
Effect of the HDAC Inhibitor on Histone Acetylation and Methyltransferases in A2780 Ovarian Cancer Cells


Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Line and Reagents
2.2. Cell Culture and Drug Treatments
2.3. Cell Viability Determination Using Trypan Blue Dye Exclusion (TBDE) Method
2.4. RNA Extraction
2.5. Human Epigenetic Chromatin Modification Enzymes RT2 Profiler PCR Array
2.6. Biological Pathway Analysis
2.7. Western Blot Analysis
2.8. Spheroid Formation
2.9. Immunofluorescence Analysis for 2D and 3D Culture
2.10. Statistical Analysis
3. Results
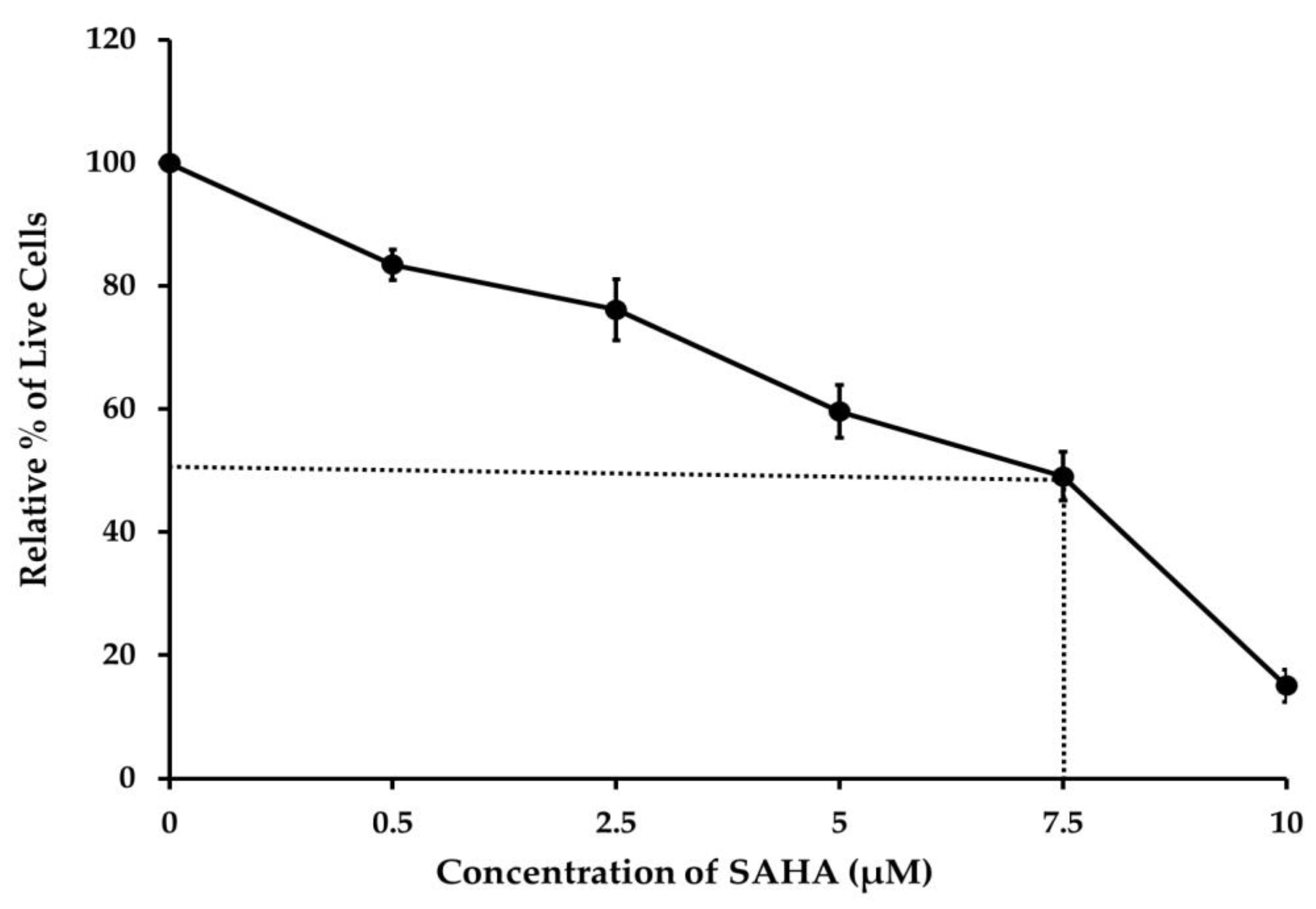
3.1. The Effect of SAHA on Reduction of A2780 Cell Viability
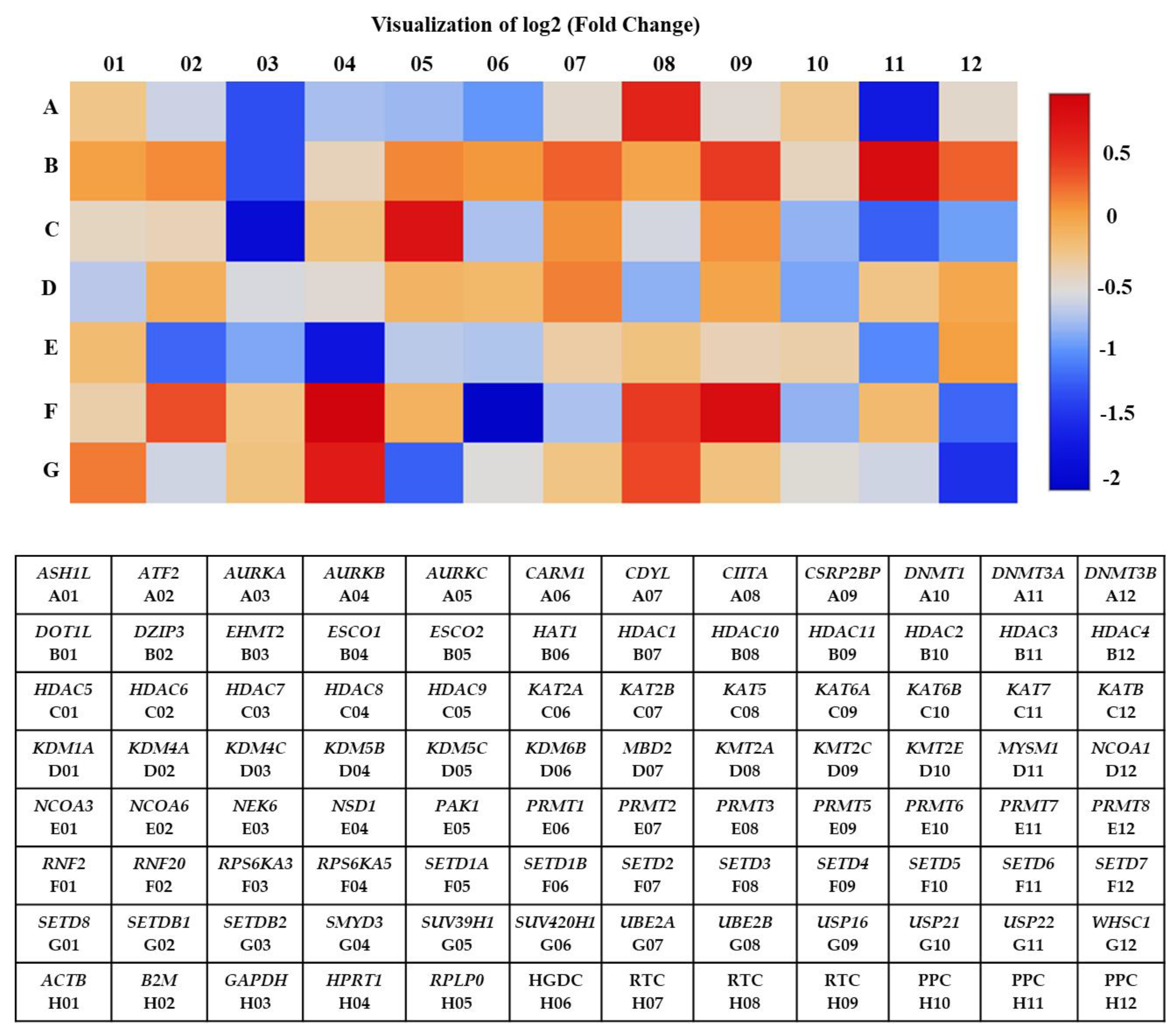
3.2. Identification of Differentially Expressed Genes in A2780 after SAHA Treatment Using Human Epigenetic Chromatin Modification Enzymes RT2 Profiler PCR Array
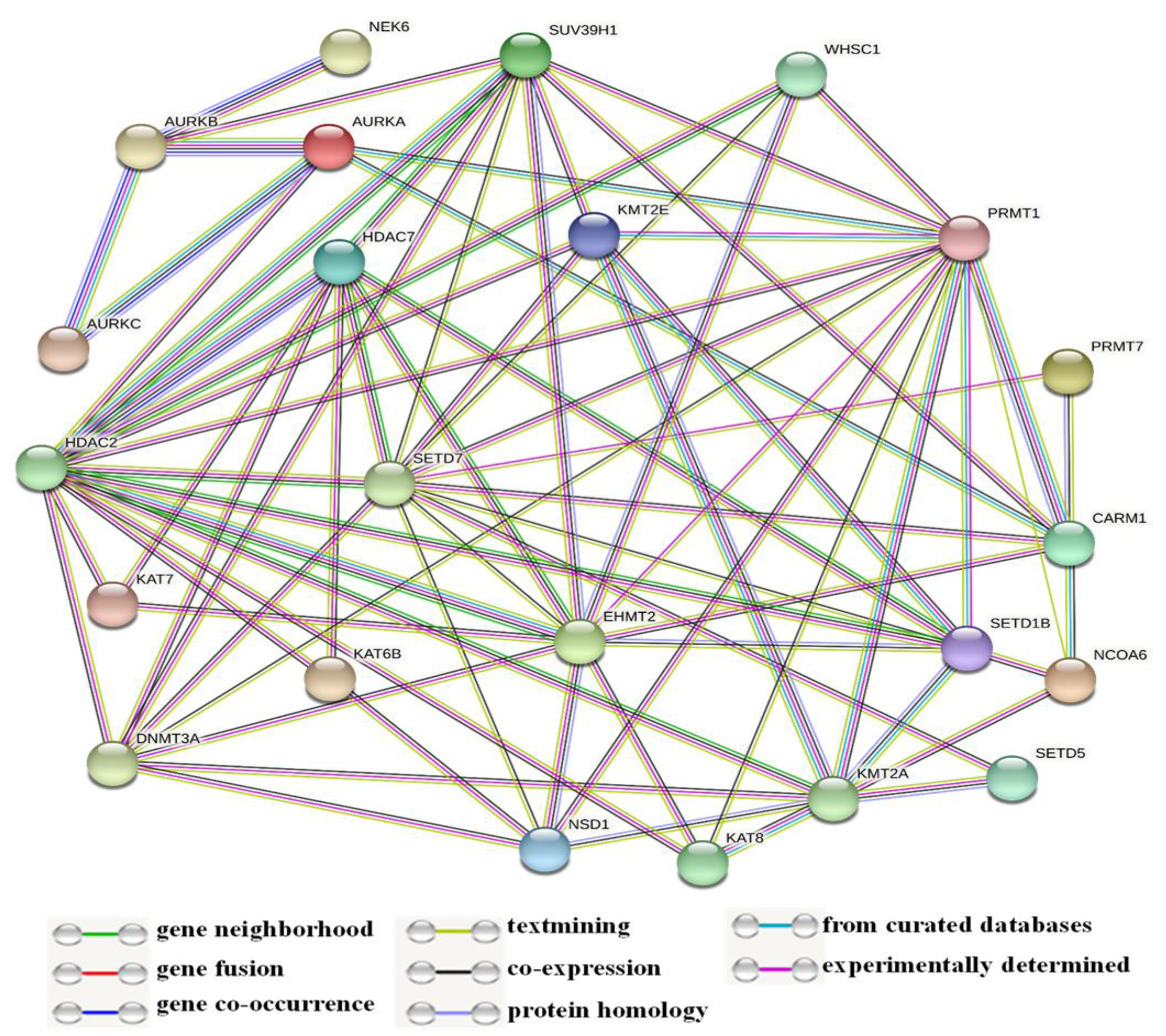
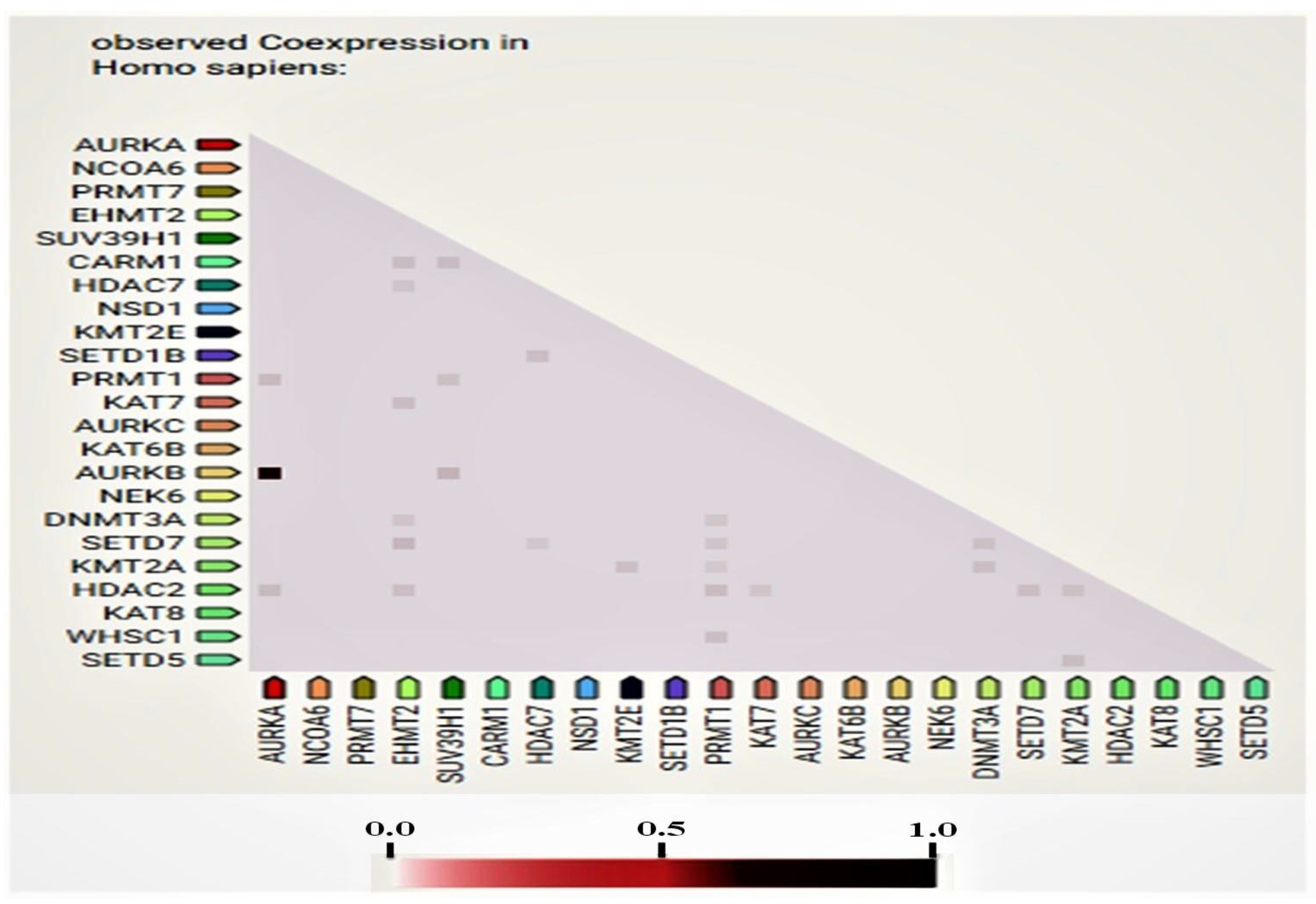
3.3. Network and Co-Expression Analysis
3.4. Up-Regulated Genes in the RT2 Profiler PCR Array Analysis
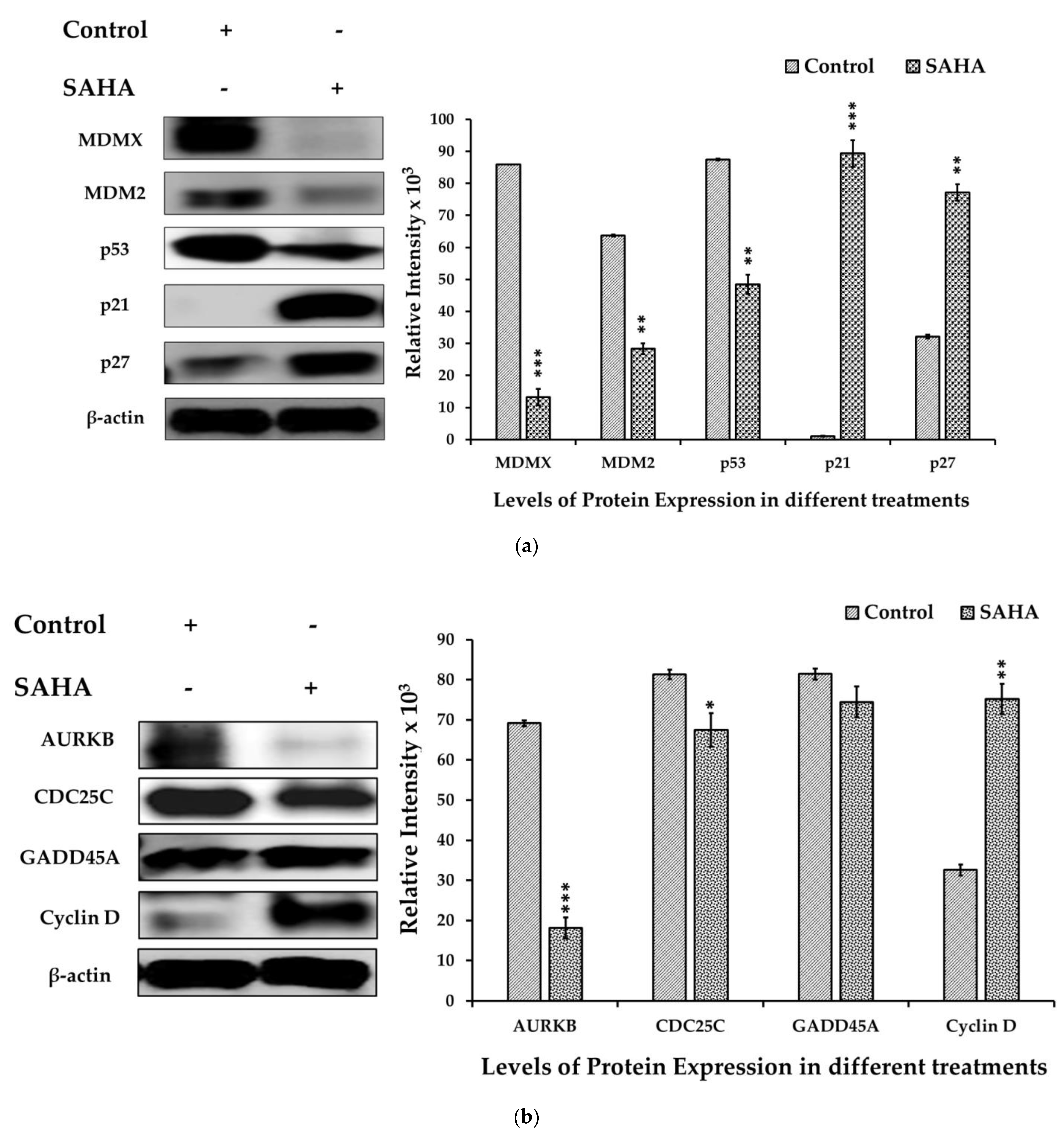
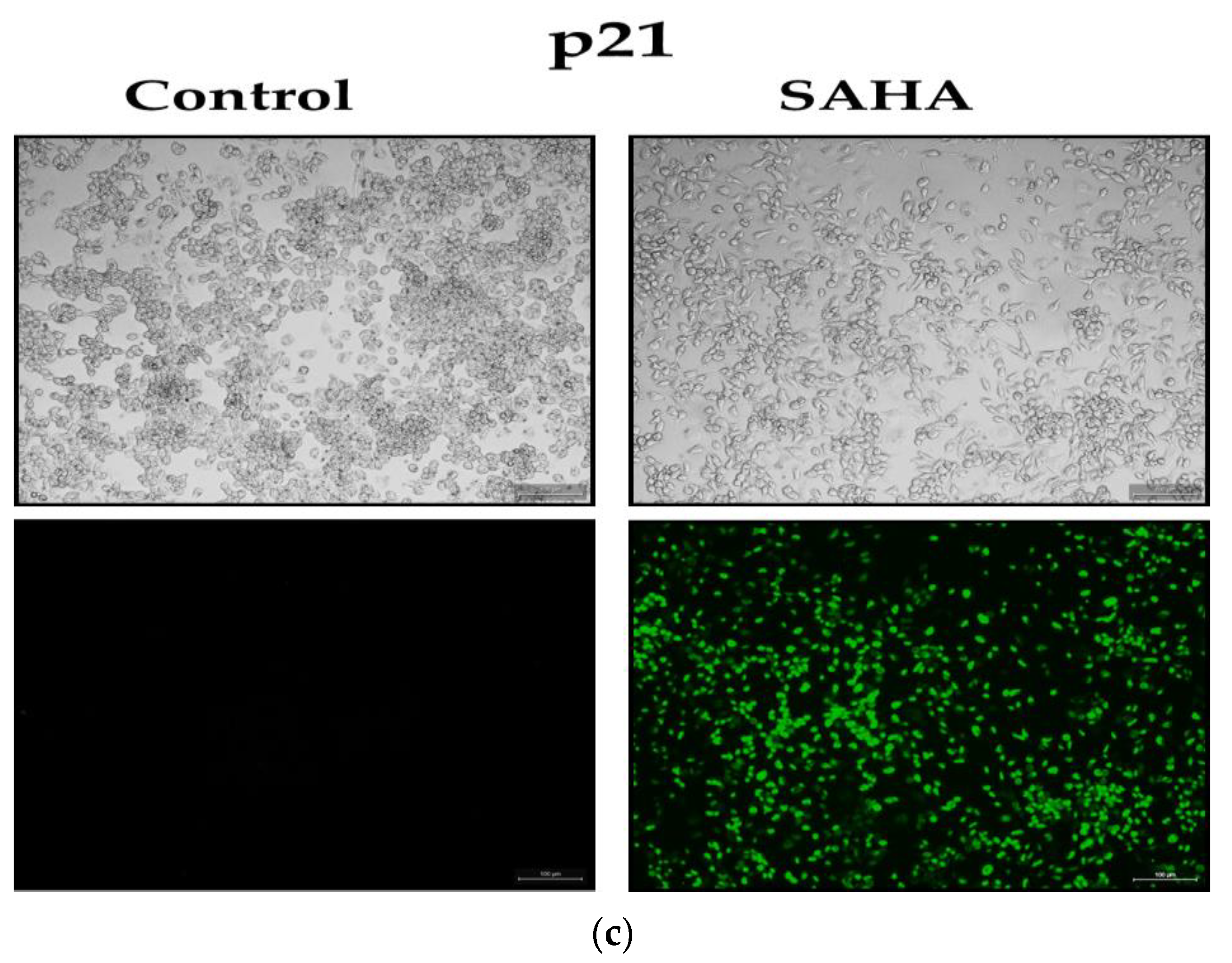
3.5. The Effect of SAHA Treatments on the Regulation of A2780 Cell Cycle Arrest
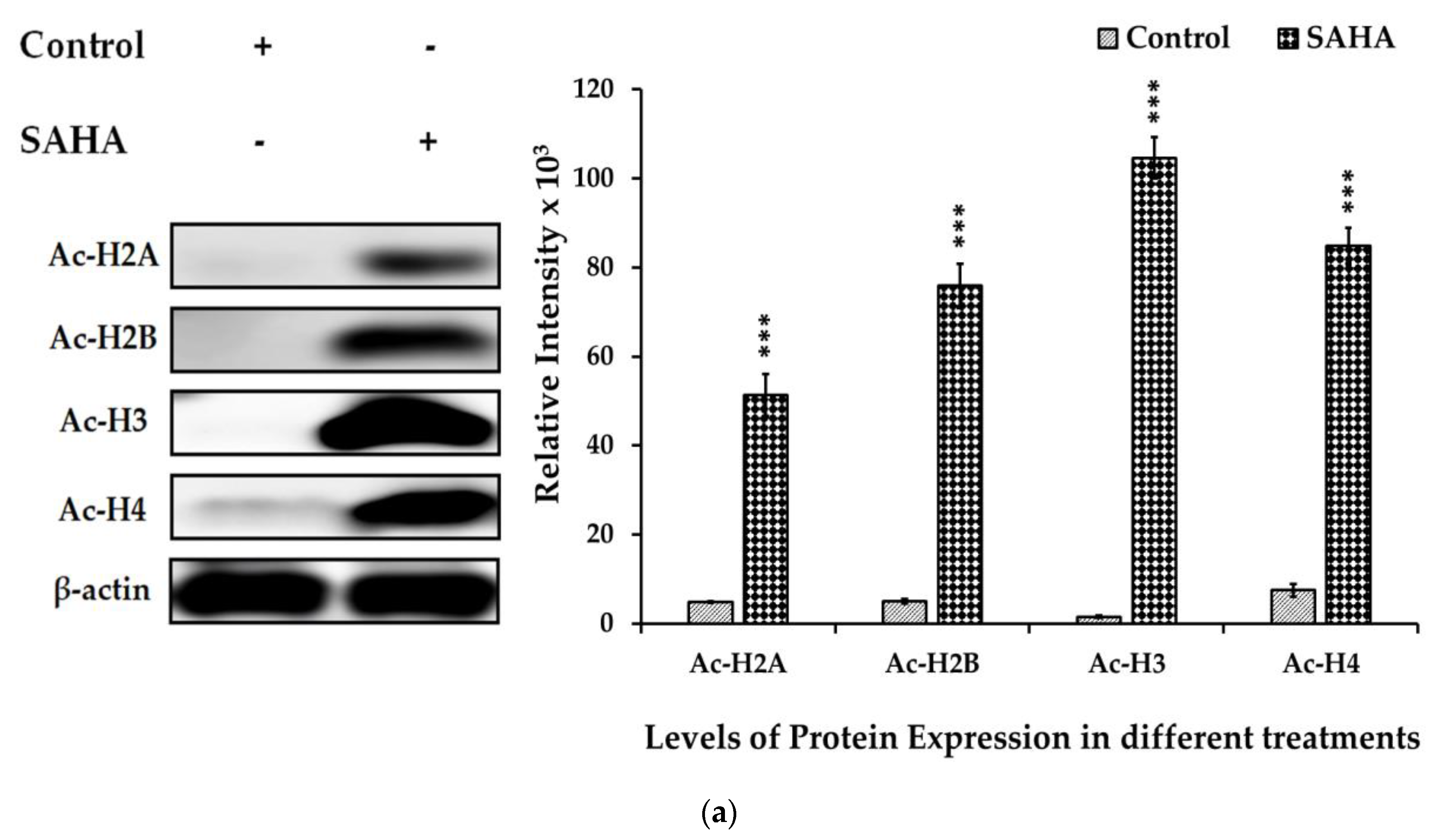
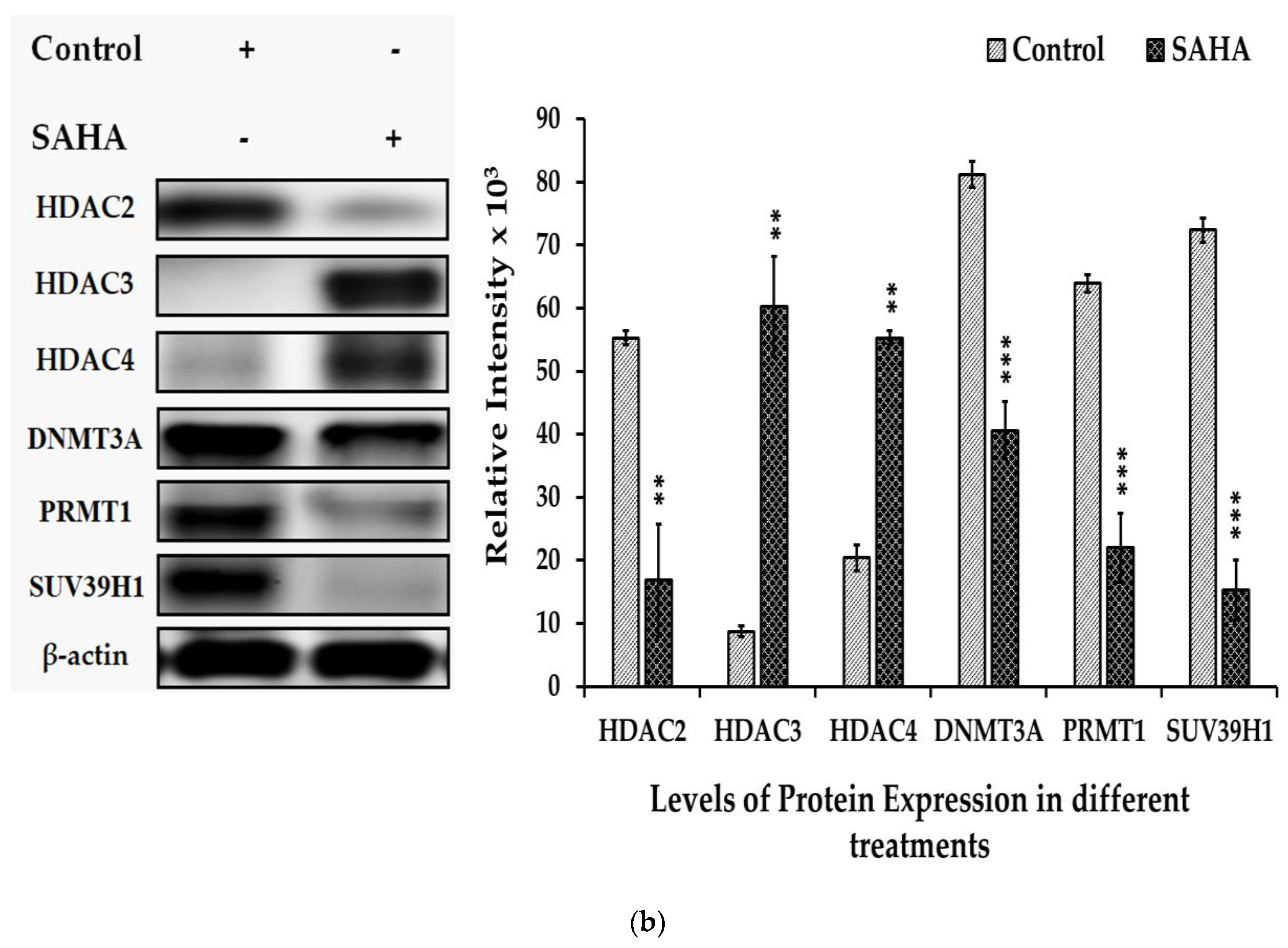
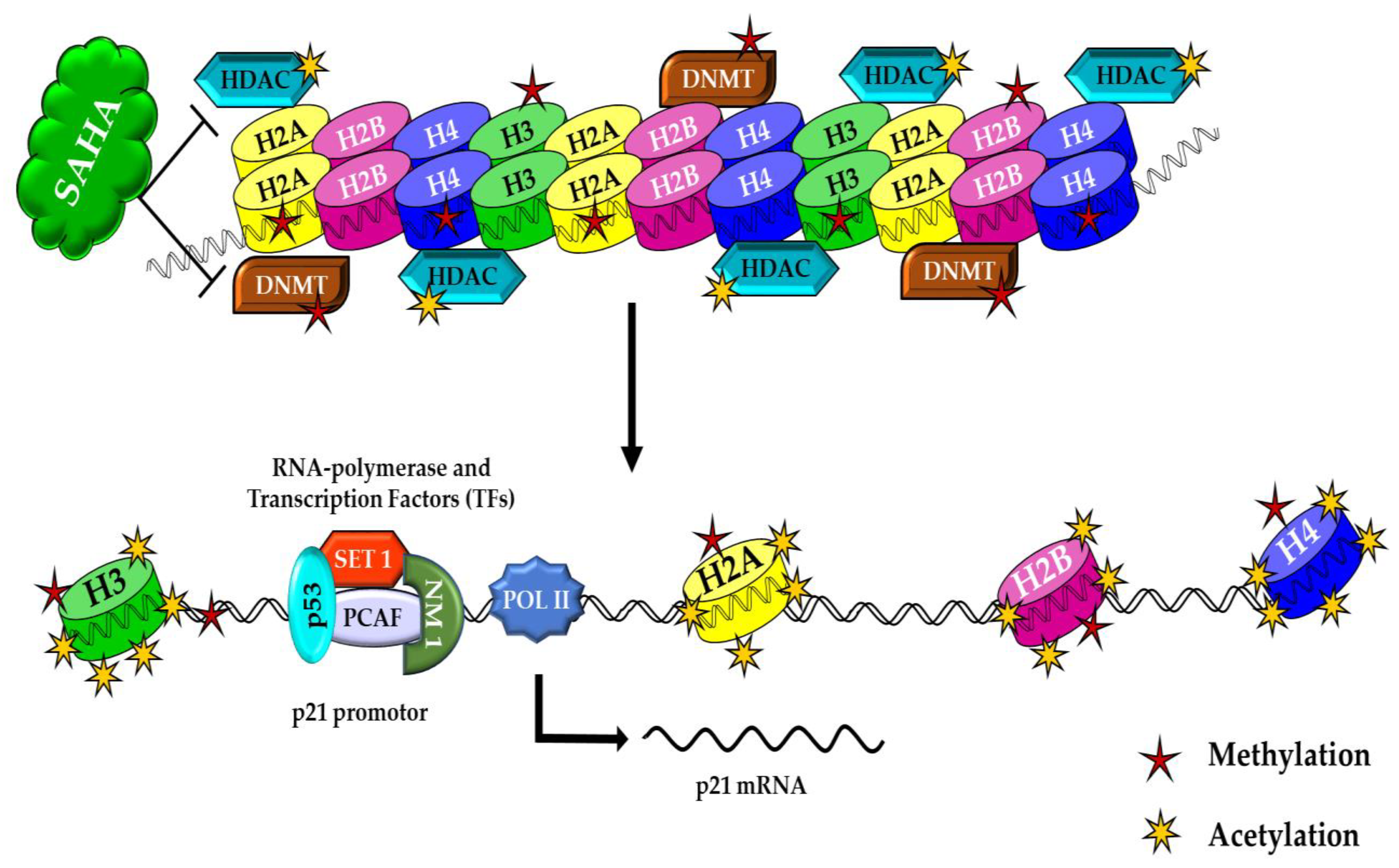
3.6. The Effects of SAHA on the Acetylation of Histones and DNMTs/HMTs Levels in A2780 Cells
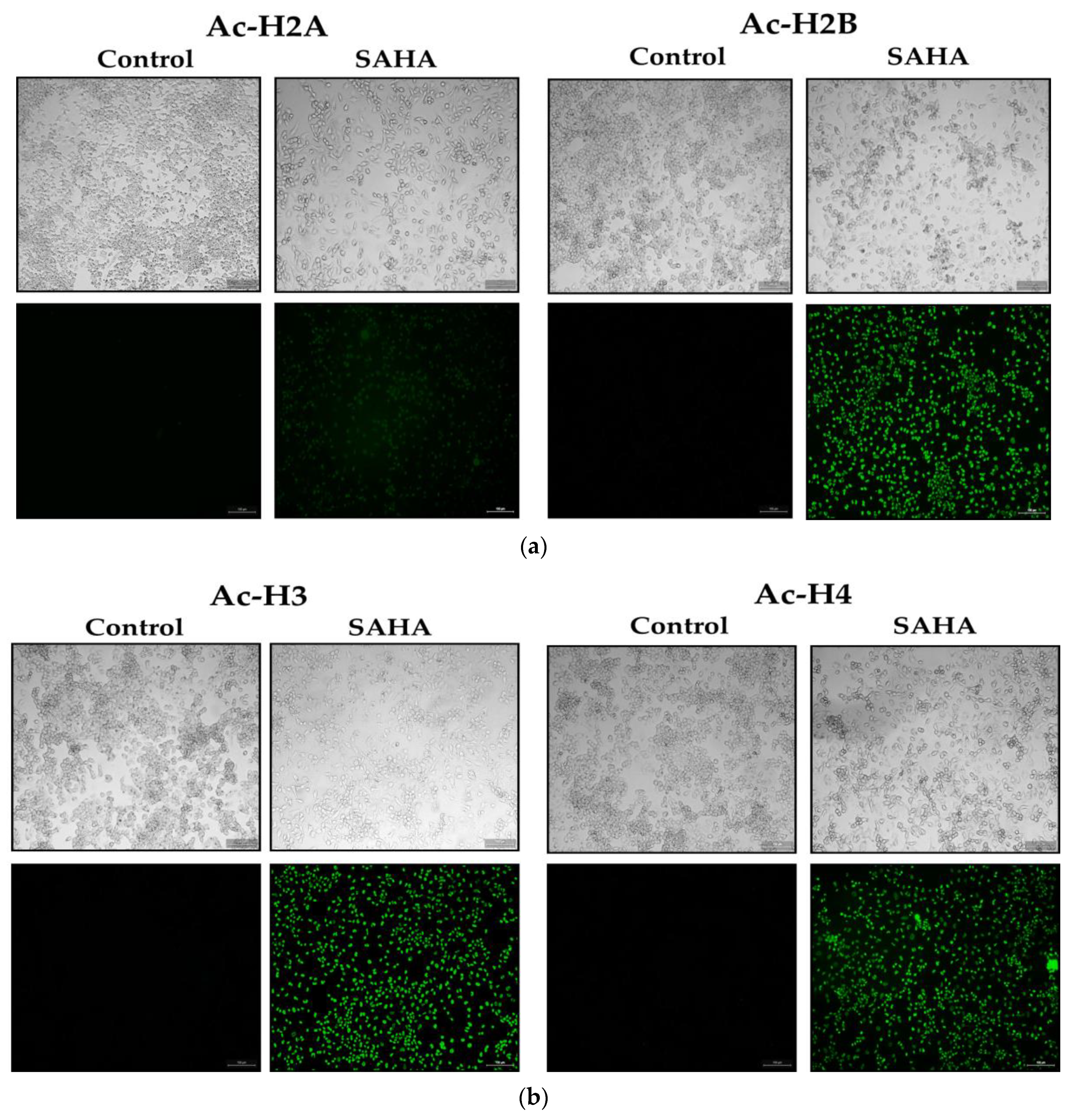
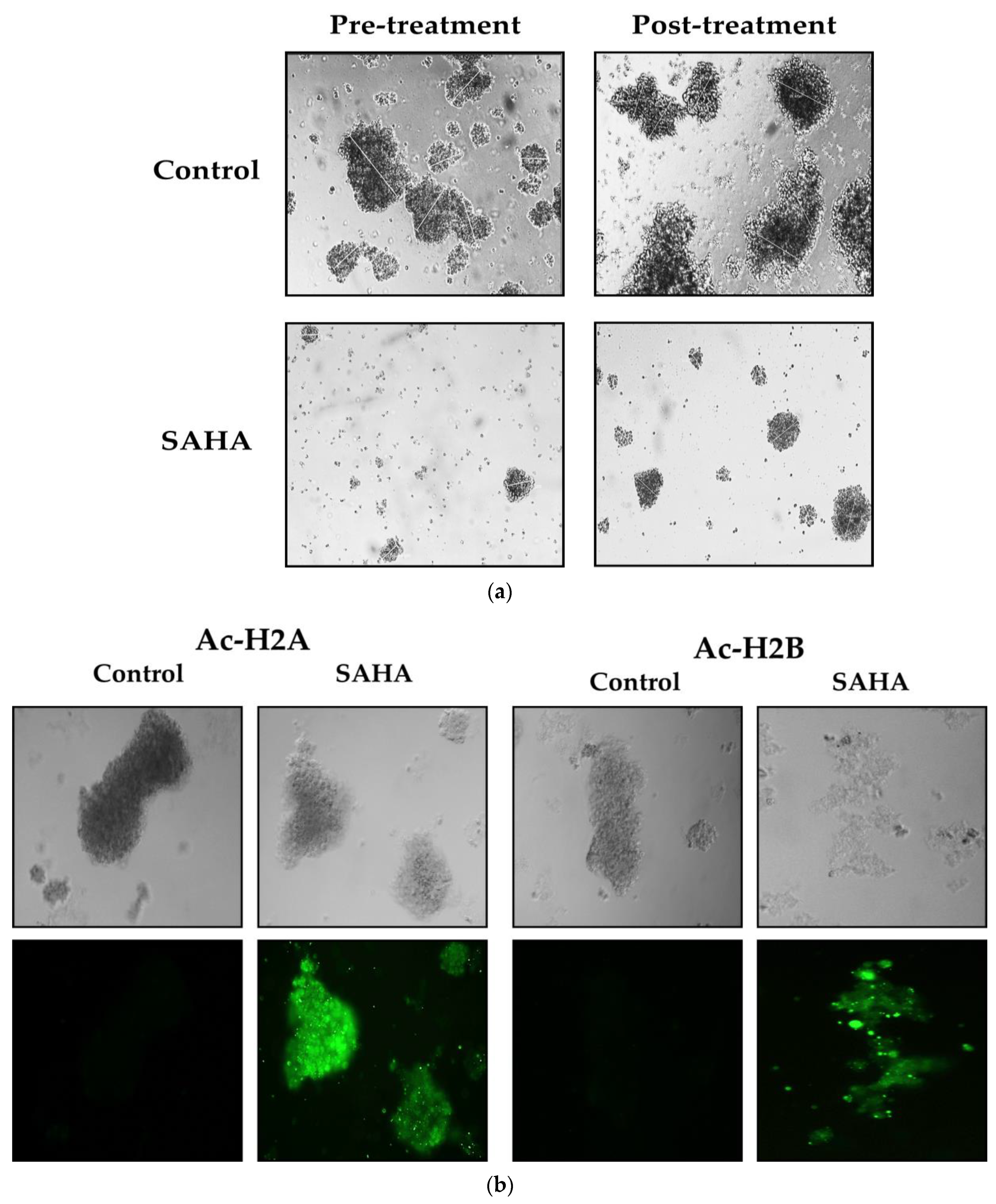
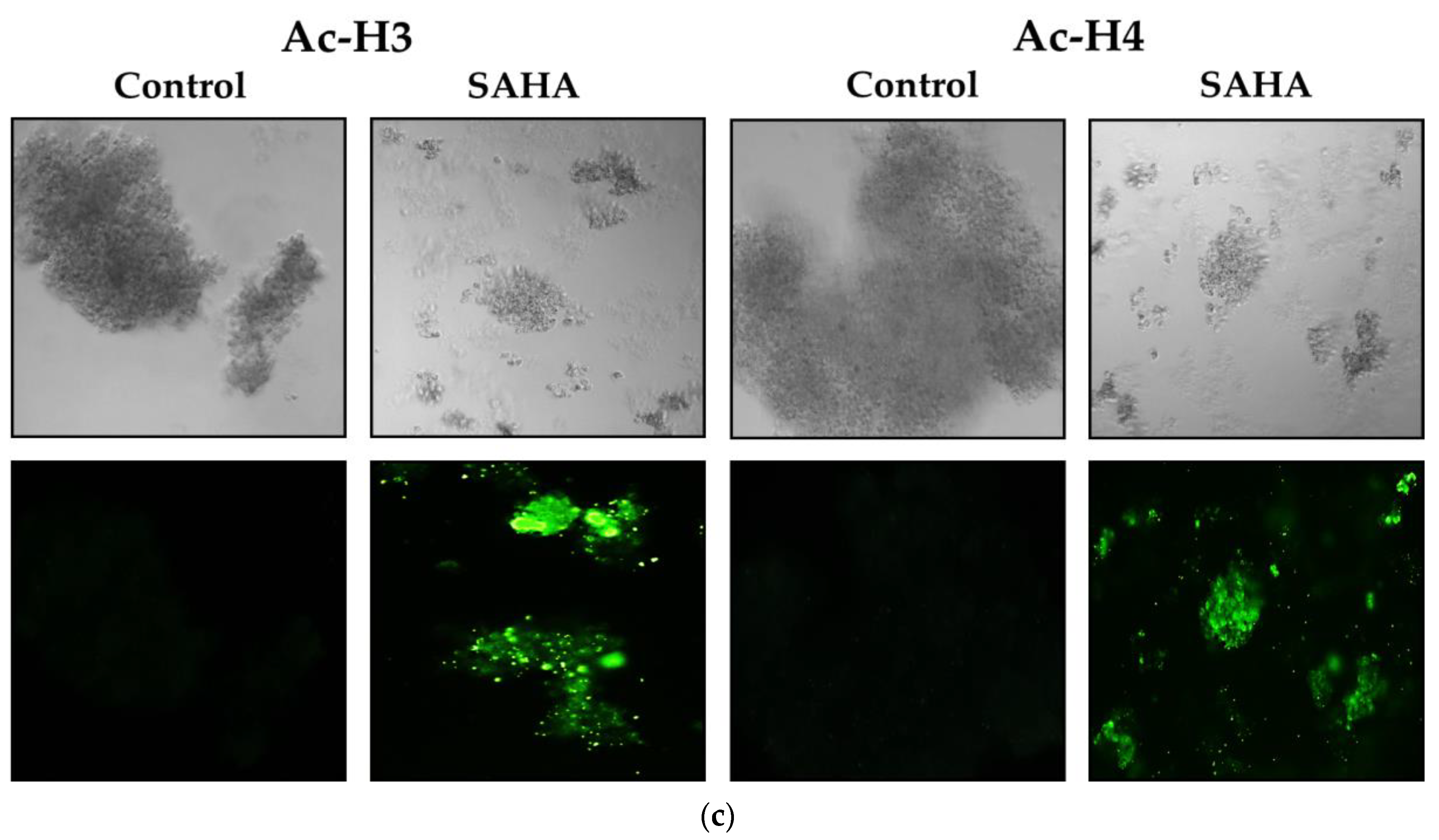
3.7. Immunofluorescence Analysis of Acetylated Histones in A2780 Cells Growing as Monolayers and Spheroids
3.8. Effect of SAHA Treatment on Spheroid Formation and Immunofluorescence Analysis of Acetylated Histones in A2780 Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- American Cancer Society. Available online: https://www.cancer.org/ (accessed on 3 March 2021).
- Yang, D.; He, Y.; Wu, B.; Deng, Y.; Wang, N.; Li, M.; Liu, Y. Integrated bioinformatics analysis for the screening of hub genes and therapeutic drugs in ovarian cancer. J. Ovarian Res. 2020, 13, 10. [Google Scholar] [CrossRef]
