
Dual Role of SIRT1 in Autophagy and Lipid Metabolism Regulation in Osteoarthritic Chondrocytes

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Tissue Samples
2.2. Primary Cultures of Normal and OA Human Articular Chondrocytes
2.3. Treatment of Normal Chondrocytes with siRNA against SIRT1 under Serum Starvation Conditions
2.4. Treatment of OA Chondrocytes with Resveratrol, Hydroxychloroquine, 3-Methyladenine and siRNA against SIRT1
2.5. Western Blot Analysis
2.6. Immunofluorescence
2.7. Immunoprecipitation
2.8. Statistical Analysis
3. Results
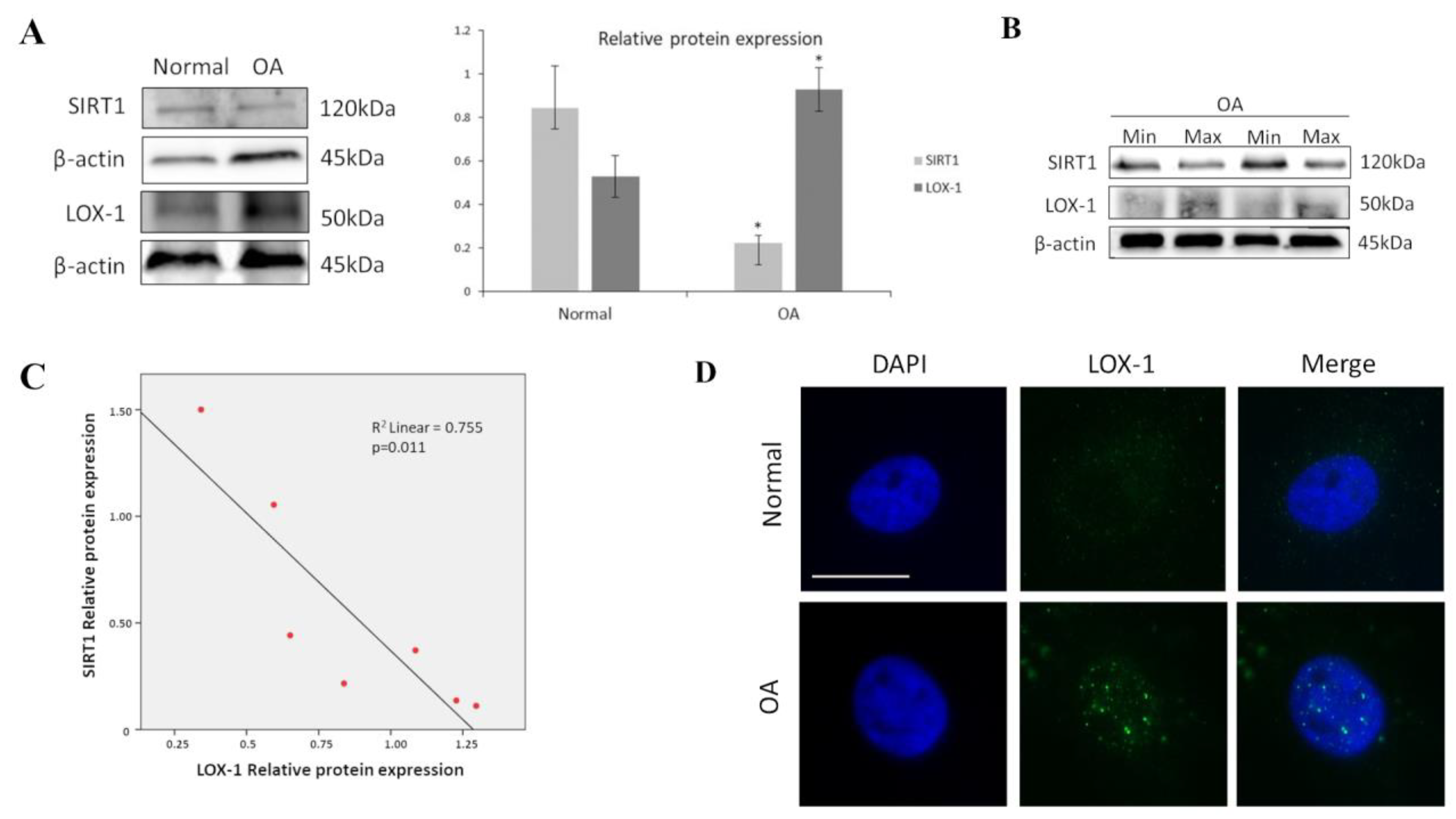
3.1. SIRT1 and LOX-1 Protein Expression Are Inversely Correlated in OA Chondrocytes
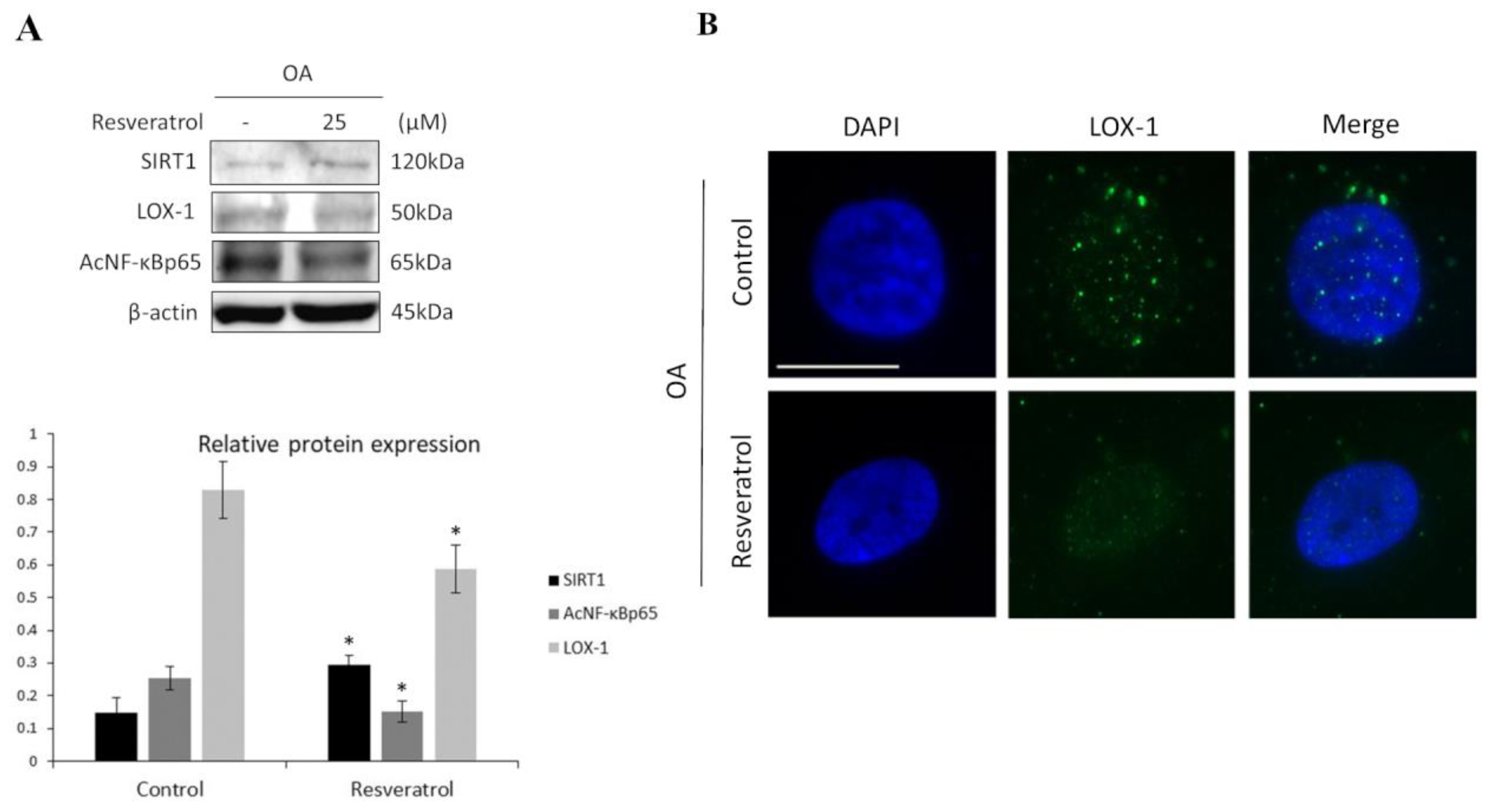
3.2. SIRT1 Regulates LOX-1 Expression via NF-κB Pathway in OA Chondrocytes
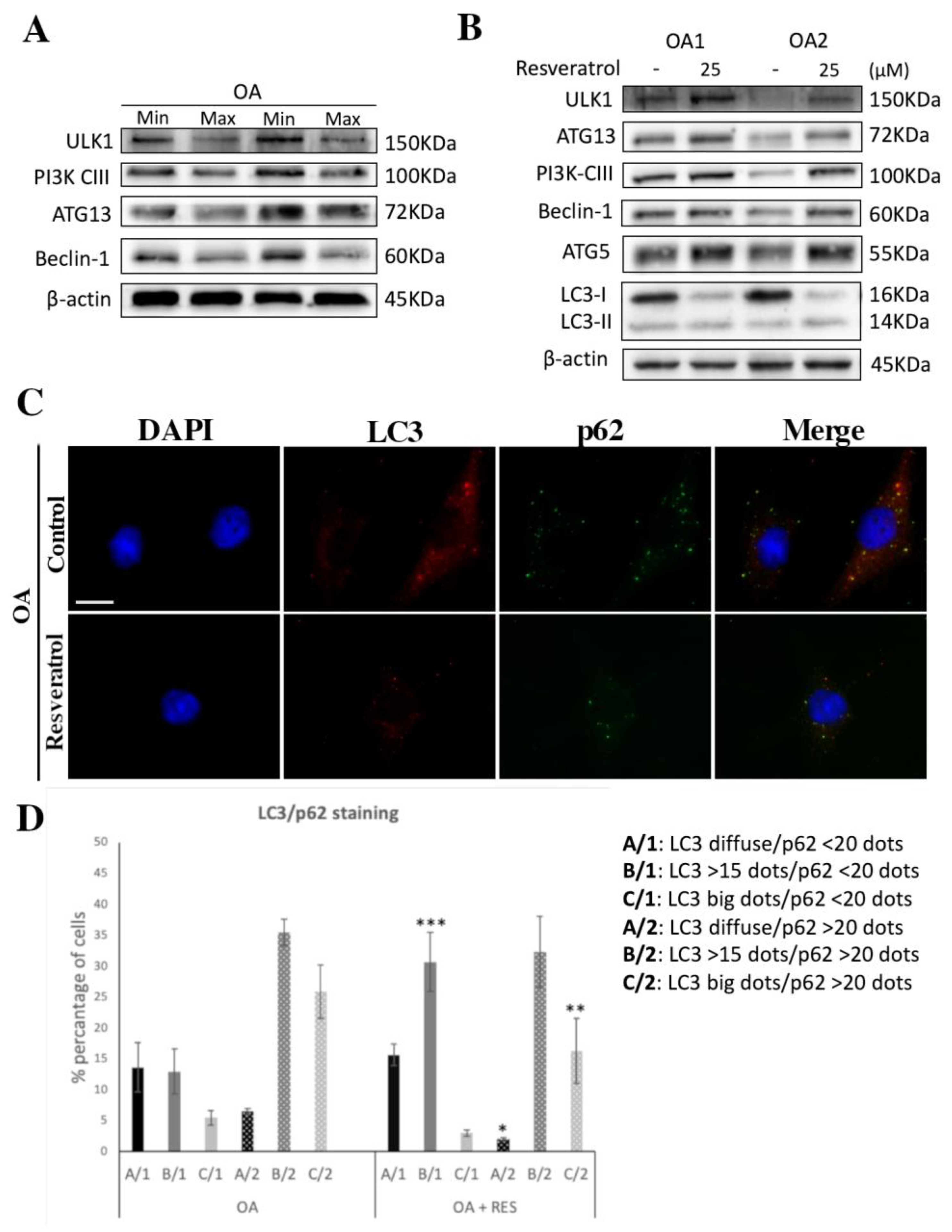
3.3. Autophagy Induction through SIRT1 in Resveratrol-Treated OA Chondrocytes
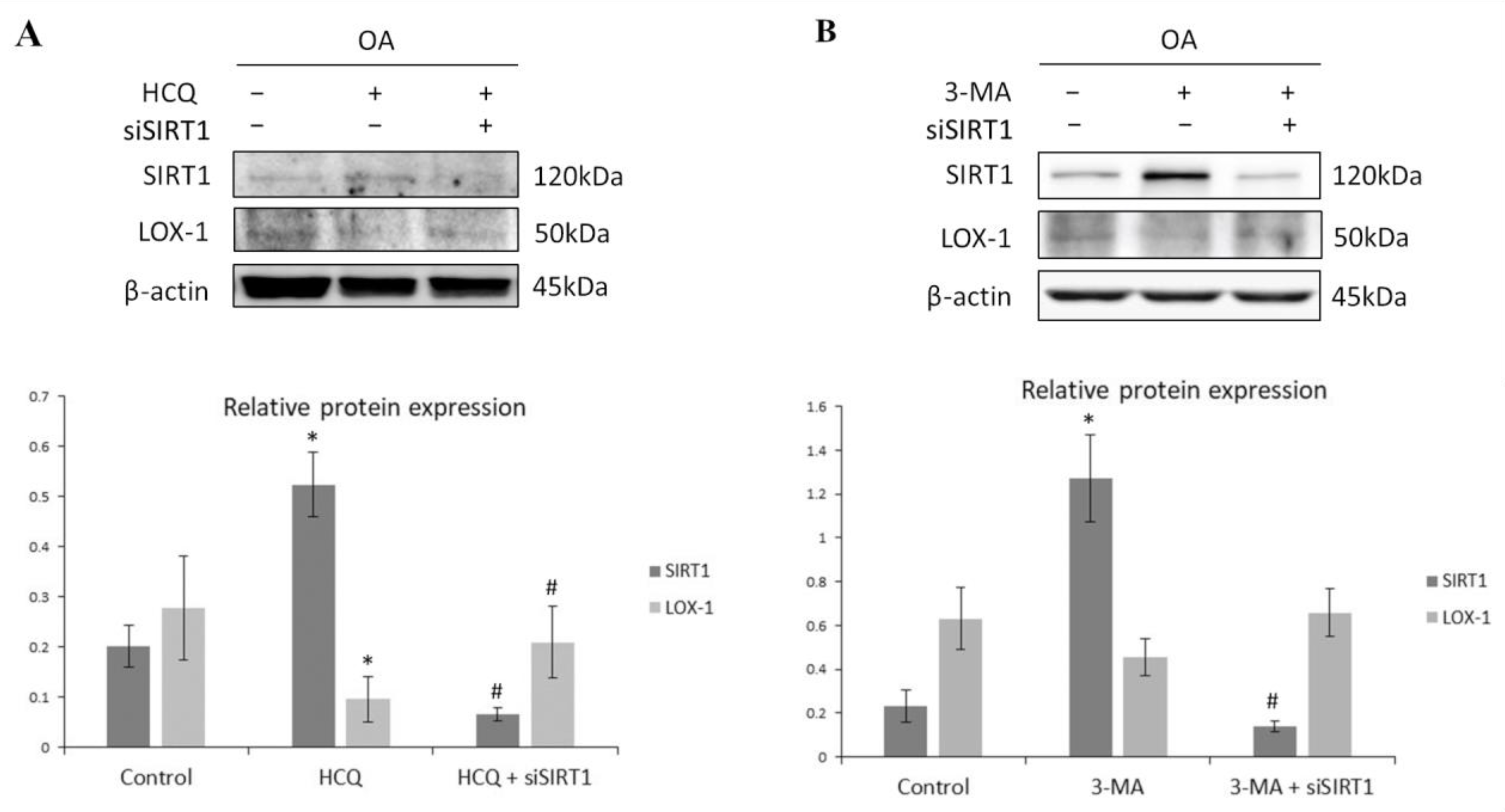
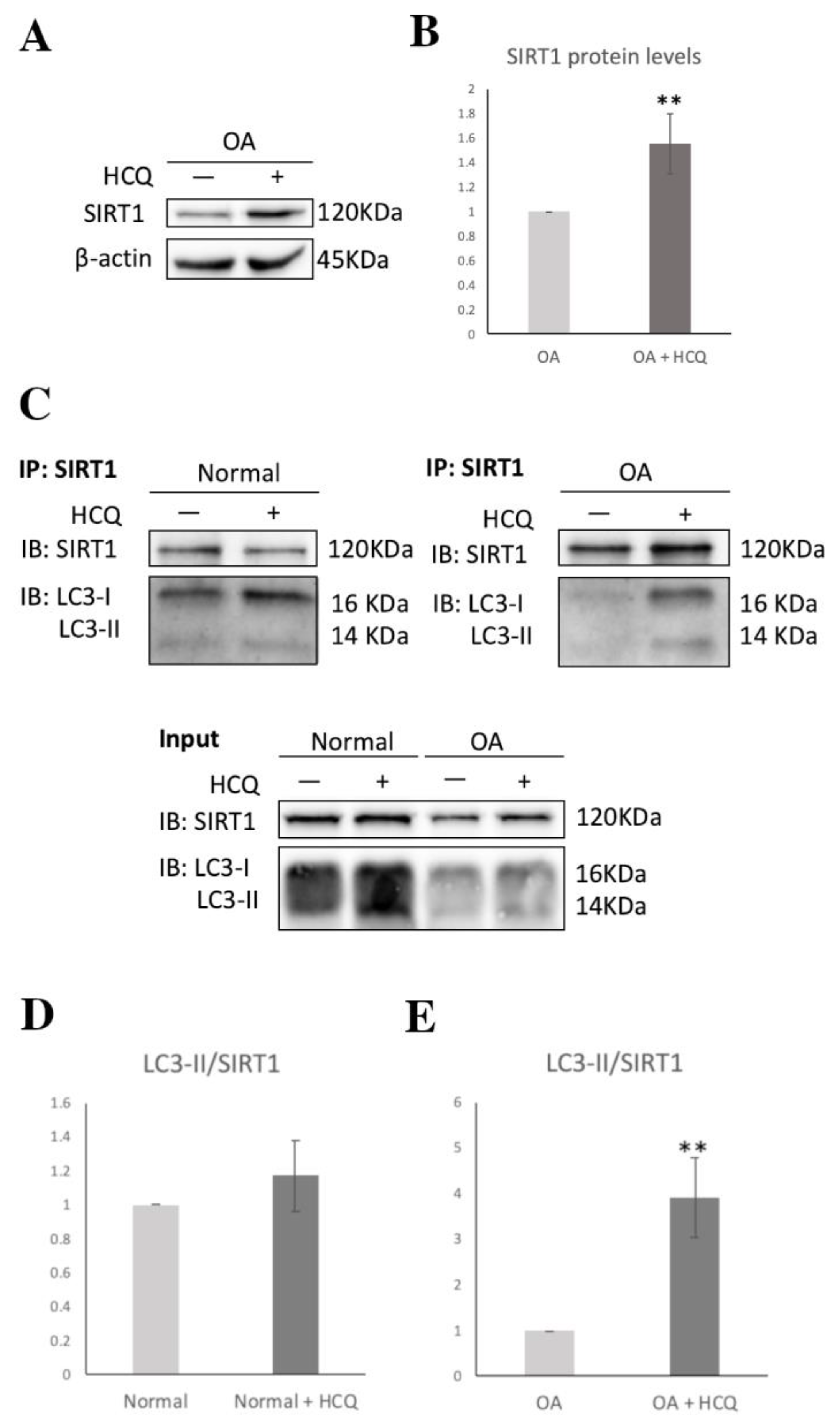
3.4. SIRT1-Mediated Regulation of LOX-1 Expression Is Autophagy-Dependent in OA Chondrocytes
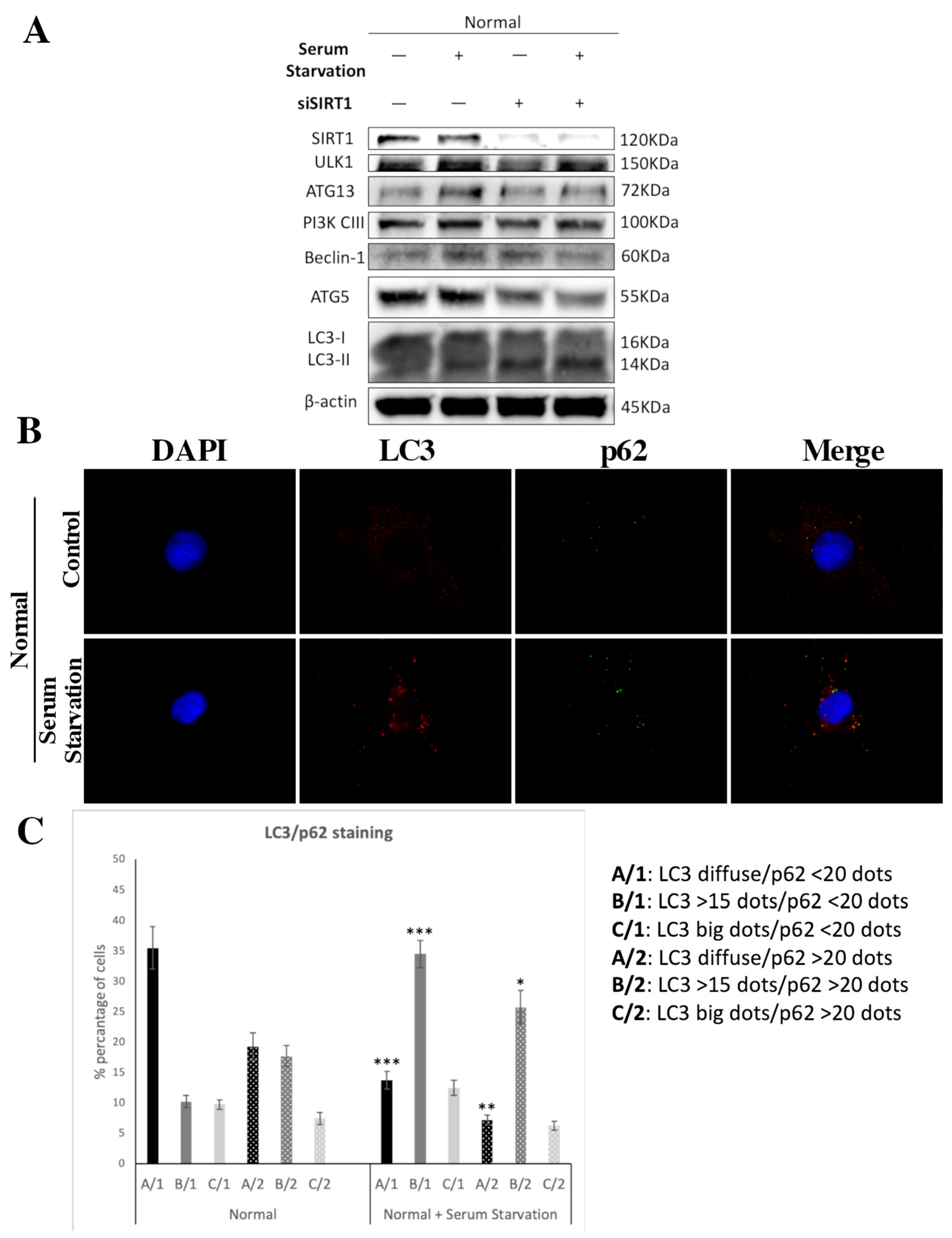
3.5. SIRT1 Is Necessary for Autophagy Induction in Articular Chondrocytes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Goldring, S.R.; Goldring, M.B. Changes in the osteochondral unit during osteoarthritis: Structure, function and cartilage–bone crosstalk. Nat. Rev. Rheumatol. 2016, 12, 632–644. [Google Scholar] [CrossRef]
- Harrell, C.R.; Markovic, B.S.; Fellabaum, C.; Arsenijevic, A.; Volarevic, V. Mesenchymal stem cell-based therapy of osteoarthritis: Current knowledge and future perspectives. Biomed. Pharmacother. 2019, 109, 2318–2326. [Google Scholar] [CrossRef] [PubMed]
- Cross, M.; Smith, E.; Hoy, D.; Nolte, S.; Ackerman, I.; Fransen, M.; Bridgett, L.; Williams, S.; Guillemin, F.; Hill, C.L.; et al. The global burden of hip and knee osteoarthritis: Estimates from the global burden of disease 2010 study. Ann. Rheum. Dis. 2014, 73, 1323–1330. [Google Scholar] [CrossRef] [PubMed]
- Grässel, S.; Muschter, D. Recent advances in the treatment of osteoarthritis. F1000Research 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Loeser, R.F.; Collins, J.A.; Diekman, B.O. Ageing and the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2016, 12, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Zhai, G. Alteration of Metabolic Pathways in Osteoarthritis. Metabolites 2019, 9, 11. [Google Scholar] [CrossRef]
