12.3(8,085,389 − 8,589,571) × 1 | 20p12.3 (PLCB1) | Deletion | 0.504 | 78 | Developmental delay |
| 24 | Male | arrXq13.1(69,228,881 − 69,240,595) × 0 | Xq13.1 (NLGN3) | Deletion | 0.012 | N/A | Developmental delay |
| 26 | Female | arrXp21.2(29,336,996 − 29,372,188) × 1 | Xp21.2 (CDKL5) | Deletion | 0.035 | N/A | Developmental delay |
| 36 | Male | arrXp22.33(1 − 2,196,782) × 0 | Xp22.33 (NLGN4) | Deletion | 2.200 | N/A | Developmental delay |
| 37 | Female | arr8q21.2q21.13(51,301,121 − 54,915,042) × 1 | 8q21.2q21.13 (TCF4) | Deletion | 3.610 | 19 | Developmental delay |
| 38 | Male | arrXq13.1q13.3(70,749,306 − 74,335,167) × 2 | Xq13.1q13.3 (NLGN3) | Duplication | 3.590 | 72 | Developmental delay |
| 39 | Male | arr17q25.3 (77,856,839 − 78,293,128) × 2.95 | 17q25.3 (NF1) | Duplication | 0.436 | N/A | Developmental delay |
| arrXp22.31q28(6,980,000 − 155,270,000) × 1.1 | Xp22.31q28 (NLGN4) | Duplication | 148.290 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.-L.; Chuang, C.-K.; Tu, R.-Y.; Chiu, H.-C.; Lo, Y.-T.; Chang, Y.-H.; Chen, Y.-J.; Chou, C.-L.; Wu, P.-S.; Chen, C.-P.; et al. Increased Diagnostic Yield of Array Comparative Genomic Hybridization for Autism Spectrum Disorder in One Institution in Taiwan. Medicina 2022, 58, 15. https://doi.org/10.3390/medicina58010015
Lee C-L, Chuang C-K, Tu R-Y, Chiu H-C, Lo Y-T, Chang Y-H, Chen Y-J, Chou C-L, Wu P-S, Chen C-P, et al. Increased Diagnostic Yield of Array Comparative Genomic Hybridization for Autism Spectrum Disorder in One Institution in Taiwan. Medicina. 2022; 58(1):15. https://doi.org/10.3390/medicina58010015
Chicago/Turabian StyleLee, Chung-Lin, Chih-Kuang Chuang, Ru-Yi Tu, Huei-Ching Chiu, Yun-Ting Lo, Ya-Hui Chang, Yen-Jiun Chen, Chao-Ling Chou, Peih-Shan Wu, Chih-Ping Chen, and et al. 2022. "Increased Diagnostic Yield of Array Comparative Genomic Hybridization for Autism Spectrum Disorder in One Institution in Taiwan" Medicina 58, no. 1: 15. https://doi.org/10.3390/medicina58010015
APA StyleLee, C.-L., Chuang, C.-K., Tu, R.-Y., Chiu, H.-C., Lo, Y.-T., Chang, Y.-H., Chen, Y.-J., Chou, C.-L., Wu, P.-S., Chen, C.-P., Lin, H.-Y., & Lin, S.-P. (2022). Increased Diagnostic Yield of Array Comparative Genomic Hybridization for Autism Spectrum Disorder in One Institution in Taiwan. Medicina, 58(1), 15. https://doi.org/10.3390/medicina58010015