- Uchida, S.; Hara, K.; Kobayashi, A.; Otsuki, K.; Yamagata, H.; Hobara, T.; Suzuki, T.; Miyata, N.; Watanabe, Y. Epigenetic Status of Gdnf in the Ventral Striatum Determines Susceptibility and Adaptation to Daily Stressful Events. Neuron 2011, 69, 359–372. [Google Scholar] [CrossRef]
- Lubin, F.D. Epigenetic gene regulation in the adult mammalian brain: Multiple roles in memory formation. Neurobiol. Learn. Mem. 2011, 96, 68–78. [Google Scholar] [CrossRef]
- He, D.Y.; Neasta, J.; Ron, D. Epigenetic regulation of BDNF expression via the scaffolding protein RACK1. J. Biol. Chem. 2010, 285, 19043–19050. [Google Scholar] [CrossRef] [PubMed]
- Allfrey, V.G.; Faulkner, R.; Mirsky, A.E. Acetylation and Methylation Of Histones And Their Possible Role In The Regulation Of Rna Synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Drummond, D.C.; Noble, C.O.; Kirpotin, D.B.; Guo, Z.; Scott, G.K.; Benz, C.C. Clinical development of histone deacetylase inhibitors as anticancer agents. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 495–528. [Google Scholar] [CrossRef]
- Umamaheswari, N.; Thiagarajan, V.; Vijayaraghavan, R.; Shila, S.; Periannan, R.; Rathinavelu, A. Cell Cycle Arrest and Cytotoxic Effects of SAHA and RG7388 Mediated through p21WAF1/CIP1 and p27KIP1 in Cancer Cells. Medicina 2019, 55, 30. [Google Scholar] [CrossRef]
- Madeleine, D.; Rakshandra, T.; Xiao, N.; Chunlei, Z.; Parul, H.; Cecilia, K.; Judy, H.; Chiao, J.F.R.; Justin, L.R.; Victoria, M.R.; et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 2007, 109, 31–39. [Google Scholar] [CrossRef]
- Yuka, S.; Yoshinori, N.; Takeshi, I.; Tatsuya, S.; Masahiko, M.; Toshitaka, M.; Seitaro, M. Effects of FK228, a novel histone deacetylase inhibitor, on human lymphoma U-937 cells in vitro and in vivo. Biochem. Pharmacol. 2002, 64, 1079–1090. [Google Scholar] [CrossRef]
- Gui, C.Y.; Ngo, L.; Xu, W.S.; Richon, V.M.; Marks, P.A. Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1. Proc. Natl. Acad. Sci. USA 2004, 101, 1241–1246. [Google Scholar] [CrossRef] [PubMed]
- Aimée, M.D.; Adrian, B. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef]
- Bilian, J.; Keith, D.R. DNA Methyltransferases (DNMTs), DNA Damage Repair, and Cancer. Adv. Exp. Med. Biol. 2013, 754, 3–29. [Google Scholar] [CrossRef]
