Abstract
One of the most common malignant tumors worldwide is lung cancer, and it is associated with the highest death rate among all cancers. Traditional treatment options for lung cancer include radiation, chemotherapy, targeted therapy, and surgical resection. However, the survival rate is low, and the outlook is still dreadfully dire. The pursuit of a paradigm change in treatment approaches is, therefore, imperative. Tyrosine kinases (TKs), a subclass of protein kinases, regulate vital cellular function by phosphorylating tyrosine residues in proteins. Mutations, overexpression, and autocrine paracrine stimulation can transform TKs into oncogenic drivers, causing cancer pathogenesis. Tyrosine kinase inhibitors (TKIs) have emerged as an attractive targeted therapy option, especially for non-small cell lung cancer (NSCLC). However, resistance to TKIs, and adverse cardiovascular effects such as heart failure, atrial fibrillation, hypertension, and sudden death, are among the most common adverse effects of TKIs. There is increasing interest in plant-derived natural products in the hunt for powerful chemosensitizer and pathway modulators for enhancing TKI activity and/or overcoming resistance mechanisms. This highlights the mechanism of TKs’ activation in cancer, the role of TKIs in NSCLC mechanisms, and the challenges posed by TKI-acquired resistance. Additionally, we explored various plant-derived natural products’ bioactive compounds with the chemosensitizer and pathway-modulating potential with TKs’ inhibitory and anticancer effects. Our review suggests that a combination of natural products with TKIs may provide a novel and promising strategy for overcoming resistance in lung cancer. In future, further preclinical and clinical studies are advised.
1. Introduction
Plants are essential sources of medications for treating a variety of diseases, and, since ancient times, people have used treatments based on medicinal plants to treat a wide range of diseases [1]. Numerous traditional and contemporary medical systems make use of various plant parts, such as stems, bark, leaves, flowers, seeds, and many more. Various plant extracts have been employed as fitness promoters in Saudi Arabia, India, China, the United States, Germany, and France in recent years. Out of the 300,000 plant species, only 15% have been processed to regulate their pharmacological characteristics, as was anticipated. Therefore, it is more encouraged to produce innovative medications using natural resources. Plants include a variety of vital compounds, including essential amino acids, lipids, vitamins, minerals, carbs, fiber, flavonoids, and other phenolic compounds [2]. Natural sources provide the model for modern synthesized drugs and drug-like chemicals. Numerous studies have been conducted on immunomodulators derived from natural sources to prevent diseases and modify immune function [3,4].
Cancer is a multifaceted disease characterized by the uncontrolled growth and spread of abnormal cells, which can invade surrounding tissues and metastasize to distant organs [5]. It remains a significant global health challenge, accounting for approximately 10 million deaths annually, making it one of the leading causes of mortality worldwide. Advances in understanding the molecular mechanisms underlying cancer have revealed its complexity, driven by genetic, epigenetic, and environmental factors. Recent research highlights the intricate interplay between cancer cells and the tumor microenvironment, including immune cells, stromal components, and extracellular matrix, which collectively influence tumor progression and response to therapy [6].
The formation of reactive oxygen and nitrogen species, which can directly cause DNA damage in epithelial cells, is a significant mechanism by which chronically activated neutrophils and macrophages directly contribute to the malignant conversion of epithelial cells [7,8]. DNA damage generated by external stimuli is a major element in the transformation of normal cells into cancer cells, and the cell response to DNA damage is an early event aimed at preventing carcinogenesis [9]. Therefore, DNA damage repair activity has a dual purpose in malignancies. Previous research has demonstrated that boosting DNA repair activity can suppress carcinogenesis or enhance apoptosis. However, it is unclear whether DNA repair activity plays a significant role in the promotion of cancer cells, and how medications with higher DNA repair activity affect cancer cells [10]. A feature of cancerous cells in culture is their reduced need for serum or other growth stimuli [11].
Current trends in cancer research emphasize early detection, personalized medicine, and novel therapeutic approaches. The development of liquid biopsies, which analyze circulating tumor DNA and other biomarkers, has revolutionized cancer diagnostics by enabling the non-invasive and real-time monitoring of disease progression. Additionally, targeted therapies, including immune checkpoint inhibitors and small-molecule inhibitors, have transformed treatment paradigms for several cancer types, offering improved outcomes for patients. The integration of artificial intelligence and machine learning into cancer genomics and drug discovery is further accelerating the development of precision oncology. These advancements, along with a deeper understanding of cancer biology, offer hope for better prevention, diagnosis, and treatment strategies in the fight against cancer [12].
Lung cancer is the greatest cause of cancer-related mortality worldwide, accounting for over 25% of all cancer deaths. Despite recent advancements in diagnosis and treatment, the five-year survival rate remains low. Lung cancer is widely classified into two types [13]. One of the key signaling enzymes in cell signal transduction is protein tyrosine kinase (TK), which catalyzes the transfer of ATP-γ-phosphate to the substrate protein’s tyrosine residues, phosphorylating it and controlling a number of physiological and biochemical processes, including cell growth, differentiation, and death. TK expression abnormalities are closely linked to tumor invasion, metastasis, and angiogenesis and typically result in disorders of cell proliferation. Many TKs are currently being used as targets in antitumor drug screening. Tyrosine kinase inhibitors (TKIs) decrease tyrosine kinase phosphorylation and compete with ATP for the ATP binding site of TK, which stops the growth of cancer cells [14].
The development, spread, invasion, and metastasis of cancers are all linked to aberrant TK activation brought on by mutations, translocations, or amplifications. Furthermore, in cancer, wild-type TKs may also serve as essential nodes for pathway activation. As such, tyrosine kinases have emerged as key targets for drug development. The purpose of a tyrosine kinase inhibitor (TKI) is to prevent the corresponding kinase from catalyzing phosphorylation [15].
One promising treatment strategy for non-small cell lung cancer (NSCLC) is targeted therapy targeting the epidermal growth factor receptor (EGFR). However, one of the biggest obstacles to its clinical management is still its resistance to EGFR-tyrosine kinase inhibitors (EGFR-TKIs) [16]. TKIs have changed the way that many solid cancers are treated and significantly increased patient survival and quality of life. In this paper, we summarized the roles of TKs in cancer, TKI treatment pathways, and TKI-acquired resistance mechanisms. We also go over a number of natural compounds that may interfere with TKs and how they work in tandem with chemotherapy to treat cancer.
2. Molecular Pathogenesis of Lung Cancer
Lung cancer remains the leading cause of cancer-related deaths globally [17,18], with a notably poor prognosis despite advancements in diagnostic tools and treatment options [17]. Lung cancer metastasis is the process by which the initial malignant tumor in the lung leaves the main site and spreads at a distance in a variety of ways. Metastasis occurs most commonly in the brain, bone, lymph nodes, and liver. Lung cancer metastasis is a highly complex process that involves the lung cancer microenvironment and lung cancer stem cells (LCSCs) as well as a variety of processes such as EMT development, angiogenesis, and lymphangiogenesis. These influencing factors are related to several non-coding RNAs (ncRNAs), associated factors of lung cancer metastasis, and multiple signaling pathways [19]. The disease is primarily categorized into two main types: non-small cell lung cancer (NSCLC), which accounts for approximately 85% of cases, and small cell lung cancer (SCLC), comprising the remaining 15%. The predominant risk factor for lung cancer is smoking; however, other factors such as exposure to environmental pollutants, including fine particulate matter, also contribute to its development [17,18].
Lung cancer develops due to a combination of genetic mutations and environmental factors, with smoking being the most significant cause, responsible for over 80% of cases. Cigarette smoke contains carcinogens that damage DNA and induce mutations in key oncogenes and tumor suppressor genes. Additionally, exposure to secondhand smoke, air pollution, and occupational carcinogens such as asbestos, arsenic, and radon gas significantly increases the risk of lung cancer. Genetic predisposition and familial history also play a role in susceptibility. Non-smokers may develop lung cancer due to environmental factors, chronic inflammation, or genetic alterations, including EGFR mutations and ALK rearrangements [17].
Lung cancer is broadly categorized into two main types, non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC), based on the cellular and molecular characteristics of the tumor (Figure 1). These classifications are essential for determining appropriate treatment strategies and understanding disease prognosis.
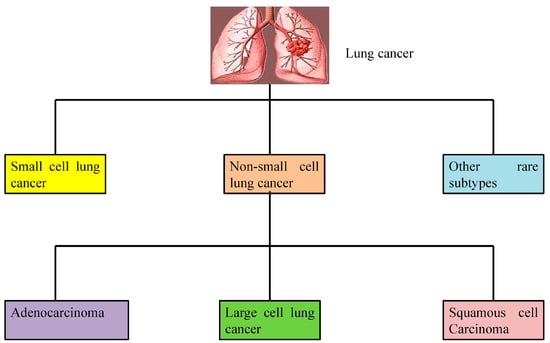
Figure 1.
Types of lung cancer. Lung cancer is broadly classified into three categories including small cell lung cancer, non-small cell lung cancer, and other rare subtypes. Non-small lung cancer is further divided into squamous cell carcinoma, large cell lung carcinoma, and adenocarcinoma.
2.1. Non-Small Cell Lung Cancer (NSCLC)
NSCLC carcinogenesis is a complex process comprising various causes, stages, and processes, similar to that of other malignant tumors. Avoidable and unavoidable risk factors are two further categories into which the etiology of NSCLC can be divided. Inhaled tobacco use is the most well-known preventable risk factor for non-small cell lung cancer. Alcohol consumption, secondhand smoke exposure, asbestos, radon, arsenic, chromium, nickel, ionizing radiation exposure, and polycyclic aromatic hydrocarbons are other causes of lung cancer. The tumor, node, metastasis (TNM) stage, the patient’s performance status, and any comorbidities all affect the prognosis of non-small cell lung cancer. Patients with subpar performance status have a lower chance of surviving. Weight loss and reduced appetite are other indicators of poor prognosis [18].
It has been demonstrated that squamous cell carcinoma (SCC), a subtype of NSCLC, has a stronger association with chronic obstructive pulmonary disease (COPD) (e.g., risk, lower overall survival, severity), as well as an increased risk of SCLC with the presence of COPD independent of smoking status. However, no biological relationship has been found [13].
NSCLC is the most common kind of lung cancer, accounting for 85% of all cases. NSCLC consists of lung adenocarcinoma, lung squamous cell carcinoma (LSCC), and lung giant cell carcinoma. Adenocarcinoma develops in the distal airway and is not associated with smoking. LSCC develops in the proximal airway and is more aggressive and strongly linked to smoking than adenocarcinoma. Large cell carcinoma develops in the distal airway, and the cancer cell mass is greater than in the other two forms of NSCLC. Large cell carcinoma is an aggressive tumor [20].
2.1.1. Adenocarcinoma
Adenocarcinoma is the most common form of NSCLC and is frequently diagnosed in non-smokers and younger individuals. It typically originates in the outer regions of the lung and arises from glandular cells that secrete mucus. Adenocarcinoma is often associated with genetic mutations such as EGFR (epidermal growth factor receptor), ALK (anaplastic lymphoma kinase), and KRAS (Kirsten rat sarcoma viral oncogene homolog), making it a target for precision therapies [21].
2.1.2. Squamous Cell Carcinoma (SCC)
Usually linked to smoking, this subtype develops in the central regions of the lungs, often near the bronchial tubes. It arises from the epithelial cells lining the airways and is characterized by a slower growth rate compared to other subtypes [22]. Squamous cell lung cancers frequently develop in the main airway, such as the left or right bronchus, or in the center of the lung. Smoking is the primary cause of cellular change. Smoking is linked to about 90% of lung cancer patients in women and 80% of cases in men. Smoking has a stronger correlation with SCC than any other kind of NSCLC. Age, family history, secondhand smoking exposure, and occupational exposure to minerals, metal particles, or asbestos are additional risk factors for lung squamous cell carcinoma [23].
2.1.3. Large Cell Carcinoma
This is the least common subtype of NSCLC and is known for its aggressive nature and rapid growth. It can appear in any part of the lung and often presents diagnostic challenges due to its lack of distinct features. The diagnosis of LCLC is primarily based on post-operative pathological examination rather than biopsy and cytological examination. It is known to be an undifferentiated non-small cell carcinoma because it lacks the cellular and structural characteristics associated with adenocarcinoma or squamous cell carcinoma. Additionally, there is evidence that LCLC is more common in older adults (>60), that it is often found in males and smokers, and that it frequently presents as a large mass with central necrosis [24].