- Sellam, J.; Berenbaum, F. Is osteoarthritis a metabolic disease? Jt. Bone Spine 2013, 80, 568–573. [Google Scholar] [CrossRef]
- Dickson, B.M.; Roelofs, A.J.; Rochford, J.J.; Wilson, H.M.; De Bari, C. The burden of metabolic syndrome on osteoarthritic joints. Arthritis Res. Ther. 2019, 21, 289. [Google Scholar] [CrossRef] [PubMed]
- Courties, A.; Sellam, J.; Berenbaum, F. Metabolic syndrome-associated osteoarthritis. Curr. Opin. Rheumatol. 2017, 29, 214–222. [Google Scholar] [CrossRef]
- Zheng, H.; Chen, C. Body mass index and risk of knee osteoarthritis: Systematic review and meta-analysis of prospective studies. BMJ Open 2015, 5, e007568. [Google Scholar] [CrossRef] [PubMed]
- Goutas, A.; Syrrou, C.; Papathanasiou, I.; Tsezou, A.; Trachana, V. The autophagic response to oxidative stress in osteoarthritic chondrocytes is deregulated. Free Radic. Biol. Med. 2018, 126, 122–132. [Google Scholar] [CrossRef]
- Feng, L.; Feng, C.; Wang, C.X.; Xu, D.Y.; Chen, J.J.; Huang, J.F.; Tan, P.L.; Shen, J.M. Circulating microRNA let-7e is decreased in knee osteoarthritis, accompanied by elevated apoptosis and reduced autophagy. Int. J. Mol. Med. 2020, 45, 1464–1476. [Google Scholar] [CrossRef]
- Yorimitsu, T.; Klionsky, D.J. Autophagy: Molecular machinery for self-eating. Cell Death Differ. 2005, 12, 1542–1552. [Google Scholar] [CrossRef]
- Doherty, J.; Baehrecke, E.H. Life, death and autophagy. Nat. Cell Biol. 2018, 20, 1110–1117. [Google Scholar] [CrossRef]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Petiot, A.; Ogier-Denis, E.; Blommaart, E.F.; Meijer, A.J.; Codogno, P. Distinct Classes of Phosphatidylinositol 3′-Kinases Are Involved in Signaling Pathways That Control Macroautophagy in HT-29 Cells. J. Biol. Chem. 2000, 275, 992–998. [Google Scholar] [CrossRef]
- Mizushima, N.; Yamamoto, A.; Hatano, M.; Kobayashi, Y.; Kabeya, Y.; Suzuki, K.; Tokuhisa, T.; Ohsumi, Y.; Yoshimori, T. Dissection of Autophagosome Formation Using Apg5-Deficient Mouse Embryonic Stem Cells. J. Cell Biol. 2001, 152, 657–668. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Valenti, M.T.; Dalle Carbonare, L.; Zipeto, D.; Mottes, M. Control of the Autophagy Pathway in Osteoarthritis: Key Regulators, Therapeutic Targets and Therapeutic Strategies. Int. J. Mol. Sci. 2021, 22, 2700. [Google Scholar] [CrossRef]
- Duan, R.; Xie, H.; Liu, Z.Z. The Role of Autophagy in Osteoarthritis. Front. Cell Dev. Biol. 2020, 8, 608388. [Google Scholar] [CrossRef]