- Sarah, H.; Karolina, L.; Nicole, S.; Meghan, L.; Sarah, R.; Sibaji, S. Use of Epigenetic Drugs in Disease: An Overview. Genet. Epigenet. 2014, 6, 9–19. [Google Scholar] [CrossRef]
- Qilian, Y.; Yuqing, Y.; Nianxin, Z.; Kexin, T.; Wayne, B.L.; Bonnie, L.; Wei, W.; Lian, X.; Zhengnan, Y.; Shuang, H.; et al. Epigenetics in ovarian cancer: Premise, properties, and perspectives. Mol. Cancer 2018, 17, 109. [Google Scholar] [CrossRef]
- Yutaka, K. Epigenetic Cross-Talk between DNA Methylation and Histone Modifications in Human Cancers. Yonsei Med. J. 2009, 50, 455–463. [Google Scholar] [CrossRef]
- Mohsen, K.A.; Atosa, E.Z.; Yu, M.; Susanne, J.H.; Vijverberg, Z.Z.; Zahidul, K.; Syed, S.; Lorenz, K.; LiFu, H.; Vladana, V.; et al. Genomic DNA Hypomethylation by Histone Deacetylase Inhibition Implicates DNMT1 Nuclear Dynamics. Mol. Cell. Biol. 2011, 31, 4119–4128. [Google Scholar] [CrossRef]
- Lindsey, A.A.; Trevor, D.; Moira, R.; Mike, F. The p21WAF1/CIP1 Promoter Is Methylated in Rat-1 Cells: Stable Restoration of p53-Dependent p21WAF1/CIP1 Expression after Transfection of a Genomic Clone Containing the p21WAF1/CIP1 Gene. Mol. Cell. Biol. 2000, 20, 1291–1298. [Google Scholar] [CrossRef][Green Version]
- Huang, L.; Sowa, Y.; Sakai, T.; Pardee, A.B. Activation of the p21WAF1/CIP1 promoter independent of p53 by the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) through the Sp1 sites. Oncogene 2000, 19, 5712–5719. [Google Scholar] [CrossRef]
- Kim, Y.K.; Han, J.-W.; Na Woo, Y.; Chun, J.K.; Yoo, J.Y.; Cho, E.-J.; Hong, S.; Lee, H.Y.; Lee, Y.-W.; Lee, H.-W. Expression of p21WAF1/Cip1 through Sp1 sites by histone deacetylase inhibitor apicidin requires PI 3-kinase–PKCε signaling pathway. Oncogene 2003, 22, 6023–6031. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Mizuno, T.; Sowa, Y.; Orita, T.; Yoshino, T.; Okuyama, Y.; Fujita, T.; Ohtani, F.N.; Matsukawa, Y.; Tokino, T.; et al. Butyrate activates the WAF1/Cip1 gene promoter through Sp1 sites in a p53-negative human colon cancer cell line. J. Biol. Chem. 1997, 272, 22199–22206. [Google Scholar] [CrossRef]
- Sowa, Y.; Orita, T.; Minamikawa-Hiranabe, S.; Mizuno, T.; Nomura, H.; Sakai, T. Sp3 but not Sp1, mediates the transcriptional activation of the p21WAF1/Cip1 gene promotor by histone deacetylase inhibitor. Cancer Res. 1999, 59, 4266–4370. [Google Scholar] [PubMed]