2.2. Small Cell Lung Cancer (SCLC)
SCLC represents about 15% of lung cancer cases and is strongly associated with heavy smoking. It is characterized by small, round, or oval-shaped cells that grow rapidly and form large tumors. SCLC is highly aggressive, with a tendency to metastasize early to other organs, such as the brain, liver, and bones. Due to its rapid progression, SCLC is typically staged as limited-stage or extensive-stage disease, which guides treatment strategies [25]. Small cell lung cancer (SCLC) accounts for 13% of all incidences and is strongly linked to tobacco use. The specific molecular pathways underlying SCLC pathogenesis are currently unknown. However, significant genetic and molecular changes have been observed in SCLC, including the development of autocrine growth loops, proto-oncogene activation, and the deletion or inactivation of tumor suppressor genes [26].
Approximately 70% of patients with SCLC had distant metastases at the time of first diagnosis. At the same time, SCLC spread to a variety of metastatic locations, including the lungs, liver, brain, bone, adrenal glands, and lymph nodes. SCLC is a kind of extremely aggressive and invasive neuroendocrine carcinoma with poor differentiation and prognosis. The histocytological study of clinical samples obtained through needle biopsy and bronchoscopy is a regularly used procedure in clinical diagnosis. Tumor metastasis occurs in four stages: invasion, penetration into the circulatory system, crossing the brain barrier, and brain colonization. During tumor invasion, tumor cells penetrate the basement membrane and transform from an in situ to an invasive tumor. During the epithelial–mesenchymal transition (EMT) process, epithelial cells lose their adhesion capacity and gain the ability of mesenchymal cells to invade, metastasize, and resist drugs [27].
Autocrine growth factors, such as neuroendocrine-regulating peptides (for example, bombesin/gastrin-releasing peptide), are prevalent in SCLC. The Myc family’s dominant oncogenes are routinely overexpressed in both SCLC and NSCLC, although the K-RAS oncogene is never mutated in SCLC but is in 30% of NSCLC. The most common genetic disorders include tumor suppressor genes (TSGs). The TSG p53 is mutated in more than 90% of SCLCs and more than 50% of NSCLCs; the retinoblastoma TSG is inactivated in over 90% of SCLCs but only 15% of NSCLCs; and p16, the other component of the retinoblastoma/p16 pathway, is almost never abnormal in SCLC but inactivated in more than 50% of NSCLCs. The FHIT TSG is inactivated in 50–70% of lung cancer [28].
2.3. Other Rare Subtypes
In addition to NSCLC and SCLC, there are rare subtypes of lung cancer, including carcinoid tumors, which are slow-growing neuroendocrine tumors, and pleural mesothelioma, which arises from the lining of the lungs and is commonly linked to asbestos exposure [29].
3. Treatment Strategies for Lung Cancer
Recent advancements in treatment strategies have shown promise in improving patient outcomes. Understanding these types is critical for optimizing treatment approaches, including surgery, chemotherapy, targeted therapy, and immunotherapy. The treatment of lung cancer by these approaches, either solely or in a combination of these approaches, depends on the stage and type of lung cancer. Research continues to explore molecular subtypes and biomarkers for better classification and personalized treatment options [29]. In NSCLC, the integration of immunotherapies, particularly immune checkpoint inhibitors targeting the PD-1/PD-L1 pathway, has led to significant and sustained responses in patients without targetable oncogenic driver alterations. Additionally, the exploration of therapeutic cancer vaccines combined with immune checkpoint inhibitors is underway to enhance antitumor immunity. For SCLC, ongoing research into its molecular characteristics and immunologic microenvironment aims to identify novel therapeutic targets, potentially leading to more effective treatments in the future [30,31].
4. EGFR Signaling: Molecular Insights and Clinical Advances
Numerous growth factors and cytokines that alter the course and behavior of cancer cells are also secreted by tumor-associated myeloid cells. These include transforming growth factor β (TGF-β), Interleukin-6 (IL-6), and Interleukin-10 (IL10) and glycoprotein nonmetastatic melanoma protein B (GPNMB), hepatocyte growth factor (HGF), and epidermal growth factor (EGF) [32,33]. As a common mitogenic factor, epidermal growth factor (EGF) promotes the growth of various cell types, particularly fibroblasts and epithelial cells, by activating the EGF receptor (EGFR/ErbB), which, in turn, triggers intracellular signaling. EGF promotes the growth of numerous cell types in tissue culture, such as keratinocytes and transformed cells. A fraction of NSCLC was identified to have active mutations of the epidermal growth factor receptor (EGFR) gene, and tumors with EGFR mutations were extremely responsive to EGFR-TKI [34].
By attaching itself to its membrane receptor, i.e., EGFR, EGF produces mitogenic effects. This receptor can then control cell growth, proliferation, differentiation, and survival, among other biological processes. Since EGFRs are expressed in a range of human tissues, such as fibroblasts, endothelial cells, and the majority of epithelial tissues, EGF is essential for wound healing and tissue integrity [35].
EGFR plays a pivotal role in the pathogenesis of lung cancer, particularly in NSCLC. EGFR is a transmembrane tyrosine kinase receptor that regulates cell proliferation, survival, and differentiation through downstream signaling pathways such as phosphatidylinositol 3-kinase/protein kinase B (PI3K-AKT), mitogen-activated protein kinase (MAPK), and the Janus kinase/Signal transducer and activator of transcription (JAK-STAT). Mutations in the EGFR gene, commonly found in exons 19 and 21, are prevalent in a subset of NSCLC patients, especially in non-smokers and individuals of Asian descent. These activating mutations result in constitutive receptor signaling, driving oncogenesis and tumor progression [36].
Targeting EGFR mutations has revolutionized lung cancer treatment through the development of EGFR tyrosine kinase inhibitors (TKIs). First- and second-generation TKIs, such as erlotinib, gefitinib, and afatinib, have demonstrated significant efficacy in patients with EGFR-mutant NSCLC. However, resistance often emerges due to secondary mutations, such as T790M in exon 20. Third-generation TKIs, like osimertinib, have been developed to overcome these resistance mechanisms and have become the standard of care for patients with advanced EGFR-mutant NSCLC. Recent studies have also explored the role of EGFR in the tumor microenvironment and immune evasion, opening avenues for combination therapies with immune checkpoint inhibitors. Advances in liquid biopsy technologies further enhance the detection of EGFR mutations, enabling the real-time monitoring of treatment response and resistance development [37,38].
5. Targeting Tyrosine Kinases (TKs) in Cancer Therapy: Molecular Mechanisms and Drug Development Strategies
Numerous physiological responses, including growth, differentiation, proliferation, survival, apoptosis, migration, and cell cycle regulation, are drastically altered when kinase activity is deregulated [39]. TKs are a type of protein kinase that is important for hematopoiesis. They include discoidin domain receptor (DDRs), erythropoietin-producing human hepatocellular (Eph) receptors, proto-oncogene c-Src (SRC), spleen tyrosine kinase (SYK), Fms-like tyrosine kinase 3 (FLT3), Janus kinase (JAK), and others. The development of hematological malignancies has long been linked to the disruption of TK signaling [40]. TKs make up a significant percentage of all oncoproteins and are involved in the transformation of numerous malignancies. As a result, finding and creating therapeutic agents for conditions associated with aberrant TK activation brought on by increased expression, mutation, or autocrine stimulation that results in aberrant downstream oncogenic signaling has become a key focus for cancer treatment [41].
The development, spread, invasion, and metastasis of cancers are all linked to aberrant TK activation brought on by mutations, translocations, or amplifications. Furthermore, in cancer, wild-type tyrosine kinases may also serve as essential nodes for pathway activation. As such, tyrosine kinases have emerged as key targets for drug development. The purpose of a tyrosine kinase inhibitor (TKI) is to prevent the corresponding kinase from catalyzing phosphorylation [15].
TKs selectively phosphorylate tyrosine residues in various substrates. When ligands attach to the extracellular domain of receptor tyrosine kinases, they become active. Extracellular signaling chemicals called ligands, such as PDGF, EGF, and others, cause receptor dimerization, with the exception of the insulin receptor. The methods used by various ligands to attain the stable dimeric conformation vary. In certain conditions, two ligands bind simultaneously to two receptors (2:2 ligand–receptor complex), which offers the most straightforward mechanism of receptor dimerization (e.g., VEGF and VEGFR). In other situations, one ligand may bind with two receptor molecules to form a 1:2 ligand–receptor complex, such as growth hormone and growth hormone receptor [41].
There are 90 distinct TK genes in the human genome; 58 of them are receptor types, which are divided into 20 subfamilies, and 32 are non-receptor types, which are divided into 10 subfamilies [40]. The two main categories of tyrosine kinases are non-receptor tyrosine kinase (NRTK), which includes SRC, ABL, FAK, and Janus kinase, and receptor tyrosine kinase (RTK), which includes EGFR, PDGFR, FGFR, and the IR. In addition to being transmembrane receptors on the cell surface, the receptor tyrosine kinases are also enzymes with kinase activity. The receptor tyrosine kinase’s structural arrangement includes a cytoplasmic section with a TK domain, a single-pass transmembrane hydrophobic helix, and a multidomain extracellular ligand for expressing ligand specificity. Both the N and C terminal ends of the kinase domain contain regulatory sequences [41,42,43].
Understanding the role and importance of TKs in cancer was made possible by the discovery that the SRC oncogene possesses changing non-receptor tyrosine kinase (NRTK) activity and that protein–tyrosine kinase (PTK) activities are associated with viral transforming proteins [41,44]. Targeting TKs represents an important approach of oncology. Advances in molecular biology, structural bioinformatics, and next-generation sequencing can open new realms of rational drug designing [14].
6. TKs in Lung Cancer: Molecular Pathways and Clinical Implications
Phosphatases are enzymes that are implicated in the removal of a phosphate group from a protein. However, kinases are enzymes that transfer a phosphate group to a target molecule and cause the phosphorylation of proteins. By controlling many signaling pathways in response to external stimuli, these two enzymatic activities are essential to the cell [40]. One of the pivotal molecular mechanisms underlying NSCLC progression is the dysregulation of TK signaling pathways. In NSCLC, the aberrant activation of receptor and non-receptor TKs contributes to tumorigenesis, metastasis, and resistance to conventional therapies [36]. TKs implicated in NSCLC can be classified into two broad categories: receptor tyrosine kinases (RTKs) and non-receptor tyrosine kinases (NRTKs). RTKs, such as EGFR, anaplastic lymphoma kinase (ALK), and fibroblast growth factor receptor (FGFR), are often mutated or overexpressed in NSCLC [41].
NSCLC carcinogenesis is a complex process comprising various causes, stages, and processes, similar to that of other malignant tumors. Its primary pathomechanism is the activation of a proto-oncogene or the inactivation of an anti-oncogene in response to a variety of circumstances, including low levels or poor DNA repair ability [45]. In human malignancies, four major mechanisms contribute to constitutive RTK activation: gain-of-function mutations, genomic amplification, chromosomal rearrangements, and/or autocrine activation. When ligands are present, intermolecular dimerization happens, activating RTKs by phosphorylating the receptor’s C-terminal tail and activating kinases. Usually without a ligand, the mutations cause the constitutive activation of the RTK. Increased local receptor concentration is caused by the overexpression of RTKs, which frequently occurs as a result of the genomic amplification of the RTK gene [46].
Chromosomal rearrangements culminate in the creation of a hybrid fusion oncoprotein made up of the RTK and the fusion partner, a separate protein. RTKs are often activated by receptor-specific ligands. Growth factor ligands bind to extracellular areas of RTKs, activating the receptor via ligand-induced receptor dimerization and/or oligomerization [47]. Most RTKs’ structural alterations allow for the trans-autophosphorylation of each TKD and the release of cis-autoinhibition [48]. This conformational shift enables the TKD to adopt an active configuration. RTK autophosphorylation recruits and activates a diverse set of downstream signaling proteins with Src homology-2 (SH2) or phosphotyrosine-binding (PTB) domains. These domains bind to certain phosphotyrosine residues within the receptor and engage downstream mediators, propagating crucial physiological signaling pathways [49].
RTKs are activated by intermolecular dimerization in the presence of ligands, which causes kinase activation and the phosphorylation of the receptor’s C-terminal tail. The alterations cause the RTK to be activated constitutively, even when no ligand is present. The overexpression of RTKs, sometimes caused by the chromosomal amplification of the RTK gene, leads to an increase in the local concentration of receptors. Depending on where the chromosomal breakpoint is, these RTK fusion proteins might be either membrane-bound or cytoplasmic. In both cases, the kinase domain becomes activated. In the absence of ligands, the duplication of the tyrosine kinase domain may result in the formation of an intramolecular dimer, which would activate RTK. The enhanced local ligand concentration activates the RTK, leading to RTK dimerization, enhanced kinase activity, and the phosphorylation of the receptor’s C-terminal tail [46].