- Vasheghani, F.; Zhang, Y.; Li, Y.H.; Blati, M.; Fahmi, H.; Lussier, B.; Roughley, P.; Lagares, D.; Endisha, H.; Saffar, B.; et al. PPARγ deficiency results in severe, accelerated osteoarthritis associated with aberrant mTOR signalling in the articular cartilage. Ann. Rheum. Dis. 2015, 74, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Czaja, M.J. Autophagy in health and disease. 2. Regulation of lipid metabolism and storage by autophagy: Pathophysiological implications. Am. J. Physiol. Cell Physiol. 2010, 298, C973–C978. [Google Scholar] [CrossRef]
- Ward, C.; Martinez-Lopez, N.; Otten, E.G.; Carroll, B.; Maetzel, D.; Singh, R.; Sarkar, S.; Korolchuk, V.I. Autophagy, lipophagy and lysosomal lipid storage disorders. Biochim. Biophys. Acta 2016, 1861, 269–284. [Google Scholar] [CrossRef]
- Singh, R. Hypothalamic lipophagy and energetic balance. Aging 2011, 3, 934. [Google Scholar] [CrossRef][Green Version]
- Liu, K.; Czaja, M.J. Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ. 2013, 20, 3–11. [Google Scholar] [CrossRef]
- Shen, P.; Zhu, Y.; Zhu, L.; Weng, F.; Li, X.; Xu, Y. Oxidized low density lipoprotein facilitates tumor necrosis factor-α mediated chondrocyte death via autophagy pathway. Mol. Med. Rep. 2017, 16, 9449–9456. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hermonat, P.L.; Zhu, H.; Cao, M.; Mehta, J.L. LOX-1 transcription. Cardiovasc. Drugs Ther. 2011, 25, 393–400. [Google Scholar] [CrossRef]
- Feng, Y.; Cai, Z.R.; Tang, Y.; Hu, G.; Lu, J.; He, D.; Wang, S. TLR4/NF-κB signaling pathway-mediated and oxLDL-induced up-regulation of LOX-1, MCP-1, and VCAM-1 expressions in human umbilical vein endothelial cells. Genet. Mol. Res. 2014, 13, 680–695. [Google Scholar] [CrossRef] [PubMed]
- Barreto, J.; Karathanasis, S.K.; Remaley, A.; Sposito, A.C. Role of LOX-1 (Lectin-Like Oxidized Low-Density Lipoprotein Receptor 1) as a Cardiovascular Risk Predictor. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 153–166. [Google Scholar] [CrossRef]
- Simopoulou, T.; Malizos, K.N.; Tsezou, A. Lectin-like oxidized low density lipoprotein receptor 1 (LOX-1) expression in human articular chondrocytes. Clin. Exp. Rheumatol. 2007, 25, 605–612. [Google Scholar]
- Deng, Z.; Li, Y.; Liu, H.; Xiao, S.; Li, L.; Tian, J.; Cheng, C.; Zhang, G.; Zhang, F. The role of sirtuin 1 and its activator, resveratrol in osteoarthritis. Biosci. Rep. 2019, 39, BSR20190189. [Google Scholar] [CrossRef]
- Lee, S.H.; Lee, J.H.; Lee, H.Y.; Min, K.J. Sirtuin signaling in cellular senescence and aging. BMB Rep. 2019, 52, 24–34. [Google Scholar] [CrossRef]