- Han, J.W.; Ahn, S.H.; Park, S.H.; Wang, S.Y.; Bae, G.U.; Seo, D.W.; Kwon, H.K.; Hong, S.; Lee, H.Y.; Lee, Y.W.; et al. Apicidin a histone deacetylase inhibitor, inhibits proliferation of tumor cells via induction of p21WAF1. Cancer Res. 2000, 60, 6068–6074. [Google Scholar]
- Li, G.-C.; Zhang, X.; Pan, T.-J.; Chen, Z.; Ye, Z.-Q. Histone deacetylase inhibitor trichostatin A inhibits the growth of bladder cancer cells through induction of p21WAF1 and G1 cell cycle arrest. Int. J. Urol. 2006, 13, 581–586. [Google Scholar] [CrossRef]
- Richon, V.M.; Sandhoff, T.W.; Rifkind, R.A.; Marks, P.A. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc. Natl Acad. Sci. USA 2000, 97, 10014–10019. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. Cancer cell cycles. Science 1996, 274, 1672–1677. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Zhu, J.; Wang, X.; Zheng, N.; Tu, C.; Qu, J.; Ren, X. Mapping dynamic histone modification patterns during arsenic-induced malignant transformation of human bladder cells. Toxicol. Appl. Pharmacol. 2018, 355, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Dowdy, S.C.; Podratz, K.C.; Jin, F.; Attewell, J.R.; Eberhardt, N.L.; Jiang, S.-W. Histone Deacetylase Inhibitors Decrease DNA Methyltransferase-3B Messenger RNA Stability and Down-regulate De novo DNA Methyltransferase Activity in Human Endometrial Cells. Cancer Res. 2005, 65, 2684–2689. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and DNMT3B are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Hsieh, C.L. In vivo activity of murine de novo methyltransferases, Dnmt3a and DNMT3B. Mol. Cell. Biol. 1999, 19, 8211–8218. [Google Scholar] [CrossRef] [PubMed]
- Qiang, M.; Denny, A.; Lieu, M.; Carreon, S.; Li, J. Histone H3K9 modifications are a local chromatin event involved in ethanol-induced neuroadaptation of the NR2B gene. Epigenetics 2011, 6, 1095–1104. [Google Scholar] [CrossRef] [PubMed]
- Tiffani, D.M.B.; Subhash, C.P. Emerging role of epigenetic mechanisms in alcohol addiction. Alcohol. Clin. Exp. Res. 2017, 41, 666–680. [Google Scholar] [CrossRef]
- Gore, S.D.; Hermes-DeSantis, E.R. Enhancing survival outcomes in the management of patients with higher-risk myelodysplastic syndromes. Cancer Control 2009, 16, 2. [Google Scholar] [CrossRef] [PubMed]