EGFR mutations, particularly in exons 19 and 21, are associated with oncogenic signaling and poor prognosis. These mutations lead to the constant activation of downstream pathways like RAS-RAF-MEK-ERK, which promote uncontrolled cell proliferation and survival. Similarly, ALK gene rearrangements and FGFR amplifications also contribute to tumor growth and invasion through the activation of PI3K-AKT-mTOR and MAPK pathways [41,50]. Many reports have shown that the interplay between cancer-related pathways and TK signaling pathways contributes to tumor progression and complicates treatment due to resistance mechanisms.
In addition to RTKs, non-receptor tyrosine kinases, such as Src family kinases and Janus kinases (JAKs), play a significant role in NSCLC. The aberrant activity of these kinases is implicated in enhancing cell motility, invasion, and resistance to apoptosis. Src family kinases, in particular, are known to promote EMT and increase metastatic potential, making them attractive targets for therapeutic interventions [51]. Beyond their involvement in intracellular signaling pathways, TKs significantly influence the tumor microenvironment by modulating angiogenesis, immune cell infiltration, and inflammatory responses. In lung cancer, VEFGR-mediated signaling induces tumor vascularization and metastasis. Additionally, TKs have been reported for their ability to upregulate immune checkpoint molecules, suppress antitumor activity, and promote immune evasion, which contributes to the resistance towards treatment. Advanced techniques including liquid biopsy and NGS have been known for their real-time detection of TK alteration, leading to improvement in patient stratification and treatment monitoring and the early identification of resistance [41,46,50].
7. Advances in Targeted Therapy for Lung Cancer: Tyrosine Kinase Inhibitors (TKIs) and Resistance Pathways
Malignancy can result from the mutations, overexpression, and autocrine paracrine stimulation of TKs, even if their activity is strictly controlled in healthy cells. Because selective TKIs can prevent constitutive oncogenic activation in cancer cells, they are seen as a promising strategy for novel genome-based therapies. The recognition of TK dysregulation as a key driver in NSCLC has led to the development of tyrosine kinase inhibitors (TKIs), which have significantly improved patient outcomes, particularly in cases with EGFR mutations. TKIs have been reported to be more successful in managing NSCLC, and its application in other types of lung cancer, such as SCLC, is more explored. However, progress in SCLC is limited. TKIs are classified into various categories on the basis of their generation (first, second, or third), mode of action (competitive, allosteric, or multi-targeted), and target (receptor or non-receptor). First-generation EGFR inhibitors like gefitinib and erlotinib, and second-generation inhibitors such as afatinib, target the kinase domain of EGFR, preventing its activation and downstream signaling. However, resistance to these therapies often develops due to secondary mutations, such as the T790M mutation, which limits the efficacy of these first-line agents [52].
To overcome this challenge, third-generation TKIs like osimertinib have been developed to specifically target EGFR mutations, including the T790M mutation. These inhibitors offer prolonged progression-free survival and are currently considered the standard of care for EGFR-mutant NSCLC. Similarly, ALK inhibitors, including crizotinib, alectinib, and brigatinib, have shown efficacy in patients with ALK-rearranged NSCLC, providing a significant survival benefit [53]. Figure 2 shows the classification of TKIs into first-, second-, and third-generation inhibitors (Figure 2).
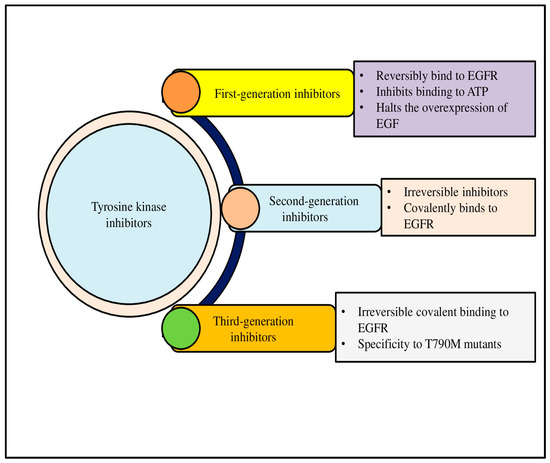
Figure 2.
Tyrosine kinases (TKs) are crucial signaling proteins in cells that have a range of biological functions, such as promoting cell migration and proliferation. One systemic cancer therapeutic approach is the inhibition of angiogenic TKs. Based on generation, three kinds of antiangiogenic Tyrosine Kinase Inhibitors have been approved for use in patient therapy.
Despite these advancements, resistance to TKIs remains a major hurdle. Mechanisms such as the acquisition of secondary mutations in the kinase domain, activation of bypass signaling pathways, and epithelial-to-mesenchymal transition contribute to treatment failure. To address this, combination therapies that target multiple signaling pathways or incorporate immune checkpoint inhibitors are being explored in clinical trials [54].
TKs are pivotal enzymes involved in cellular signaling processes that regulate cell growth, differentiation, survival, and metabolism. The dysregulation of TK activity is commonly associated with various cancers, where the aberrant activation of these kinases contributes to oncogenesis, tumor progression, and metastasis. Targeting TKs in malignant cells can disrupt the cell signaling pathways implicated in tumor growth [41]. A promising therapeutic approach for NSCLC involves targeted therapy targeting EGFR with precision therapies [16].
8. Mechanisms of Action and Molecular Targets of TKIs
Given their role in driving tumor progression, aberrant TKs have become central therapeutic targets. The purpose of a TKI is to prevent the corresponding kinase from catalyzing phosphorylation [15]. By targeting aberrant TKs, TKIs block phosphorylation, disrupt signaling, and induce apoptosis in malignant cells. Over the past few decades, TKIs have emerged as a cornerstone in the treatment of several malignancies, offering targeted therapeutic strategies with the potential for better specificity and fewer side effects compared to conventional chemotherapy [41]. TKIs can be bivalent, allosteric, or ATP-competitive [55].
TKIs inhibit TK activity by competitively binding to the ATP binding site. They can also influence activity by binding to other sites, which may block access to the Cdc37-Hsp90 chaperone system and, ultimately, inhibit downstream signaling pathways such as PI3K/Akt and Raf/MeK/Erk. TKIs have the ability to contend with ATP for tyrosine kinases’ ATP binding site. This stops signal transduction pathways from becoming phosphorylated and activated. TKIs can also attach to the human epidermal growth factor receptor (EGFR) kinase area. TKIs also exert their effects by binding to allosteric sites. Therefore, they disrupt the access to chaperone systems including Cdc37-HSP90 and, thereby, they interfere with kinase activity and function. In brief, TKs rely on the molecular chaperone system for cellular stability, but TKIs can prevent them from accessing it [14].
RTKs, such as EGFR, human epidermal growth factor receptor 2 (HER2), and VEGFR, are frequently mutated or overexpressed in various kinds of cancer, leading to uncontrolled signaling and tumor growth. In contrast, NRTKs, such as Src family kinases and Janus kinases (JAKs), are known to be implicated in cancer cell survival, migration, and invasion [56]. TKIs are designed to inhibit the kinase activity of these dysregulated TKs, preventing the phosphorylation of tyrosine residues on substrate proteins and, thus, halting the downstream signaling pathways that drive cancer cell proliferation. From this point of view, TKIs can be broadly categorized into small molecules and monoclonal antibodies. Small-molecule TKIs, such as imatinib, gefitinib, and erlotinib, typically target the intracellular kinase domains of RTKs, while monoclonal antibodies, such as trastuzumab and cetuximab, bind to the extracellular ligand-binding domains of RTKs, preventing receptor activation [57].
EGFR is a transmembrane glycoprotein which comprises an extracellular ligand-binding domain and an intracellular tyrosine kinase domain that modulates the main signaling pathways regulating cell growth and survival. Oncogenic mutations in EGFR are frequently associated with NSCLC and are responsible for aberrant cellular proliferation. When the EGFR binds to its ligand, intrinsic tyrosine/kinase activity causes autophosphorylation, which sets off a number of signal transduction cascades. The persistent activation of certain downstream target sequences is believed to contribute to more aggressive tumor behaviors. Mutations in epidermal growth factor receptor are commonly associated with various lung malignancies. Tyrosine kinase inhibitors significantly increase the responsiveness of lung adenocarcinomas with mutant epidermal growth factor receptors, but they do not seem to improve survival for unselected individuals [58].
9. Therapeutic Role of TKIs Across Cancer Types Including Lung Cancer
Several TKIs have demonstrated significant efficacy in clinical settings, particularly in cancers driven by specific mutations or the overexpression of TKs. Imatinib, the first TKI approved for clinical use, targets the BCR-ABL fusion protein in chronic myelogenous leukemia (CML), providing a revolutionary treatment option for CML patients with Philadelphia chromosome-positive tumors. Other TKIs, such as gefitinib and erlotinib, target EGFR mutations in NSCLC, leading to improved progression-free survival and overall response rates in patients harboring sensitive EGFR mutations [59].
In breast cancer, trastuzumab targets HER2-positive tumors, offering a significant survival benefit for patients with HER2 amplification. Other HER2-targeting TKIs, such as lapatinib, provide an option for patients who develop resistance to trastuzumab. Similarly, TKIs targeting vascular endothelial growth factor receptor (VEGFR), such as sorafenib and sunitinib, are used to treat renal cell carcinoma and hepatocellular carcinoma by inhibiting angiogenesis, a process essential for tumor growth and metastasis [60].
It is believed that more aggressive tumor behaviors result from the constitutive or persistent activation of certain downstream target sequences. Mutations in EGFR have been reported in relation to various lung malignancies. TKIs significantly increase the responsiveness of lung adenocarcinomas with mutant EGFR, but they do not seem to improve survival for unselected individuals [58].
Recent efforts have been made to evaluate aberrant kinases including FGFR1, AXL, and IGF-1R as potential therapeutic targets for the treatment of SCLC [43,45]. In NSCLC, multiple oncogenic drivers such as EGFR, AK, ROS1, MET, and BRAF have led to the approval of various TKIs with improved survival and disease control [44,46]. Targeted therapy against EGFR has been considered to be a promising treatment option for NSCLC, especially in patients with EGFR mutations. Moreover, their clinical benefits have a significant limitation on the eventual development of resistance. This resistance is a significant challenge [16]. Numerous studies have demonstrated that blocking TKs in cancerous cells with monoclonal antibodies, interfering RNAs, and/or tiny kinase inhibitors reduces cell survival and proliferation, causing growth arrest and apoptosis [61]. Clinical trials linked to various TKIs are provided in Table 1.

Table 1.
FDA-approved TKIs with their clinical trial numbers and outcomes of these trials are provided.
10. Challenges for Implicating TKIs in Lung Cancer: Sensitivity and Resistance
Drug resistance can be classified as intrinsic (primary) or acquired (secondary) resistance to antiangiogenic targeted therapies. Intrinsic resistance occurs when cancer cells are naturally insensitive to TKIs and do not respond to the drug, whereas acquired resistance occurs when cancer cells respond to TKI treatment initially but subsequently relapse as the drug loses efficacy over time due to the acquisition of various resistance mechanisms [67]. In aberrantly active EGFR, through mechanisms comparable to wild-type receptors, mutant EGFRs (caused by the deletion of exon 19 or punctual mutation in exon 21 known as L858R) exhibit higher levels and durations of EGFR activation. RAS/RAF/MEK/MAPK, phosphoinositide 3-kinase (PI3K)/AKT, and STAT3/STAT5 pathways can all be activated by mutated EGFR (Figure 3a) [68].
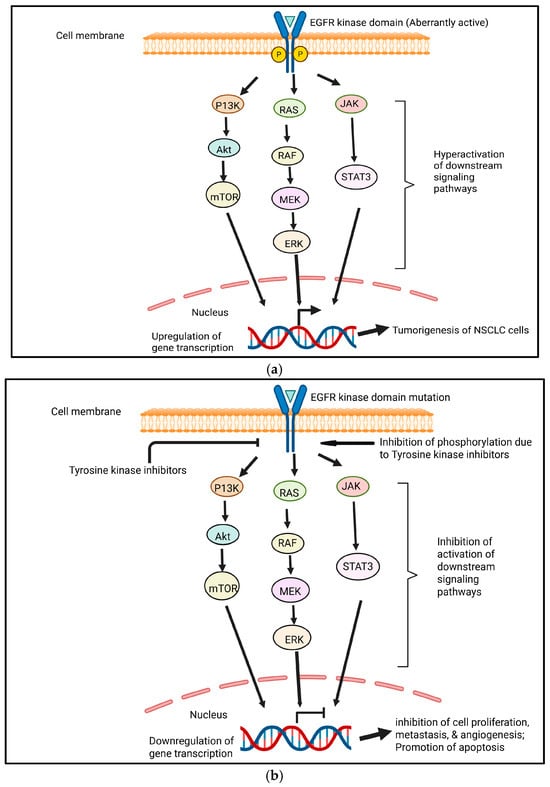
Figure 3.