- Dali-Youcef, N.; Lagouge, M.; Froelich, S.; Koehl, C.; Schoonjans, K.; Auwerx, J. Sirtuins: The ‘magnificent seven’, function, metabolism and longevity. Ann. Med. 2009, 39, 335–345. [Google Scholar] [CrossRef]
- Grabowska, W.; Sikora, E.; Bielak-Zmijewska, A. Sirtuins, a promising target in slowing down the ageing process. Biogerontology 2017, 18, 447–476. [Google Scholar] [CrossRef] [PubMed]
- Portal-Núñez, S.; Esbrit, P.; Alcaraz, M.J.; Largo, R. Oxidative stress, autophagy, epigenetic changes and regulation by miRNAs as potential therapeutic targets in osteoarthritis. Biochem. Pharmacol. 2016, 108, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Liao, F.X.; Huang, F.; Ma, W.G.; Qin, K.P.; Xu, P.F.; Wu, Y.F.; Wang, H.; Chang, Y.; Yin, Z.S. The New Role of Sirtuin1 in Human Osteoarthritis Chondrocytes by Regulating Autophagy. Cartilage 2019. Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yang, H.; Hu, B.; Zhang, M. Sirt1 regulates apoptosis and extracellular matrix degradation in resveratrol-treated osteoarthritis chondrocytes via the Wnt/β-catenin signaling pathways. Exp. Ther. Med. 2017, 14, 5057–5062. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.H.; Chiua, Y.C.; Wu, C.H.; Jou, I.M.; Tu, Y.K.; Hung, C.H.; Hsieh, P.L.; Tsai, K.L. Homocysteine causes dysfunction of chondrocytes and oxidative stress through repression of SIRT1/AMPK pathway: A possible link between hyperhomocysteinemia and osteoarthritis. Redox Biol. 2018, 15, 504–512. [Google Scholar] [CrossRef]
- Caramés, B.; Taniguchi, N.; Otsuki, S.; Blanco, F.J.; Lotz, M. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum. 2010, 62, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Wang, W.; Zhang, H.; Hu, Y.; Wang, M.; Yin, Z. The dual role of autophagy in chondrocyte responses in the pathogenesis of articular cartilage degeneration in osteoarthritis. Int. J. Mol. Med. 2013, 32, 1311–1318. [Google Scholar] [CrossRef]
- Wang, F.; Liu, J.; Chen, X.; Zheng, X.; Qu, N.; Zhang, B.; Xia, C. IL-1β receptor antagonist (IL-1Ra) combined with autophagy inducer (TAT-Beclin1) is an effective alternative for attenuating extracellular matrix degradation in rat and human osteoarthritis chondrocytes. Arthritis Res. Ther. 2019, 21, 171. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Mitter, S.K.; Qi, X.; Beli, E.; Rao, H.V.; Ding, J.; Ip, C.S.; Gu, H.; Akin, D.; Dunn, W.A., Jr.; et al. Oxidative stress-mediated NFκB phosphorylation upregulates p62/SQSTM1 and promotes retinal pigmented epithelial cell survival through increased autophagy. PLoS ONE 2017, 12, e017194. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, S.R.; Mizushima, N. Monitoring and Measuring Autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef]