- Kavanaugh, S.M.; White, L.A.; Kolesar, J.M. Vorinostat: A novel therapy for the treatment of cutaneous T-cell lymphoma. Am. J. Health Syst. Pharm. 2010, 67, 793–797. [Google Scholar] [CrossRef]
- Santi, D.V.; Norment, A.; Garrett, C.E. Covalent bond formation between a DNA-cytosine methyltransferase and DNA containing 5-azacytosine. Proc. Natl. Acad. Sci. USA 1984, 81, 6993–6997. [Google Scholar] [CrossRef]
- Ghoshal, K.; Majumder, S.; Jacob, S.T. Analysis of promoter methylation and its role in silencing metallothionein I gene expression in tumor cells. Methods Enzymol. 2002, 353, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Phillips, D.L.; Ferguson, A.T.; Nelson, W.G.; Herman, J.G.; Davidson, N.E. Synergistic activation of functional estrogen receptor (ER)-alpha by DNA methyltransferase and histone deacetylase inhibition in human ER-alpha-negative breast cancer cells. Cancer Res. 2001, 61, 7025–7029. [Google Scholar] [PubMed]
- Nicolai, M.; Marco, B.; Frank, R.; Christian, S.; Nobuyuki, O.; Ming, Z.; Walter, J.; Koichi, N. Hypermethylation and histone deacetylation lead to silencing of the maspin gene in human breast cancer. Biochem. Biophys. Res. Commun. 2002, 297, 125–128. [Google Scholar] [CrossRef]
- Januchowski, R.; Dabrowski, M.; Ofori, H.; Jagodzinski, P.P. Trichostatin A down-regulate DNA methyltransferase 1 in Jurkat T cells. Cancer Lett. 2007, 246, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Jing-Ni, O.; Jérôme, T.; Alexander, U.; Nadine, P.; Keisuke, S.; Mohsen, K.; Tomas, J.E.; Moshe, S. Histone deacetylase inhibitor trichostatin A induces global and gene-specific DNA demethylation in human cancer cell lines. Biochem. Pharmacol. 2007, 73, 1297–1307. [Google Scholar] [CrossRef]
- Kalle, A.M.; Wang, Z. Differential effects of two HDAC inhibitors with distinct concomitant DNA hypermethylation or hypomethylation in breast cancer cells. bioRxiv 2019, preprint. [Google Scholar] [CrossRef]
- Starlard, D.A.; Kutanzi, K.; Tryndyak, V.; Word, B.; Lyn-Cook, B. Restoration of the methylation status of hypermethylated gene promoters by microRNA-29b in human breast cancer: A novel epigenetic therapeutic approach. J. Carcinog. 2013, 12, 15. [Google Scholar] [CrossRef]