(a) EGFR pathways in NSCLC tumorigenesis. Aberrantly active EGFR leads to the continuous activation of signaling pathways, which results in the tumorigenesis of NSCLC cells. Created in https://BioRender.com. (b) The general inhibition of EGFR pathways due to tyrosine kinase inhibitors. Small-molecule tyrosine kinase inhibitors inhibit phosphorylation, resulting in the inhibition of the activation of downstream signaling pathways. This leads to the downregulation of genes involved in tumorigenesis. Cell proliferation, metastasis, and angiogenesis become inhibited. However, apoptosis becomes promoted. Created in https://BioRender.com.
Because TKIs target important pathways involved in cancer proliferation, survival, and metastasis, they have greatly improved the prognosis for patients with advanced NSCL [69]. EGFR TKIs have demonstrated efficacy in managing EGFR-mutated lung cancer after several years of usage [68] (Figure 3b). On the other hand, resistance and subsequent disease progression are nearly always associated with VEGF-targeted TKI treatment. The immunosuppressive nature of the tumor microenvironment in lung cancer often prevents the efficacy of TKIs and also promotes resistance by interfering with signaling pathways. The various mechanisms that underlie TKI resistance, such as the upregulation of alternative proangiogenic pathways, EMT, efflux pumps that lower intracellular drug concentrations, lysosomal sequestration, changes in the tumor microenvironment, and genetic factors, make it difficult to mitigate drug resistance [67].
Lung cancer, particularly NSCLC, is characterized by a considerable frequency of heterogeneity, and this heterogeneity presents a significant obstacle to the uniform efficacy of TKIs. An excellent illustration is the well-known targeted treatment for NSCLC known as EGFR-TKI, which blocks overexpressed EGFR. The identification of EGFR-activating mutations marked a mile stone in the development of EGFR-TKIs, as these mutations were found to predict differential treatment response [70]. Regretfully, after a median of 9 to 14 months, resistance has been observed in EGFR-mutant patients on EGFR-TKI therapy [71,72]. In NSCLC, acquired resistance against first-generation EGFR-TKIs has been primarily linked to the acquisition of T790M mutation, which alters the kinase domain and inhibits drug-binding affinity [70]. The histological transformation from NSCLC to the SCLC phenotype has been reported to be an important mechanism of resistance, which makes therapeutic approaches more complicated.
Although TKIs show an effective initial response, their long-term potential is often limited due to the development of resistance, which is a significant challenge. Resistance to TKIs can occur through several mechanisms, including the development of secondary mutations in the kinase domain, activation of alternative signaling pathways, and upregulation of drug efflux pumps. For example, in NSCLC, resistance to first-generation EGFR TKIs is often associated with the acquisition of the T790M mutation, which alters the binding affinity of these inhibitors. In addition, intratumoral heterogeneity and clonal evaluation may promote the outgrowth of resistant subclonal populations characterized by additional oncogenic alterations beyond EGFR, such as MET amplification and HER2 mutations. To overcome resistance, third-generation EGFR TKIs, such as osimertinib, have been developed to specifically target the T790M mutation and exhibit improved clinical outcomes [73].
In addition, combination therapies that pair TKIs with other therapeutic modalities, such as immune checkpoint inhibitors or chemotherapy, are being actively explored to overcome resistance and improve treatment efficacy. Clinical trials are also investigating the potential of novel TKIs targeting less explored tyrosine kinases and the development of personalized medicine approaches to tailor TKI treatment based on specific molecular profiles [74]. Since resistance mechanisms in advanced lung cancer are dynamic and adaptive, ongoing molecular monitoring is necessary to modify therapeutic approaches in real time.
11. Expanding Therapeutic Landscapes of Kinases: Complexities and Challenges in Designing Novel TKIs
In addition to their established role in cancer, protein kinases have been implicated in a wide range of diseases, including as immunological disorders, skeletal and craniosynostosis disorders, hematological and vascular disorders, neurological disorders, multiorgan disorders, and endocrine and metabolic disorders. Many of these diseases are caused by mutations in members of the kinase gene family. As a result, TKIs now are recognized as valuable therapeutic agents outside of oncology. For example, tofacitinib is the first TKI approved for the treatment of inflammatory disorders [75].
Protein kinases have, thus, emerged as important pharmacological targets. Currently, there are approximately 250 kinase therapeutic candidates under clinical investigation, and 37 small TKIs have been approved for clinical use globally. A study examined the target spectrum of 243 clinically tested kinase drugs using chemical proteomics and provided a comprehensive data resource outlining the target landscape of 243 clinically tested KIs [76].
The landscape of TKIs is evolving rapidly, highlighting the need for continued research to discover new targets and develop TKIs capable of treating a broader range of solid tumors. However, there are several challenges such as enhanced structural diversity, improving kinase selectivity, and minimizing the off-toxicity profiles of existing kinase inhibitors [77].
Moreover, with increasing resistance mutations, there is a growing interest in the development of fourth-generation TKIs that exploit novel mechanisms of action. The successful establishment of such TKIs may require the exploration of unique binding modes, rigorous clinical validation, and the identification of appropriate patient populations [78]. Expanding the clinical applications of TKIs into non-oncology domains and establishing the effective combination approaches for cancer treatments require specifically designed TKIs. This undoubtedly presents a demand for more advanced research in medicinal chemistry, structural biology, and rational drug designing to ensure the discovery of safe, selective, and efficacious TKIs [75].
12. Plant-Derived Natural Products in Cancer Therapy: Modulating Apoptosis, Cell Signaling, and Chemo-Resistance
Numerous natural compounds with a wide range of structures are produced by plants. In contrast to the “primary metabolites,” which are necessary for the growth and development of plants, these products are often referred to as “secondary metabolites”; these natural compounds play crucial roles in how plants interact with their biotic and abiotic surroundings. For instance, they can function as hormones or signal molecules, floral colors that draw pollinators, or defensive substances against infections and herbivores. Natural products have a significant cultural influence in addition to their physiological role in plants. Throughout human history, they have been utilized as pharmaceuticals, condiments, and pigments [79].
As a rich source of therapeutically relevant biomolecules for the creation of new medications, natural products and secondary metabolites have already been mentioned. Obesity, diabetes, and cardiovascular disease are just a few of the chronic diseases that are greatly increased by poor eating. Chronic disease will most likely be exacerbated by food deficits resulting from an inadequate diet. Diets heavy in sodium and low in whole grains, fruit, vegetables, nuts, and seeds have been linked to higher death rates [80].
When compared to synthetic compounds, naturally occurring phytochemicals and products have been determined to be somewhat safe for human consumption. They are also reasonably non-toxic, affordable, and available in an ingestible form [81]. Over centuries, numerous plants and their products have been incorporated into traditional remedies, many of which have inspired modern pharmaceuticals [82]. It has previously been mentioned that plant-derived natural products and secondary metabolites are recognized as important sources of pharmacological compounds for drug development. New medications have been transformed by antibiotics (like penicillin, tetracycline, and erythromycin), antiparasitics (like avermectin), antimalarials (like quinine and artemisinin), lipid control agents (like lovastatin and analogs), immuno-suppressants for organ transplants (like cyclosporin and rapamycins), and anticancer medications (like doxorubicin and paclitaxel) [5].
A significant portion of our daily diet consists of fruits and vegetables, which are rich in polyphenolic chemicals with beneficial biological and pharmacological properties [83]. Because they have fewer adverse effects and can help reduce resistance to cancer treatment, medicinal plants or their bioactive constituents are dynamic sources of medications [84].
One of the leading causes of mortality worldwide is cancer. Current treatment methods, such as chemotherapy and radiation therapy, have a number of negative health impacts on individuals. According to this perspective, the bioactive component of natural products is essential for managing disease since it inhibits and activates biological processes like inflammation, oxidative stress, and cell signaling molecules. Natural products can be useful adjuvants or a kind of supporting therapy, but they are not a replacement for medications [85].
Because of their great effectiveness and minimal toxicity, natural products are gaining more and more attention. Numerous active components derived from herbs are frequently employed as antitumor medicines, which can increase anticancer efficacy and lessen adverse effects [86]. The US Food and Drug Administration (FDA) has licensed numerous medications derived from plants for use in cancer therapy, including taxanes like paclitaxel and vinca alkaloids like vinblastine. Dietary supplements are another type of natural product that is commonly used by cancer patients, but they lack the FDA-reviewed evidence of safety and efficacy—whether linked to survival, palliation, symptom reduction, and/or immune system enhancement—that is required for therapeutic approval. Nearly half of cancer patients in the United States report that they began using new dietary supplements after receiving their diagnosis. Oncologists face challenges when advising patients on which supplements are safe and effective to use to treat cancer or the adverse effects of cancer therapy [87].
In lung cancer specifically, several natural compounds have shown promise and potential antitumor activity with a favorable safety profile, while the combinatorial action of an anticancer drug with a natural compound provides synergistic action which helps boost the overall therapeutic action against cancer cells. In cancer, there is a dysregulation of apoptosis that facilitates the survival of the cancer cell, resulting in the progression of cancer. Many cancer drugs cause mutations of genes that regulate cancer and should kill the cancer cell but lead to chemoresistance. Many bioactive natural molecules modulate cancer-related cell signaling pathways, restore apoptosis, and exert cytotoxic effects in the target tumor cells. The importance of these compounds is emerging in many therapies developed with dual action, often including a natural compound [88].
13. Targeting TKI-Resistant Lung Cancer: Therapeutic Promise of Plant-Derived Natural Products
According to a recent review, 67 recognized natural compounds have the ability to fight cancer’s resistance to EGFR-TKIs through at least 30 pathways, primarily ROS, PD-L1, EGFR, MAPK, mTOR, HSP90, JNK, PTEN, and FOXO [86]. Additionally, various previous studies have documented the role of natural products such as TKIs against multiple kinds of cancers (Table 2). EGFR-TKI resistance is one of the biggest issues in cancer treatment, especially in NSCLC. Even though several studies, both in preclinical and clinical trials, have shown the promising therapeutic effects of polyphenolic compounds in anticancer therapy, the function of the natural compounds in TKI-resistant (TKIR) lung cancer remains poorly studied. Polyphenolic substances, including equol, kaempferol, resveratrol, ellagic acid, p-Coumaric acid, hesperidin, and gallic acid, were significantly reported to reduce cancer growth in TKIR cell H1993 while preserving TKIS cell H2073. When combined, our research offers a basic yet significant screening of possible natural chemicals for the development of anticancer drugs, particularly to overcome TKI resistance in advanced lung cancer. As a result, giving polyphenolic substances to patients with lung cancer may be a powerful adjuvant therapeutic approach that supports long-term TKI treatment [89].
Table 2.
The efficacy of natural products such as tyrosine kinase inhibitors (TKIs) against a variety of malignancies has been shown in multiple studies, underscoring their promise for cancer treatment.
The therapeutic benefits of EGFR-TKIs are enhanced by bioactive substances found in natural products such as alkaloids, saponins, terpenoids, polyphenols, resins, nucleosides, and quinones with significant tyrosine kinase inhibitory activity. By blocking the phosphorylation process and interfering with the signal transmission in the pathway, they can prevent the excessive activation of tyrosine kinase and cure cancer by competing with the ligand and ATP for binding sites on the kinase. Curcumin, resveratrol, ginsenosides, astragaloside IV, cucurbitacin D and cucurbitacin B, apigenin, quercetin, betulinic acid, β-elemene, licochalcone, sulforaphane, EGCG, and shikonin are among the natural chemicals that have significant glycolysis-inhibiting properties. These substances target a variety of glycolytic components and offer promising treatment options for EGFR-TKI resistance and cancer cell growth inhibition.