- Ting, L.; Jing, Z.; Kangdi, L.; Lingnan, D.; Hongxiang, W. Combination of an Autophagy Inducer and an Autophagy Inhibitor: A Smarter Strategy Emerging in Cancer Therapy. Front. Pharmacol. 2020, 11, 408. [Google Scholar] [CrossRef]
- Sacitharan, P.K.; Bou-Gharios, G.; Edwards, J.R. SIRT1 directly activates autophagy in human chondrocytes. Cell Death Discov. 2020, 6, 41. [Google Scholar] [CrossRef]
- He, L.; Zhang, J.; Zhao, J.; Ma, N.; Kim, S.W.; Qiao, S.; Ma, X. Autophagy: The Last Defense against Cellular Nutritional Stress. Adv. Nutr. 2018, 9, 493–504. [Google Scholar] [CrossRef]
- Matsuzaki, T.; Matsushita, T.; Takayama, K.; Matsumoto, T.; Nishida, K.; Kuroda, R.; Kurosaka, M. Disruption of Sirt1 in chondrocytes causes accelerated progression of osteoarthritis under mechanical stress and during ageing in mice. Ann. Rheum. Dis. 2014, 73, 1397–1404. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, T.; Sasaki, H.; Takayama, K.; Ishida, K.; Matsumoto, T.; Kubo, S.; Matsuzaki, T.; Nishida, K.; Kurosaka, M.; Kuroda, R. The overexpression of SIRT1 inhibited osteoarthritic gene expression changes induced by interleukin-1β in human chondrocytes. J. Orthop. Res. 2013, 31, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Gagarina, V.; Gabay, O.; Dvir-Ginzberg, M.; Lee, E.J.; Brady, J.K.; Quon, M.J.; Hall, D.J. SirT1 enhances survival of human osteoarthritic chondrocytes by repressing protein tyrosine phosphatase 1B and activating the insulin-like growth factor receptor pathway. Arthritis Rheum. 2010, 62, 1383–1392. [Google Scholar] [CrossRef] [PubMed]
- Gabay, O.; Oppenhiemer, H.; Meir, H.; Zaal, K.; Sanchez, C.; Dvir-Ginzberg, M. Increased Apopototic Chondrocytes in Articular Cartilage of Adults Heterozygous SirT1 Mice. Ann. Rheum. Dis. 2012, 71, 613–616. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Liu-Bryan, R. Effect of nicotinamide riboside on cartilage matrix homeostasis. Osteoarthr. Cartil. 2019, 27, S148. [Google Scholar] [CrossRef]
- Oh, H.; Kwak, J.S.; Yang, S.; Gong, M.K.; Kim, J.H.; Rhee, J.; Kim, S.K.; Kim, H.E.; Ryu, J.H.; Chun, J.S. Reciprocal regulation by hypoxia-inducible factor-2α and the NAMPT-NAD(+)-SIRT axis in articular chondrocytes is involved in osteoarthritis. Osteoarthr. Cartil. 2015, 23, 2288–2296. [Google Scholar] [CrossRef]