- Zhou, Q.; Agoston, A.T.; Atadja, P.; Nelson, W.G.; Davidson, N.E. Inhibition of histone deacetylases promotes ubiquitin-dependent proteasomal degradation of DNA methyltransferase 1 in human breast cancer cells. Mol. Cancer Res. 2008, 6, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Chuang, L.S.; Ian, H.I.; Koh, T.W.; Ng, H.H.; Xu, G.; Li, B.F. Human DNA-(cytosine-5) methyltransferase- PCNA complex as a target for p21WAF1. Science 1997, 277, 1996–2000. [Google Scholar] [CrossRef]
- Ryu, T.Y.; Kim, K.; Son, M.-Y.; Min, J.-K.; Kim, J.; Han, T.-S.; Kim, D.-S.; Cho, H.-S. Downregulation of PRMT1, a histone arginine methyltransferase, by sodium propionate induces cell apoptosis in colon cancer. Oncol. Rep. 2019, 41, 1691–1699. [Google Scholar] [CrossRef] [PubMed]
- Mirza, S.; Sharma, G.; Parshad, R.; Gupta, S.D.; Pandya, P.; Ralhan, R. Expression of DNA methyltransferases in breast cancer patients and to analyze the effect of natural compounds on DNA methyltransferases and associated proteins. J. Breast Cancer 2013, 16, 23–31. [Google Scholar] [CrossRef]
- Fuks, F.; Burgers, W.A.; Brehm, A.; Hughes-Davies, L.; Kouzarides, T. DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat. Genet. 2000, 24, 88–91. [Google Scholar] [CrossRef]
- Rea, S.; Eisenhaber, F.; O’Carroll, D.; Strahl, B.D.; Sun, Z.W.; Schmid, M.; Opravil, S.; Mechtler, K.; Ponting, C.P.; Allis, C.D.; et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 2000, 406, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Manuel, M.M.; Beat, F.; Lenka, B.; Glen, L.; Tom, W.M. A two-state activation mechanism controls the histone methyltransferase Suv39h1. Nat. Chem. Biol. 2016, 12, 188–193. [Google Scholar] [CrossRef]
- Nakayama, J.-I.; Rice, J.C.; Strahl, B.D.; Allis, C.D.; Grewal, S.I.S. Role of Histone H3 Lysine 9 Methylation in Epigenetic Control of Heterochromatin Assembly. Science 2001, 292, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, Y.; Hieda, M.; Nishioka, Y.; Matsumoto, A.; Higashi, S.; Kimura, H.; Yamamoto, H.; Mori, M.; Matsuura, S.; Matsuura, N. Cancer-associated upregulation of histone H3 lysine 9 trimethylation promotes cell motility in vitro and drives tumor formation in vivo. Cancer Sci. 2013, 104, 889–895. [Google Scholar] [CrossRef]