13.1. Alkaloids
13.1.1. Capsaicin
Capsaicin is a bioactive alkaloid and is found in Capiscum annum L. Capsaicin exhibits significant antimetastatic effects in human fibrosarcoma (HT-1080 cells) by targeting EGFR-dependent signaling pathways. It inhibits the EGF-induced activation of MMP-2 and MMP-9, which are implicated in extracellular remodeling and tumor invasion [90]. Mechanistically, capsaicin suppresses EGFR-mediated downstream signaling cascades, including FAK/AkT, PKC/Raf/ERK, and p38 MAPK pathways, as well as AP-1 transcriptional activity, leading to a marked reduction in MMP-9 expression and tumor cell migration [91].
13.1.2. Oxymatrine
Oxymatrine is derived from Sophora flavescens and it exhibits potent anticancer activities against various malignancies. In human malignant glioma (U251MG) cells, it was found to inhibit cell growth, induce the arrest of the cell cycle at the G0/G1 phase, and suppress the expression of key cell cycle regulatory proteins, thereby hindering tumor proliferation [92]. In gastric cancer cells, oxymatrine decreased the proliferation and invasion of gastric cells by inhibiting the EGFR/Cyclin D1/CDK4/6, EGFR/Akt, and MEK-1/ERK1/2/MMP2 pathways by inhibiting EGFRp-Tyr845. This results in reduced cancer cell proliferation and invasion, underscoring oxymatrine’s potential as an EGFR-targeted therapeutic agent [93].
13.1.3. Tatrandrine
Tatrandrine is an alkaloid and is obtained from Stephania tetrandra S. Moore. Tatrandrine has demonstrated promising anticancer activities. In human colorectal adenocarcinoma (HT29), it effectively inhibits the phosphorylation of EGFR and its downstream signaling pathways, thereby suppressing tumor cell growth and survival mechanisms [94].
13.2. Flavonoids
13.2.1. Apigenin
Apigenin, an active component of many Chinese medicinal herbs, has been widely studied for its anticancer properties and underlying mechanisms of action. Under in vivo conditions, apigenin is often conjugated to a glycoside in its native state, and apigenin was categorized as a class II medicine under the Biopharmaceutical Classification System [129]. Traditionally used in herbal medicines, apigenin has gained attention for its broad pharmacological activities. Among flavonoids, apigenin is recognized for its antioxidant, anti-proliferative, and carcinogenic effects. In addition to such benefits, the tumor-suppressing capability of apigenin has been documented in both historical and contemporary studies. Apigenin exhibits antitumor activity towards a variety of cancerous tumors using both in vitro cell lines and in vivo mice models [84].
Apigenin, which is technically known as 4′,5,7-trihydroxyflavone, is a member of the flavone family and is widely available in fruits, vegetables, and drinks. The foods rich in apigenin include celery, parsley, artichokes, and oregano, as well as beverages like red wine and beer. The dried flowers of Matricaria recutita L. are used to make chamomile tea, which is a rich source of apigenin. The chamomile flower head contains around 16.8% apigenin [130]. Because of its antioxidant and anti-inflammatory activities [131,132], its ability to lower blood pressure [133], and its antibacterial and antiviral properties [134], apigenin has been considered a multifunctional therapeutic agent.
Apigenin and cetuximab have been shown to inhibit the expression of p-STAT3, p-Akt, p-EGFR, and Cyclin D1 [135]. Apigenin and gefitinib’s combined effects on non-small cell lung cancer with a mutated epidermal growth factor receptor (EGFR) were assessed. Apigenin and gefitinib were found to inhibit several oncogenic drivers, including HIF-1α, EGFR, and c-Myc, and to decrease the expression of the MCT1 and Gluts proteins. As a result, treating lung cancer with apigenin and gefitinib together offers an appealing alternative therapeutic option for acquired resistance to epidermal growth factor receptor TKIs [136].
Apigenin can potentiate the antitumor effect of chemotherapeutic agents and/or alleviate the side effects of many anticancer agents. Due to the fact that TKIs are mostly metabolized by CYP3A4 enzymes and that apigenin could alter enzymatic activity, potential drug interactions could be expected following their co-administration [137]. Apigenin and kaempferol have been shown to inhibit EGFR, HER2, and MEK1, potentially contributing to the systemic prevention of metastatic colorectal cancer (mCRC). Furthermore, kaempferol and apigenin both exhibited dose-dependent antiangiogenic effects [138]. By specifically targeting RTKs, apigenin can influence lung cancer cell cycle progression, apoptosis, and EMT, highlighting the importance of flavonoid-mediated RTK inhibition [139,140]. Furthermore, it has been discovered that apigenin inhibits VEGF expression in human umbilical artery endothelial cells (HUA-EC) via HIF-1α. It was demonstrated that apigenin inhibits VEGF expression by degrading HIF-1α and interfering with Hsp90 activity [141]. Apigenin suppressed Akt and p70S6K1 activation, which may have been a consequence of VEGF suppression. Additionally, it has been demonstrated that apigenin in particular lowers the quantity of HIF bound to p300 and HIF-1α expression in lung cancer cells (NCI-H157) [142].
13.2.2. Baicalein
The Src family of kinases includes the widely expressed non-receptor TKs (Src tyrosine kinases). Although Src family kinases are well known for their functions in the development of cancer, they also play a part in chemotaxis, proliferation, and signaling pathways linked to inflammation. Following the activation of multiple receptor types, Src is essential for attracting a variety of cell signaling molecules, which causes the production of several cytokines, including IL-6 [143]. There have been reports of baicalein’s many biological effects. In addition to exhibiting antiviral and antitumor actions, it is well known for its anti-inflammatory, antipyretic, and antihypersensitivity qualities. Human gastric, colon, hepatoma, pancreatic, and prostate cancer cells have all been shown to undergo apoptosis when exposed to baicalein. It has also been demonstrated to target metastasis and tumor angiogenesis [144].
Baicalein reduces the growth of tumor cells in non-small cell lung cancer (NSCLC) via inducing apoptosis, according to research by Leung and coworkers. This effect is associated with changes in the regulation of cell cycles and the altered expression of apoptotic regulatory proteins, including p53, caspase-3, and the bcl-2/bax ratio [97]. Because flavonoids may lower reactive oxygen species, which damage cells and tissues and raise the risk of inflammatory disorders, they are useful in the treatment of a variety of diseases. The cytotoxicity and anti-inflammatory properties of two flavonoids, baicalin and baicalein, which are present in the roots of Scutellaria baicalensis (S. baicalensis) and the leaves of Thymus vulgaris and Oroxylum indicum, were examined. Baicalein was shown to be the more potent substance, exhibiting greater inhibitory effects on Src tyrosine kinase and cytokine IL-6 production. Baicalein was shown to be the more potent substance, exhibiting greater inhibitory effects on Src tyrosine kinase and cytokine IL-6 production [145]. Additionally, baicalin and baicalein from S. baicalensis have shown possible inhibitory effects against EGFR tyrosine kinase activity in experiments [146].
13.2.3. Curcumin
Diferuloylmethane, or curcumin, is extracted from Curcuma longa’s rhizome. Curcumin exhibits anti-NSCLC properties by modulating caspase-3 activity and miR-192-5p expression, leading to phosphoinositide 3-kinase (PI3K)/Akt signaling pathway inhibition and apoptosis [147]. By inhibiting the Wnt/β-catenin pathway, which is triggered by the metastasis-associated protein-1 (MTA-1), curcumin also inhibits the growth and invasion of NSCLC [148]. MTA-1 facilitates the invasion and metastasis of NSCLC cells [149]. The PI3K/Akt signaling pathway in NSCLC cells is suppressed and curcumin’s effects on cell viability and death are amplified by miR-192-5p mimics, but anti-miR-192-5p mimics have the opposite effect [147].
Additionally, curcumin therapy has been documented for its potential to suppress the development of NSCLC by lowering MTA-1. Curcumin induces autophagy in NSCLC, and this effect is reversed by autophagy inhibitor 3-methyladenine (3-MA). This effect exhibits the role of autophagy in anticancer activity [150]. Curcumin potentiates the therapeutic effects of gefitinib in TKI-resistant NSCLC. By lowering EGFR phosphorylation and raising EGFR degradation, curcumin causes apoptosis in TKI-resistant NSCLC cells, hence preventing the formation of cancer [151]. Most significantly, curcumin and EGFR-TKI therapy together significantly suppresses the development of NSCLC by lowering EGFR, c-MET, and cyclin D1 expression. Through the regulation of mitogen-activated protein kinase activity, the combination therapy improves intestinal epithelial cells’ survival rate and reduces intestinal mucosal damage [152].
In addition, as compared to curcumin or gefitinib therapy alone, the combination of the two induces significant autophagy activation, autophagic cell death, and autophagy-mediated apoptosis. Autophagic cell death brought on by therapy is lessened by pharmacological autophagy inhibitors such as bafilomycin A1 or 3-MA, beclin-1 or autophagy-related 7 knockdown, or both [152]. Frontline therapy with EGFR-TKIs includes afatinib and erlotinib. When erlotinib and curcumin are administered together, they cause apoptosis and increase IκB expression, which limits nuclear factor-κB (NF-κB) to the cytoplasm and inhibits its capacity to bind DNA. This drastically reduces the viability of NSCLC cells [153].
By reducing EGFR, surviving expression, and blocking NF-κB activity in erlotinib-resistant NSCLC cells, the combo therapy also markedly promotes apoptosis [154]. There is already a patent for the use of curcumin and afatinib together to treat NSCLC that is resistant to gefitinib and erlotinib (CN105476996A). For NSCLC, curcumin also reverses chemotherapy resistance. HIF-1α has been linked, in a recent study, to the development of chemotherapeutic resistance in cancer; consequently, cisplatin resistance may be reversed by targeting HIF-1α using RNA-interference or small interfering RNA. Combining curcumin with cisplatin has been shown to significantly reduce the growth of cisplatin-resistant NSCLC cells and induce apoptosis by activating caspase-3 and encouraging HIF-1α degradation, respectively [155].
In NSCLC, curcumin also overcomes cisplatin resistance by promoting cisplatin-induced apoptosis through the production of intracellular reactive oxygen species (ROS) and the proteosomal breakdown of Bcl-2 [156]. Studies on androgen-dependent and androgen-independent prostate cancer cells have shown that curcumin can suppress EGFR activity [157]. Curcumin and beta-phenyl ethyl isothiocyanate (PEITC) treatment caused the prostate cancer line PC-3 to undergo apoptosis, resulting in caspase-3 disruption and the suppression of EGFR- and EGF-induced Akt and PI3K activation [158]. Some clinical phase trials linked with curcumin in lung cancer are provided in the Table 3.
Table 3.
Clinical phase trials of curcumin in lung cancer treatment.
13.2.4. Fisetin
With a diphenylpropane structure, fisetin (3,3′,4′,7-tetrahydroxyflavone) is a naturally occurring bioactive flavonol. Apples, strawberries, cucumbers, persimmons, and a variety of acacia plants and shrubs are common sources of fisetin. Its anti-inflammatory, anti-microbial, anticancer, and neuroprotective qualities have been demonstrated previously [162]. By modifying a number of cell signaling pathways, such as inflammation, apoptosis, angiogenesis, growth factor, transcription factor, and others, fisetin has been shown to have anticancer properties [163]. In vitro tumor angiogenesis and VEGFR expression in Y79 cells were both dose-dependently reduced by fisetin. Fisetin may, therefore, be a viable treatment option for retinoblastoma angiogenesis inhibition since it was discovered to do so by blocking the VEGF/VEGFR signaling pathway [102]. Fisetin enhanced the cytotoxic effects of erlotinib, a TKI, and reduced the capacity of H1299 cells to establish colonies on soft agar [164]. Fsetin’s antiproliferative effect, which entails causing a modest G2/M arrest and stopping the cell cycle in the G1 phase, is ascribed to its inhibition of VEGF production [165]. In non-small cell lung cancer, a novel 4′-brominated derivative of fisetin inhibits the EGFR/ERK1/2/STAT3 pathways and causes cell cycle arrest and apoptosis without having any negative effects on mice. Analogs of fisetin also inhibited the EGFR/ERK1/2/STAT3 pathways. A greater ratio of Bax to Bcl-2 expression was seen in conjunction with apoptosis triggered by fisetin analogue [166].