- Kobayashi, H.; Terauchi, K.; Yui, N.; Yatabe, K. The Nicotinamide Adenine Dinucleotide (NAD)-Dependent Deacetylase Sirtuin-1 Regulates Chondrocyte Energy Metabolism through the Modulation of Adenosine Monophosphate-Activated Protein Kinase (AMPK) in Osteoarthritis (OA). J. Arthritis 2017, 6, 238. [Google Scholar] [CrossRef]
- Sawamura, T.; Kume, N.; Aoyama, T.; Moriwaki, H.; Hoshikawa, H.; Aiba, Y.; Tanaka, T.; Miwa, S.; Katsura, Y.; Kita, T.; et al. An endothelial receptor for oxidized low-density lipoprotein. Nature 1997, 386, 73–77. [Google Scholar] [CrossRef]
- Bierna-Zeinstra, S.M.A.; Waarsing, J.H. The role of atherosclerosis in osteoarthritis. Best Pract. Res. Clin. Rheumatol. 2017, 31, 613–633. [Google Scholar] [CrossRef]
- Parhami, F.; Tintut, Y.; Patel, J.K.; Mody, N.; Hemmat, A.; Demer, L.L. Regulation of vascular calcification in atherosclerosis. Z. Kardiol. 2001, 90, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Pirillo, A.; Norata, G.D.; Catapano, A.L. LOX-1, OxLDL, and atherosclerosis. Mediat. Inflamm. 2013, 2013, 152786. [Google Scholar] [CrossRef]
- Fujita, N.; Matsushita, T.; Ishida, K.; Kubo, S.; Matsumoto, T.; Takayama, K.; Kurosaka, M.; Kuroda, R. Potential involvement of SIRT1 in the pathogenesis of osteoarthritis through the modulation of chondrocyte gene expressions. J. Orthop. Res. 2011, 29, 511–515. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal. Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Salminen, A.; Hyttinen, J.M.T.; Kauppinen, A.; Kaarniranta, K. Context-dependent regulation of autophagy by IKK-NF- B signaling: Impact on the aging process. Int. J. Cell Biol. 2012, 2012, 849541. [Google Scholar] [CrossRef] [PubMed]
- Shoelson, S.E.; Lee, J.; Yuan, M. Inflammation and the IKKβ/IκB/NF-κB axis in obesity- and diet-induced insulin resistance. Int. J. Obes. 2003, 27, S49–S52. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Signaling to NF-kappaB. Genes Dev. 2004, 18, 2195–2224. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J. Ubiquitin signalling in the NF-kappaB pathway. Nat. Cell Biol. 2005, 7, 758–765. [Google Scholar] [CrossRef]
- Lin, Y.; Bai, L.; Chen, W.; Xu, S. The NF-κB activation pathways, emerging molecular targets for cancer prevention and therapy. Expert Opin. Ther. Targets 2010, 14, 45–55. [Google Scholar] [CrossRef]
- Chen, L.F.; Greene, W.C. Regulation of distinct biological activities of the NF-κB transcription factor complex by acetylation. J. Mol. Med. 2003, 81, 549–557. [Google Scholar] [CrossRef]