- Espen, B.A.; Paula, M.D.A.; Aasa, R.S.; Katherine, L.K.; Marit, A.; Ole, P.F.C. Subcellular localization of the spindle proteins Aurora A, Mad2, and BUBR1 assessed by immunohistochemistry. J. Histochem. Cytochem. 2007, 55, 477–486. [Google Scholar] [CrossRef]
- Rong, R.; Jiang, L.Y.; Sheikh, M.S.; Huang, Y. Mitotic kinase Aurora-A phosphorylates RASSF1A and modulates RASSF1A-mediated microtubule interaction and M-phase cell cycle regulation. Oncogene 2007, 26, 7700–7708. [Google Scholar] [CrossRef] [PubMed]
- Alejandra, G.L.; Gonzalo, F.M.; Marianna, T.; David, P.; Kumiko, S.; Hiromi, O.; Marta, C.; Alba, M.; Ángel, M.R.; Guillermo, C.; et al. Aurora B Overexpression Causes Aneuploidy and p21Cip1 Repression during Tumor Development. Mol. Cell. Biol. 2015, 35, 3566–3578. [Google Scholar] [CrossRef]
- Tsou, J.-H.; Chang, K.-C.; Chang-Liao, P.-Y.; Yang, S.-T.; Lee, C.-T.; Chen, Y.-P.; Lee, Y.-C.; Lin, B.-W.; Lee, J.-C.; Shen, M.-R.; et al. Aberrantly expressed AURKC enhances the transformation and tumourigenicity of epithelial cells. J. Pathol. 2011, 225, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Kanagasabai, T.; Venkatesan, T.; Natarajan, U.; Alobid, S.; Alhazzani, K.; AlGahtani, M.; Rathinavelu, A. Regulation of cell cycle by MDM2 in prostate cancer cells through Aurora Kinase-B and p21WAF1/CIP1 mediated pathways. Cell. Signal. 2020, 66, 109435. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Accession Number | Description | Fold Change |
|---|---|---|---|
| Up-regulated genes | |||
| RPS6KA5 | NM_004755 | Ribosomal protein S6 kinase, 90 kDa, polypeptide 5 | 1.96 |
| HDAC3 | NM_003883 | Histone deacetylase 3 | 1.79 |
| HDAC9 | NM_178425 | Histone deacetylase 9 | 1.7 |
| SETD4 | NM_017438 | Su(var)3–9, enhancer-of-zeste, trithorax (SET) domain containing 4 | 1.78 |
| SMYD3 | NM_022743 | SET and Myeloid-Nervy-DEAF1 (MYND) domain containing 3 | 1.6 |
| HDAC4 | NM_006037 | Histone deacetylase 4 | 1.23 |
| Down-regulated genes | |||
| SETD1B | NM_015048 | SET domain containing 1B | 0.24 |
| HDAC7 | NM_001098416 | Histone deacetylase 7 | 0.27 |
| NSD1 | NM_022455 | Nuclear receptor binding SET, domain protein 1 | 0.29 |
| DNMT3A | NM_022552 | DNA (cytosine-5-)-methyltransferase 3 alpha | 0.31 |
| WHSC1 | NM_007331 | Wolf-Hirschhorn syndrome candidate 1 | 0.35 |