13.2.5. Formononetin
One of the main biomolecules extracted from red clover and the Chinese plant Astragalus membranaceus is formononetin, a methoxylated isoflavone (7-hydroxy-3-(4-methoxypheny)-4H-1-benzopyran-4-one), which is a member of the isoflavonoid group of phytoestrogens. Formononetin exerts anticancer effects by modulating several cellular functions and molecular signaling pathways in various malignancies [167]. In NSCLC cell lines (HCC827 (EGFR Del E746-A750), H1975 (EGFR L858R/T790M), H3255 (EGFR L858R), A549 (EGFR WT), and H1299 (EGFR WT), formononetin was shown to promote the efficacy of EGFR-TKI by modulating the EGFR-AKT-Mcl-1 axis in a ubiquitination-dependent manner [101]. According to in vitro research using various human cancer cell lines, formononetin inhibits the development of carcinogenesis and metastasis by several mechanisms, such as controlling transcription factors, altering epigenetic targets, controlling estrogen receptors, controlling the cell cycle, triggering apoptosis, and controlling growth and developmental signaling pathways [168].
Inspired by the binding mechanism of lapatinib to EGFR, several novel formononetin compounds were designed and synthesized. Compound 4v showed the strongest anti-EGFR and anti-proliferation effect against the MDA-MB-231 cell line, which was comparable to that of lapatinib, according to an in vitro EGFR and cell growth inhibition experiment. The findings of additional biological experiments showed that 4v could effectively target EGFR and, subsequently, inhibit downstream signaling pathways, including EGFR/PI3K/Akt/Bad, EGFR/ERK, and EGFR/PI3K/Akt/β-catenin, respectively, in order to induce apoptosis, limit proliferation, and migrate in MDA-MB-231 cells [169].
13.2.6. Luteolin
Common flavones like luteolin (3,4,5,7-tetrahydroxyflavone) are present in foods including celery, carrots, peppers, thyme, oregano, and more. The benefits of luteolin are diverse and include anti-inflammatory, anti-oxidant, neuroprotective, cardio-protective, and anticancer properties [170]. It has been discovered that luteolin inhibits EGFR autophosphorylation and causes EGFR degradation through the lysosomal pathway in NSCLC (NCI-H1975) and epidermoid carcinoma (A431) cells, respectively [100,171]. Numerous RTK-related components, such as VEGF, PI3K, Akt, MAPK, MMP-2, MMP-9, focal adhesion kinase (Fak), B-cell lymphoma/leukemia 2 (Bcl-2), B-cell lymphoma extra-large (Bcl-xL), nuclear factor-kappa b (NF-κB), cyclooxygenase-2 (COX-2), signal transducer and activator of transcription 3 (STAT-3), tumor necrosis factor-alpha (TNF-α), and hypoxia-inducible factor-1 alpha (HIF-1α), were all inhibited by luteolin [172,173]. By inhibiting the ROS system and VEGF-induced gastric cancer cell angiogenesis by regulating the VEGFR2 signaling pathway, luteolin lowers VEGF expression [174].
The acquired resistance of first- and second-generation EGFR-TKIs caused by the EGFR T790M mutation in NSCLC has been overcome by osimertinib, a third-generation EGFR-TKI. Osimertinib and luteolin together induced apoptosis and prevented H1975/OR cells from proliferating, migrating, and invading. By inhibiting the HGF-MET-Akt pathway, luteolin and osimertinib can work in concert to overcome MET amplification and overactivation-induced acquired resistance to osimertinib. This suggests that luteolin and osimertinib may be used clinically in patients with acquired resistance in non-small cell lung cancer [175].
A number of cellular processes that contribute to the growth and advancement of cancer cells can be inhibited by luteolin through the reduction in the activity of certain receptor tyrosine kinases (RTKs), including IGFR, EGFR, and ERs, as well as their downstream effector molecules [176]. The combined effects of luteolin and gefinitib, a selective tyrosine kinase inhibitor that suppresses both EGFR and the kinase activity of cyclin G-associated kinase (GAK), on PC-3 prostate cancer cells were investigated in 2014 [177].
13.2.7. Quercetin
Quercetin is also known as 3,3,4,5,7-pentahydroxy-2-phenylchromen-4-one, reflecting the presence of five hydroxyl groups at positions 3-,5-,7-, and 4 of its molecular structure. Quercetin inhibits cancer development and progression through the modulation of various cell signaling pathways. Quercetin exhibits several pharmacological effects, including antibacterial, anti-inflammatory, anticancer, and antioxidant properties. Extensive research over recent decades has shown that quercetin possesses anti-ulcer, anti-allergy, antitumor, antiviral, and antidiabetic properties, along with anti-hypertension, anti-infection, gastro-protection, and immuno-modulation effects [178]. Quercetin has been documented to show a notable protective effect against metronidazole-induced neuronal damage [179].
Numerous studies have highlighted quercetin’s anti-allergic, anti-inflammatory, antiviral, and anticancer effects [180,181]. Quercetin has been shown to dramatically suppress the expression of growth factor signaling molecules, including EGFR, pAKT, pGSK-3β, β-Catenin, NFκB, and cyclin D, in lung cancer cells (A549) [182]. Additionally, after quercetin therapy, the study observed a reduction in matrix metalloproteinase 2 (MMP-2) and MMP-9 expression, which was probably caused by the EGFR/Akt/β catenin signaling pathway, and had an anti-metastatic impact. Molecular docking studies have extensively investigated quercetin’s ability to bind to receptor TKs (RTKs). Quercetin has been demonstrated to bind via hydrogen bonds, hydrophobic contacts, and π-π interactions to the ATP binding pocket or active site of RTKs such as EGFR, VEGFR2, FGFR1, IGF1R, and c-MET [183,184].
Quercetin targets several kinases involved in the growth and progression of cancerous cells. Quercetin inhibits the PI3K-Akt/PKB pathway by binding to PI3Kγ (IC50 = 3.8 µM) without targeting Akt/PKB [185,186]. As a strong inhibitor of TKs, including Syk, Src, Fyn, and Lyn [187], quercetin is the most investigated flavonoid. It has additionally been demonstrated to have anti-metastasis properties on stomach cancer cell lines [188]. It was demonstrated, in a phase I clinical trial, that intravenous quercetin had an anticancer effect in cisplatin-resistant ovarian cancer, evidenced by its antiproliferative effect and the inhibition of lymphocyte TK activity [103].
13.3. Phenolic Molecules
13.3.1. Caffeic Acid
All plant species produce caffeic acid, a phenolic molecule that has anti-inflammatory, anti-carcinogenic, and antioxidant properties. It can be found in drinks like coffee, wine, and tea as well as in common medications like propolis [189]. By increasing ROS levels and compromising mitochondrial function, caffeic acid can cause cancer cells to undergo apoptosis. Caffeic acid and its derivatives in cancer therapy have an impact on molecular pathways that play a part in the progression of cancer, including PI3K/Akt and AMPK. By blocking the epithelial-to-mesenchymal transition process, caffeic acid suppresses metastasis and lessens the aggressive aggressiveness of malignancies. Notably, caffeic acid and caffeic acid phenethyl ester (CAPE) can increase cancer cells’ sensitivity to chemotherapy-induced cell death and their response to chemotherapy. Caffeic acid and CAPE have been used in combination with other antitumor agents such gallic acid and p-coumaric acid to increase their ability to suppress malignancy. Because of its low bioavailability, nanocarriers have been created to increase its capacity to suppress cancer [190].
In the therapy of cancer, it has been discovered that caffeic acid targets receptor tyrosine kinases (RTK). One type of RTK is the cell-surface receptor for epidermal growth factor, known as the EGFR. In breast cancer cells, caffeine inhibits the phosphorylation of EGFR [191]. By modifying important genes and proteins involved in cancer resistance and treatment, CAPE is a therapeutically useful chemical that helps AZD9291 treat EGFR-TKI-resistant cells [104]. CAPE and docetaxel treatment together significantly reduced the expression of SKP2, c-MYC, and phospho-EGFR (Tyr 992) proteins in NSCLC compared to either CAPE or docetaxel treatment alone [192].
13.3.2. Epigallocatechin-3-Gallate (EGCG)
Epigallocatechin-3-gallate (EGCG), the primary active component of green tea, has been shown to have preventive and therapeutic effects against various diseases. The health benefits are mainly attributed to its potent anti-inflammatory and antioxidant qualities. EGCG’s anticancer effects have been observed in multiple types of cancer and are still being investigated. It has been demonstrated that EGCG has a chemopreventive impact by blocking the initiation, promotion, and development of the carcinogenesis process [193]. EGCG, the most abundant catechins of green tea, has been widely researched in several studies for its anti-carcinogenic properties [194]. The bioavailability of chemopreventive drugs, such as EGCG, has been increased by the application of recently developed nanotechnology [195,196].
In order to help cure human lung cancer cells, an EGCG nanoemulsion (nano-EGCG) was created. The anticancer effects of this emulsion were systematically investigated. Previous studies suggest that EGCG exhibits a number of biological effects, including anti-inflammatory, antitumor, antidiabetic, and anti-obesity effects [194]. EGCG has varying effects on the few NSCLC cell lines that have been studied, despite its ability to stop the growth of small cell lung cancer cells [197]. In addition to causing cell death, EGCG may interfere with angiogenesis, migration, invasion, carcinogenic activity, and tumor formation. In vitro and in vivo studies indicate that EGCG effectively induces apoptosis and inhibits the growth in a number of cancers, including leukemia and brain, breast, kidney, and colon tumors [105]. These antitumor effects are associated with the modulation of several signaling molecules, including reactive oxygen species, NF-κB, Akt, vascular endothelial growth factor, peroxisome proliferator-activated receptor, Bcl-2, and mitogen-activated protein kinases, as well as epigenetic modification [198]. While EGCG inhibits the growth of small cell lung cancer cells, it exhibits inconsistent effects on the small number of NSCLC cell lines tested [199,200]. EGCG causes EGFR internalization and ubiquitin degradation, leading to the disruption of EGFR signaling, whereas erlotinib prevents EGFR phosphorylation and maintains EGFR at the plasma membrane [201].
13.3.3. Ellagic Acid
Ellagic acid (EA) is a thermostable polyphenolic molecule naturally occurring in a wide range of fruits and nuts, such as black currants, raspberries, strawberries, walnuts, and grapes. EA exists either in a bound form, such as ellagitannins, or in its free form like glycosides. It exhibits many advantageous pharmacological properties such as antidiabetic, antimutagenic, antibacterial, antiviral, anticancer, and chemoprotective effects [202]. EA has been found to be a naturally occurring dual inhibitor of VEGF and PDGF receptors, indicating that it has significant antiangiogenic qualities that could aid in the prevention and management of cancer [111]. Erlotinib and EA work in concert to combat the EGFR H773_V774 insH mutation [203]. The dual inhibitor role of EA suggests that EA can be used as a leading promising compound for the development of novel plant-derived TKIs in cancer therapy.
13.3.4. Gossypol
Gossypol is a polyphenol compound found in the seeds of cotton plants (Gossypium sp.) and is known for its defensive properties. It has been investigated for the treatment of gynecological diseases as well as a potential oral male contraceptive. Extensive research has documented its antitumor, antioxidant, antiviral, antibacterial, and immunomodulatory properties [204]. Gossypol exerts its anticancer action by inhibiting anti-apoptotic Bcl-2 family proteins, thereby promoting apoptosis [112]. Gossypol has demonstrated its anticancer activity across multiple human breast cancer cells (MCF-7, MDA-MB-231, MDA-MB-468, ZR-75-1, and T47D), pancreatic cancer cells (BxPC-3 and MIA PaCa-2), human colon cancer cells (COLO 225), human cervical cancer cells (HeLa and SiHa cell lines), non-small cell lung cancer (NSCLC) cell lines (H1975), human lung cancer cell lines (H1299 and H358), and prostate cancer cells [205,206].
Although a number of EGFR inhibitors have been used to treat NSCLC, their effectiveness has decreased due to the introduction of EGFRL858R/T790M resistance mutations. The EGFR T790 M mutation is the primary mechanism of EGFR-TKI resistance and complicates treatment in NSCLC patients with activating EGFR mutations. Additionally, resistance to EGFR-TKIs is conferred by YAP/TAZ. By targeting both YAP/TAZ and EGFRL858R/T790M, gossypol emerges as a promising therapeutic candidate for overcoming EGFR-TKI resistance [206]. Gossypol inhibited cell proliferation and induced the apoptosis of NSCLC cells by targeting EGFRL858R/T790M mutation [207].
13.3.5. Honokiol
Honokiol is a biphenolic compound isolated from Magnolia species. It exhibits significant anticancer activity in NSCLC. Honokiol has been documented to downregulate the P13K/AkT/mTOR signaling pathways by decreasing AkT phosphorylation and upregulating pTEN expression. Thereby, it impairs cancer cell survival and proliferation [113]. Moreover, honokiol inhibits STAT3 phosphorylation irrespective of EGFR mutation status, suggesting broad-spectrum efficacy. The critical role of STAT3 in mediating the effects of honokiol is further underscored by findings that STAT3 knockdown abolishes honokiol-induced anti-proliferative and anti-metastatic activities [114]. Through simultaneously targeting STAT3 and P13K/AkT/mTOR pathways, honokiol represents a multitargeted agent for combating NSCLC progression and metastasis.