- Ghisays, F.; Brace, C.S.; Yackly, S.W.; Kwon, H.J.; Mills, K.F.; Kashentseva, E.; Dmitriev, I.P.; Curiel, D.T.; Imai, S.I.; Ellenberger, T. The N-Terminal Domain of SIRT1 Is a Positive Regulator of Endogenous SIRT1-Dependent Deacetylation and Transcriptional Outputs. Cell Rep. 2015, 10, 1665–1673. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, W.; Pan, H.; Feldser, H.G.; Lainez, E.; Miller, C.; Leung, S.; Zhong, Z.; Zhao, H.; Sweitzer, S.; et al. SIRT1 Activators Suppress Inflammatory Responses through Promotion of p65 Deacetylation and Inhibition of NF-κB Activity. PLoS ONE 2012, 7, e46364. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, T.; Sawamura, T.; Furutani, Y.; Matsuoka, R.; Yoshida, M.C.; Fujiwara, H.; Masaki, T. Structure and chromosomal assignment of the human lectin-like oxidized low-density-lipoprotein receptor-1 (LOX-1) gene. Biochem. J. 1999, 339, 177–184. [Google Scholar] [CrossRef]
- Kattoor, A.L.; Goel, A.; Mehta, J.L. LOX-1: Regulation, Signaling and Its Role in Atherosclerosis. Antioxidants 2019, 8, 218. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Zhang, W.; Cui, Z.M.; Cui, S.Y.; Fan, J.B.; Zhu, X.H.; Liu, W. Resveratrol alleviates the interleukin-1β-induced chondrocytes injury through the NF-κB signaling pathway. J. Orthop. Surg. Res. 2020, 15, 424. [Google Scholar] [CrossRef]
- Borriello, A.; Bencivenga, D.; Caldarelli, I.; Tramontano, A.; Borgia, A.; Zappia, V.; Della Ragione, F. Resveratrol: From basic studies to bedside. Cancer Treat. Res. 2014, 159, 167–184. [Google Scholar] [CrossRef]
- Qin, N.; Wei, L.; Li, W.; Yang, W.; Cai, L.; Qian, Z.; Wu, S. Local intra-articular injection of resveratrol delays cartilage degeneration in C57BL/6 mice by inducing autophagy via AMPK/mTOR pathway. J. Pharmacol. Sci. 2017, 134, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Cai, L.; Zhang, Y.; Cui, L.; Shen, G. Intra-articular resveratrol injection prevents osteoarthritis progression in a mouse model by activating SIRT1 and thereby silencing HIF-2α. J. Orthop. Res. 2015, 33, 1061–1070. [Google Scholar] [CrossRef]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef]
- Lizaso, A.; Tan, K.T.; Lee, Y.H. β-adrenergic receptor-stimulated lipolysis requires the RAB7-mediated autolysosomal lipid degradation. Autophagy 2013, 9, 1228–1243. [Google Scholar] [CrossRef]