| AURKA | NM_003600 | Aurora kinase A | 0.41 |
| EHMT2 | NM_006709 | Euchromatic histone-lysine N-methyltransferase 2 | 0.41 |
| KAT7 | NM_007067 | K(lysine) acetyltransferase 7 | 0.43 |
| SUV39H1 | NM_003173 | Suppressor of variegation 3–9 homolog 1 (Drosophila) | 0.43 |
| NACOA6 | NM_014071 | Nuclear receptor coactivator 6 | 0.44 |
| SETD7 | NM_030648 | SET domain containing (lysine methyltransferase) 7 | 0.44 |
| PRMT7 | NM_019023 | Protein arginine methyltransferase 7 | 0.49 |
| CARM1 | NM_199141 | Coactivator-associated arginine methyltransferase 1 | 0.51 |
| KAT8 | NM_032188 | K(lysine) acetyltransferase 8 | 0.53 |
| KMT2E | NM_182931 | Myeloid/lymphoid or mixed-lineage leukemia 5 (trithorax homolog, Drosophila) | 0.54 |
| NEK6 | NM_014397 | NIMA (never in mitosis gene a) -related kinase 6 | 0.54 |
| KMT2A | NM_005933 | Myeloid/lymphoid or mixed-lineage leukemia (trithorax homolog, Drosophila) | 0.56 |
| KAT6B | NM_012330 | K(lysine) acetyltransferase 6B | 0.57 |
| SETD5 | NM_001080517 | SET domain containing 5 | 0.57 |
| AURKC | NM_003160 | Aurora kinase C | 0.58 |
| AURKB | NM_004217 | Aurora kinase B | 0.59 |
| PRMT1 | NM_001536 | Protein arginine methyltransferase 7 | 0.61 |
| HDAC2 | NM_001527 | Histone deacetylase 2 | 0.75 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Natarajan, U.; Venkatesan, T.; Rathinavelu, A. Effect of the HDAC Inhibitor on Histone Acetylation and Methyltransferases in A2780 Ovarian Cancer Cells. Medicina 2021, 57, 456. https://doi.org/10.3390/medicina57050456
Natarajan U, Venkatesan T, Rathinavelu A. Effect of the HDAC Inhibitor on Histone Acetylation and Methyltransferases in A2780 Ovarian Cancer Cells. Medicina. 2021; 57(5):456. https://doi.org/10.3390/medicina57050456
Chicago/Turabian StyleNatarajan, Umamaheswari, Thiagarajan Venkatesan, and Appu Rathinavelu. 2021. "Effect of the HDAC Inhibitor on Histone Acetylation and Methyltransferases in A2780 Ovarian Cancer Cells" Medicina 57, no. 5: 456. https://doi.org/10.3390/medicina57050456
APA StyleNatarajan, U., Venkatesan, T., & Rathinavelu, A. (2021). Effect of the HDAC Inhibitor on Histone Acetylation and Methyltransferases in A2780 Ovarian Cancer Cells. Medicina, 57(5), 456. https://doi.org/10.3390/medicina57050456