13.3.6. Magnolol
Magnolol is a neolignan and is derived from Magnolia officinalis. Magnolol has shown potent anticancer activities against various types of cancers including NSCLC. It exerts its effects by targeting multiple signaling pathways relayed by TKs, particularly the EGFR/P13K/Akt/mTOR and MAPK/ERK axes, which are frequently dysregulated in NSCLC. By blocking EGFR phosphorylation, restoring PTEN, and activating caspases, it effectively suppresses tumor growth and overcomes resistance, highlighting its potential as a supportive therapy in TKI refractory lung cancer [208].
13.4. Stilbene
Resveratrol
Resveratrol is a naturally occurring stilbene that offers several beneficial medicinal properties. Certain plants, including a variety of food sources such as peanuts, plums, apples, raspberries, blueberries, grapes, and goods made from them, are important sources of resveratrol. Many plants produce resveratrol in response to injury [209], and it has been discovered that over 70 plant species contain this natural polyphenol [210]. Resveratrol has been demonstrated to possess a number of therapeutic properties, such as powerful antioxidant activity and anti-inflammatory and antiproliferative properties [211,212,213].
Hydroxyl derivatives like oxyresveratrol exhibit similar effects [214,215]. In order to overcome gefitinib resistance, resveratrol increases intracellular gefitinib concentration, inhibits the removal of gefitinib from cells, and reduces CYP1A1 and ABCG2 expression. When gefitinib accumulates in gefitinib-resistant NSCLC cells, it causes apoptosis, autophagy, and senescence [216]. The anti-NSCLC actions of gefitinib are consequently enhanced when co-treated with resveratrol. Although it enhances the efficacy of TKIs, the direct inhibition of TKs by resveratrol has not been clearly documented.
13.5. Saponins
13.5.1. Ginsenosides
In Asian nations, Panax ginseng, commonly referred to as Korean ginseng, is a traditional medicine. Triterpenoid saponins, which are ginsenosides, are its main active ingredients. Among these, Re, a well-known ginsenoside, has demonstrated a number of biological actions, such as anti-inflammatory and anticancer qualities. Panax ginseng, a member of the Araliaceous plant groups, has long been utilized in both medicine and cooking. In East Asian countries like China and Korea, Panax ginseng has been used for preventing as well as treating cancer and other diseases [217,218].
In ginseng roots, fruits, stems, and leaves, ginsenosides are the primary saponins. To date, approximately 60 ginsenosides have been identified and isolated. Ginsenosides share a common core structure, but different sub-types vary in the arrangements of the four steroidal rings, including the A-Panaxadiol group (e.g., Rb1, Rb2, Rb3, Rc, Rd, Rg3, and Rh2), the B-Panaxatriol group (e.g., Re, Rg1, Rg2, and Rh1), and the C-Oleanolic acid group (e.g., Ro) [219].
Additionally, in NSCLC, ginsenosides enhance gefitinib’s anticancer action. In NSCLC cells, ginsenoside Rg3 has been shown to enhance the cytotoxic effects of gefitinib in a way that is dependent on both time and dosage. Gefitinib and ginsenoside Rg3 together dramatically boost apoptosis in non-small cell lung cancer cells by upregulating the pro-apoptotic protein Bax and caspase-3 activity while downregulating the anti-apoptotic protein Bcl-2 [220]. As master regulators that control several genes involved in EMT, such as E-cadherin [221], the combination therapy also prevents NSCLC cell migration by reducing the protein expression of Snail and Slug [220]. According to a study, ginsenosides inhibited the growth of NSCLC cells by activating the tumor suppressor p53-binding protein-1 and upregulating vaccinia-related kinase 1 [119]. Osimertinib is a third-generation EGFR-TKI, which has been associated with increased stemness and a higher potential for metastasis in osimertinib-resistant NSCLC cells compared with their parental cells [222].
Remarkably, ginsenoside Rg3 reduces the stemness of NSCLC cells and their resistance to osimertinib. This is evidenced by decreased stemness marker expression and reduced spheroid formation, which is reliant on the Hippo signaling pathway [118]. Ginsenosides also help patients who are resistant to chemotherapy medications like cisplatin (DDP), which is used to treat non-small cell lung cancer. Clinical studies demonstrate that ginsenoside Rg3 improves chemotherapy’s therapeutic effects in NSCLC patients. The patients receiving Rg3 therapy had longer overall survival and improved quality of life [223].
The leucocyte count is restored, the CD4/CD8 T-cell ratio is raised, and vascular endothelial growth factor (VEGF) expression is decreased in these individuals by Rg3. Many clinical studies show that combining Rg3 with chemotherapy improves the efficacy and overall survival time of patients with NSCLC. Ginsenoside may also help NSCLC patients overcome their cisplatin resistance, according to experimental studies. Ginsenoside suppresses the NRF2 pathway and makes A549/DDP cells much more sensitive to cisplatin in cisplatin-resistant NSCLC cells. NRF2 knockdown reduces ginsenosides’ synergistic effects in cells resistant to cisplatin [224]. According to prior research, Rg3 also reduced PD-L1 expression by inhibiting NF-κB p65 and Akt, which restored T cell cytotoxicity against cancer cells and reduced A549/DDP cells’ resistance to cisplatin therapy [225]. Although ginsenosides show numerous therapeutic responses, they do not directly interact or inhibit TKs, and their anticancer effects are mediated by the modulation of key cell signaling pathways.
13.5.2. Astragaloside IV
The decoctions made from the roots of Astragali Radix (Huangqi), are widely used in traditional Chinese medicine to treat cancer, inflammation, and bacterial and viral infections. Astragaloside IV (AS-IV), a cycloartane-type triterpene glycoside, is the primary active compound in Astragalus membranaceus and is responsible for its pharmacological effects. AS-IV serves as a standard marker for quality control in the Chinese pharmacopoeia. The primary active ingredient of Astragali Radix, AS-IV, can prevent cardiovascular disease [226], protect the liver [227], treat antidiabetic nephropathy [228], and have anticancer properties [120]. AS-IV enhances the sensibility of lung adenocarcinoma cells to bevacizumab by inhibiting autophagy [229]. Due to its demonstrated efficacy in a variety of cancer models of colorectal [230], liver [231], and lung [229] carcinomas, AS-IV is being explored as a promising anticancer agent. AS-IV can also be utilized to boost the sensitivity of chemotherapeutic medications and in conjunction with other anticancer drugs [232].
AS-IV has shown a good safety profile and low toxicities in experimental studies. By controlling sirtuin 6 (SIRT6), AS-IV sensitizes NSCLC to gefitinib. Inhibiting SIRT6 eliminates the sensitization effect of AS-IV in NSCLC cells, such as HCI-H1299, HCC827, and A549 cells [233]. SIRT6 is an NAD-dependent deacetylase that facilitates NSCLC. The inhibition of transforming growth factor-β1-induced EMT in NSCLC cells by SIRT6 depletion highlights how astragaloside IV enhances gefitinib’s anti-NSCLC activities. Recent studies support the potential of AS-IV as a novel anticancer therapeutic medication, as demonstrated by recently published studies showing strong antitumor efficacy [234]. However, no current report supports the direct inhibition of TKs by AS-IV, and it is grouped to be a bioactive modulator with a significant ability for the complement of TKI therapy.
13.5.3. Polyphyllin
The primary bioactive ingredient in Paris polyphylla is polyphyllin I (PPI). Recent studies on PPI across a variety of cancer types have demonstrated that PPI exerts diverse antitumor effects, including inducing cell cycle arrest, promoting cell apoptosis, triggering autophagy, inhibiting angiogenesis, enhancing tumor sensitivity to chemotherapy, and modulating immune and inflammatory responses [235]. PP I demonstrates strong antitumor action in both in vitro and in vivo models and increases the sensitivity of drug-resistant cells to gefitinib. PP I induces NSCLC cell death through multiple mechanisms, including the activation of the SAPK/JNK pathway, the suppression of p65 and DNMT1 expression [122], the downregulation of MALAT1, and the inhibition of the suppression of STAT3 phosphorylation [123]. PP I also reverses epithelial–mesenchymal transition (EMT) in HCC827 cells by inhibiting the prevention of IL-6 signaling, thereby overcoming resistance to erlotinib [236]. By blocking the PI3K/AKT/mTOR pathway, PP II can stop the proliferation and induce the apoptosis of gefitinib-resistant PC-9/ZD cells [237].
13.6. Triterpenes
13.6.1. Cucurbitacin
Cucurbitacin and its derivatives are categorized into 12 groups, including dihydrocucurbitacin B, A, B, C, D, E, I, H, Q, and R, with 40 related compounds currently recognized [238]. Among them, cucurbitacin B, a steroid with a distinct bitter taste and cytotoxic qualities, has been most widely investigated. Cucurbitacin B (C32H46O8, molecular weight 558.712 g/mol, 19-(10➝9β)-abeo-10-lanost-5-ene triterpene) is present in plants of the cucurbitaceae family as well as other plant families like Brassicaceae. Its bitterness may serve as a natural defense against parasites and predators [239].
It has shown efficacy against a number of medical conditions, including immune-related and angiogenic disorders, cell adhesion and leukemic disorders, insect infestation, carbon chloride-induced hepatotoxicity and profound cholestasis, generalized inflammation and algesia, and CD18-mediated disorders [240,241]. A chemical analog of cucurbitacin B was isolated from fruiting bodies of a mushroom “Leucopaxillus gentianeus” in another study. This analog differed from cucurbitacin B in that it lacked an oxygen substituent at carbon-16. It was then screened for antitumor toxicity based on variations in its chemical structure [242].
Through hydrophobic and electrostatic forces, cucurbitacin B binds strongly with human serum albumin and increases ibuprofen–albumin binding [243]. Cucurbitacin B rapidly altered breast cancer cell morphology by disrupting F-actin and microtubules within 15–20 min after exposure in a dose-dependent manner. These results were confirmed in naked mice, where cucurbitacin B injections administered intraperitoneally at a dose of 1 mg/kg every three weeks produced a suppression of tumor size growth of around 50% [244].
By triggering G2/M cell-cycle arrest and mitochondrial death, cucurbitacin B effectively inhibits the development of non-small cell lung cancer. The thiol antioxidant N-acetyl cysteine reduces the anti-NSCLC effects of cucurbitacin B, whereas butithione-sulfoxime, an inhibitor of glutathione formation, intensifies the anti-NSCLC effects [245]. According to another study, cucurbitacin B treatment significantly decreases the ratio of thiol and glutathione to oxidized glutathione in NSCLC cells, demonstrating that the anti-NSCLC properties of cucurbitacin B are facilitated by an impairment of the cellular redox balance [246].
In NSCLC cells, cucurbitacin B and D also conquer gefitinib resistance. By dephosphorylating oncogenic kinases and transcription factors, CIP2A hinders protein phosphatase-2A (PP2A) [247], leading to a reduction in tumor development [248]. Additionally, in NSCLC cells, the lysosomal degradation of EGFR is induced by the cucurbitacin-B-mediated suppression of the CIP2A/PP2A/Akt axis [249]. In a prior work, a solid-phase EGF-EGFR interaction test showed that cucurbitacin D overcomes gefitinib resistance by preventing EGF from binding to EGFR. Cucurbitacin D was found to directly disrupt the interaction between EGFR and EGF. As a result, cucurbitacin D reduces EGFR phosphorylation in gefitinib-resistant NSCLC cells [250].
13.6.2. Betulinic Acid and Betulin
Betulinic acid (BA), a pentacyclic triterpene molecule, can be extracted from Birch tree bark and it can also be chemically synthesized or modified through biotransformation [251]. BA exerts anticancer effects by binding to specific target molecules and modulating key cellular signaling pathways [252,253,254]. The primary anticancer mechanism of BA involves the induction of mitochondrial-mediated apoptosis [255,256,257] with no notable side effects [258,259]. BA has been reported to exert several biological effects, including the modulation of transcription factors [260,261], suppression of NF-κB and STAT3 signaling pathways [262,263], and regulation of autophagy [256].