- Schroeder, B.; Schulze, R.J.; Weller, S.G.; Sletten, A.C.; Casey, C.A.; McNiven, M.A. The Small GTPase Rab7 as a Central Regulator of Hepatocellular Lipophagy. Hepatology 2015, 61, 1896–1907. [Google Scholar] [CrossRef] [PubMed]
- Sathyanarayan, A.; Mashek, M.T.; Mashek, D.G. ATGL Promotes Autophagy/Lipophagy via SIRT1 to Control Hepatic Lipid Droplet Catabolism. Cell Rep. 2017, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Haemmerle, G.; Moustafa, T.; Woelkart, G.; Büttner, S.; Schmidt, A.; Van de Weijer, T.; Hesselink, M.; Jaeger, D.; Kienesberger, P.C.; Zierler, K.; et al. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-α and PGC-1. Nat. Med. 2011, 17, 1076–1085. [Google Scholar] [CrossRef]
- Khan, S.A.; Sathyanarayan, A.; Mashek, M.T.; Ong, K.T.; Wollaston-Hayden, E.E.; Mashek, D.G. ATGL-catalyzed lipolysis regulates SIRT1 to control PGC-1α/PPAR-α signaling. Diabetes 2015, 64, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Caramés, B.; Hasegawa, A.; Taniguchi, N.; Miyaki, S.; Blanco, F.J.; Lotz, M. Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann. Rheum. Dis. 2012, 71, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xu, C.; Johansen, T.; Berger, S.L.; Dou, Z. SIRT1—A new mammalian substrate of nuclear autophagy. Autophagy 2021, 17, 593–595. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wang, L.; Fozouni, P.; Evjen, G.; Chandra, V.; Jiang, J.; Lu, C.; Nicastri, M.; Bretz, C.; Winkler, J.D.; et al. SIRT1 is downregulated by autophagy in senescence and ageing. Nat. Cell Biol. 2020, 22, 1170–1179. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papageorgiou, A.-A.; Goutas, A.; Trachana, V.; Tsezou, A. Dual Role of SIRT1 in Autophagy and Lipid Metabolism Regulation in Osteoarthritic Chondrocytes. Medicina 2021, 57, 1203. https://doi.org/10.3390/medicina57111203
Papageorgiou A-A, Goutas A, Trachana V, Tsezou A. Dual Role of SIRT1 in Autophagy and Lipid Metabolism Regulation in Osteoarthritic Chondrocytes. Medicina. 2021; 57(11):1203. https://doi.org/10.3390/medicina57111203
Chicago/Turabian StylePapageorgiou, Aliki-Alexandra, Andreas Goutas, Varvara Trachana, and Aspasia Tsezou. 2021. "Dual Role of SIRT1 in Autophagy and Lipid Metabolism Regulation in Osteoarthritic Chondrocytes" Medicina 57, no. 11: 1203. https://doi.org/10.3390/medicina57111203
APA StylePapageorgiou, A.-A., Goutas, A., Trachana, V., & Tsezou, A. (2021). Dual Role of SIRT1 in Autophagy and Lipid Metabolism Regulation in Osteoarthritic Chondrocytes. Medicina, 57(11), 1203. https://doi.org/10.3390/medicina57111203