Due to its limited water solubility, BA remains under investigation and is not yet widely used in clinical oncology. Numerous biological activities, including antitumor, anti-inflammatory, anti-HIV, antimalarial, antibacterial, and antioxidant properties, are exhibited by BA and its derivatives (produced by modification at the C-3, C-20, and C-28 sites) [264,265,266,267]. It is possible that BA works better when combined with other anticancer medications. For example, BA suppresses the growth of the pancreatic cancer cells (PANC-1 and Mia PacA-2) by activating the AMPK pathway. Patients with lung adenocarcinoma who have an EGFR mutation are treated with EGFR-TKIs, such as erlotinib and gefitinib, as their first line of treatment. However, drug resistance typically develops within 8–11 months of treatment. Co-treatment with BA and either erlotinib or gefitinib significantly reduced the viability of drug-resistant H1975 cancer cells in a dose-dependent manner in comparison with erlotinib or gefitinib alone and increased the apoptotic cell populations [268,269]. BA was more successful in preventing PANC-1 cell growth and triggering apoptosis when coupled with gemcitabine [270]. BA exhibits cytotoxic effects that contribute to tumor suppression via kinase-dependent mechanisms, although it does not directly inhibit TK activity.
Betulin is a Triterpenoid and is found in many plants and herbs. In the human chronic myelogenous leukemia cell line, betulin modulates the mitogen-activated protein (MAP) kinase pathway with activity comparable to that of the well-known ABL1 kinase inhibitor imatinib mesylate [127].
13.6.3. Leelamine
Because of their distinct mode of action and variety of molecular structures, natural molecules are propelling the creation of novel TKIs. New advancements in the study of anticancer drug discovery are encouraged by the identification of novel TK inhibitory molecules from natural products [271]. Pine bark plants naturally produce a lysosomotropic chemical called leelamine (LEE) or dehydroabietylamine, which is a lipophilic diterpene amine. Numerous earlier investigations have revealed LEE’s possible anticancer properties. LEE was reported to modulate the STAT5 pathway in myelogenous leukemia cells, causing both autophagy and death. It has also been documented that LEE can activate PTPε in multiple myeloma cells and block the STAT3/JAK signaling pathway [128]. LEE prevents intracellular cholesterol transfer. Its lysosomotropic properties, accumulation in the lysosome, and blockage of cholesterol transport result in a lack of availability for essential activities needed for cancer cell activity, which, in turn, causes cell death [272]. Interestingly, the pyruvate dehydrogenase kinase inhibitors (PDK inhibitors) used in all of the studies looking into combination therapy including EGFR-TKIs and PDK inhibitors to overcome resistance in NSCLC were synthetic. Huzhangoside A, leelamine, and otobaphenol are examples of natural-product-based PDK inhibitors that have been shown in a prior study to promote pyruvate dehydrogenase (PDH)-activity-dependent cancer cell death [273].
14. Translational Insights into Natural-Product-Enhanced Lung Cancer Combinational Therapy
Chemotherapeutic drugs may have one or several adverse effects; therefore, there is a growing interest in the combination of chemotherapeutic drugs with fewer or minimal toxic natural molecules for the management of tumors. In order to improve tumor treatment, the therapeutic approach of combining natural compounds with chemotherapeutic drugs can lessen the development of drug resistance in tumor cells, improve the tumor-killing effect of chemotherapeutic drugs, and lessen the severe side effects of chemotherapeutic drugs on patients [274].
The viability of using EGFR-TKI in conjunction with other NSCLC treatments, such as radiation, cytotoxic chemotherapies, targeted therapies, and new immunotherapies, has been the subject of numerous clinical investigations. However, the development and application of these solutions are hampered by the substantial gap that still persists when extrapolating preclinical outcomes to clinical circumstances [275]. The ideas of synergy and combination therapy are closely related, and it has been noticed that the quality of life for patients with advanced diseases like cancer, hypertension, and asthma has been significantly improved by evolving combination therapy [276]. The combination of phytocompounds or plant extracts with complementary therapy has been widely valued, and it has been used for the treatment of cancer. It has been suggested that the specific properties of natural substances and their capacity for enhancing the effects of chemotherapeutic drugs may improve lung cancer patient outcomes and may open the door for more individualized and efficient future treatment options [277].
It has been documented that natural products reduce chemotherapy-induced tumor resistance. Hence, when used in conjunction with chemotherapy, natural products may work in concert to increase antitumor effects and may encourage tumor cell death [278]. Moreover, there are several mechanisms of action of natural products, like sensitizing cancer cells to increase the tumoricidal effect, reversing chemoresistance by inhibiting multiple targets involved in the development of drug resistance, and reducing chemotherapy-induced toxicity in non-tumoral cells by encouraging repair mechanisms, which may contribute to their synergistic effects [279].
For lung cancer, the use of natural substances in conjunction with traditional chemotherapy medications has also shown encouraging outcomes [280]. Emodin has shown synergistic benefits with enhanced anticancer activity against lung adenocarcinoma and non-small cell lung cancer when combined with the chemotherapies like sorafenib, afatinib, cisplatin, paclitaxel, gemcitabine, and endoxifen [280,281]. When paired with EGFR-TKIs, a variety of natural compounds that have strong inhibitory effects on drug-resistant cells can greatly boost the sensitivity of these cells to EGFR-TKIs [86].
Additionally, Scutellaria baicalensis and cisplatin have shown synergistic effects, preventing tumor growth in vitro (Lewis lung carcinoma cells, or LLC) and in vivo (C57BL/6J tumor-inoculated mouse model) [282]. Using H460, H1975, and H292 cell lines, the extract from Marsdenia tenacissima and gefitinib worked in concert to cause apoptosis in resistant cells. The combo treatment decreased EGFR downstream signaling pathways, according to the results of flow cytometry [283]. Through the SHH signaling system, combination therapy with sulforaphane and gefitinib dose-dependently decreases the growth of gefitinib-resistant lung cancer cells and inhibits the expression of SHH, SMO, and GLI1 [284].
Solamargine has been reported to reduce cell proliferation and increase apoptosis in the cisplatin-resistant lung cancer cell lines NCI-H1299 and NCI-H460. More significantly, solamargine and cisplatin worked in concert, each increasing the effectiveness of the other [285]. Hederagenin is a pentacyclic triterpenoid that has been shown to increase the conversion of LC3-I to LC3-II in lung cancer cells, thereby inhibiting autophagy. It has also been suggested that hederagenin and paclitaxel and cisplatin, respectively, may work in concert to increase their anticancer effects [286]. By dose-dependently inhibiting TMEM16A, narirutin, a flavonoid extracted from citrus unshiu, enhanced the anticancer activity of cisplatin when used in conjunction with cisplatin for lung cancer [287].
In fact, an effectiveness investigation in NSCLC cells shown that the medication combination of cisplatin and curcumin had better benefits [288]. By controlling the Cu-Sp1-CTR1 regulatory loop, curcumin improved the therapeutic efficacy of cisplatin in lung cancer cell lines A549, H460, and H1299, according to an in vitro study [289]. It has been reported that extracts from Terminalia bellerica and Phyllanthus emblica function in combination with doxorubicin and cisplatin to prevent the growth of human lung cancer and hepatocellular carcinoma cells [290]. The combination of 5-Demethylnobiletin and paclitaxel substantially inhibited the proliferation of lung cancer cell lines [291].
In NSCLC Aloe-emodin may increase PC9-GR cells’ sensitivity to gefitinib and reverse epithelial–mesenchymal transition (EMT) via inhibiting the PI3K/Akt/TWIS1 signal pathway [292]. When compared to TKI alone, a combination of TKI and Chinese herbal medicines increased PFS in patients with advanced NSCLC that had an EGFR mutation [293]. Yiqi Chutan Tang (YQCT) boosts gefitinib-induced apoptosis, increases autophagy, causes mild cell cycle arrest, and decreases gefitinib-induced drug resistance. These findings suggest that, when combined with gefitinib, YQCT can boost anticancer effects and decrease drug resistance at the molecular level [294].
Anticancer TKIs (nilotinib, bosutinib, dasatinib, and ponatinib) and a methoxyflavanone derivative synergistically inhibit the growth of A549 cells. When combined with nilotinib and PDMF, they caused G2/M cell cycle arrest and de novo G1 arrest. In athymic nude mice, the co-administration of PDMF and nilotinib dramatically reduced the in vivo tumorigenicity of A549 cells [295]. When paired with osimertinib, berberine selectively and synergistically reduced the lifespan of many MET-amplified osimertinib-resistant EGFR-mutant NSCLC cell lines. This was likely due to increased apoptotic induction brought on by Bim elevation and Mcl-1 reduction. Crucially, this combination was well tolerated and successfully increased the suppressive effect on the growth of MET-amplified osimertinib-resistant xenografts in nude mice [296]. The combinational therapies of TKIs or chemotherapeutic drugs and natural products in lung cancer treatment are provided in Table 4.
Table 4.
Combinational therapies of TKIs or chemotherapeutic drugs and natural products in lung cancer treatment.
15. Fourth-Generation TKIs
The development of acquired resistance to EGFR-TKIs is inevitable, which restricts their long-term efficacy. EGFR secondary mutations and bypass signaling activations are the primary mechanisms of the resistance process. A lot of work has gone into creating new fourth-generation EGFR-TKIs, even though first-, second-, and third-generation EGFR-TKIs have achieved great strides in NSCLC-targeted therapy. Allosteric inhibitors, such as EAI045, JBJ-25-02, JBJ-09-063, BLU-945, BLU-701, BDTX-1535, BBT-176, JIN-A02, TRX-221, BPI-361175, and BAY 2927088, have been discovered through preclinical drug development. These inhibitors bind to the EGFR allosteric site and cause conformational changes that reduce its affinity with ATP [86]. In order to overcome on-target resistance, fourth-generation TKIs are becoming a more intriguing therapeutic alternative. Thiazole-amid inhibitors, whose activity is mediated by the allosteric inhibition of EGFR and has a high selectivity towards mutant-EGFR, have been discovered as a result of preclinical therapeutic research. To clarify their activity in the therapeutic context as well, early phase 1/2 clinical trials are now being conducted [297]. As a fourth-generation EGFR-TKI, BLU945 selectively targets co-mutations in T790M and C797S as well as other T790M resistance mutations brought on by osimertinib resistance [298]. Although TQB3804 is undergoing phase I (NCT04128085) and phase II (NCT04180150) trials, no pertinent data have been made public as of yet; therefore, its clinical potential will have to be ascertained later. The BBT-176 was unable to avoid the fate of halting research and development, as evidenced by the termination of its clinical study. In the preclinical stage, the same company’s BBT-207, which is now being developed (NCTNCT05920135), has demonstrated outstanding antitumor effects against double mutations, including C797S, and triple mutations [299].
16. Caspase-Mediated Apoptosis Induced by Phytochemicals in Lung Cancer Models
Apoptosis or cellular death is caused by the proteolytic cleavage of millions of proteins by effector caspases, and it is a crucial mechanism for tumor suppression [300]. Multicellular organisms depend on cell death for proper development, tissue homeostasis, and integrity. Various executioner and regulatory molecule groups carefully govern apoptosis. Cell shrinkage, dynamic membrane blebbing, the condensation of chromatin material, DNA fragmentation in the nucleus, and the loss of adherence to the external matrix are characteristic mechanisms of action of apoptotic cell death. Additional biochemical changes include phosphatidylserine externalization and the activation of cysteine aspartyl proteases, or caspases, which cause cell death [301]. Although certain drugs may induce apoptosis through caspase-independent routes, caspases are crucial mediators of apoptotic signals from upstream molecules, and their activation is thought to be a hallmark of apoptosis [302].
Numerous diseases, including cancer, autoimmune diseases, neurological diseases, and cardiovascular diseases, are linked to the dysregulation of apoptosis. In apoptotic signaling cascades, initiator caspases (caspase-8 and -9) and executioner caspases (caspase-3, -6, and -7) are essential. Tumor necrosis factor (TNF) and other ligands start the extrinsic route by generating a death-inducing signaling complex (DISC), which activates caspase-8 and subsequent caspases [303].
The incapacity to undergo apoptosis in response to apoptotic stimuli is a hallmark of human cancers. It has been discovered that the disruption of initiator caspase-8 and caspase-9 expression or function may play a role in the development or progression of cancer, and that their inactivation may increase resistance to existing therapeutic strategies [304]. In one study, it was documented that curcumin did not activate Bcl-2 or Bcl-xl-transfected cells, but it did activate caspase-8 and caspase-3 in HL-60 neo cells [305]. Natural products such as tea extracts are rich in flavonoids and phenolic compounds, which exhibit anti-proliferative effects. These extracts promote apoptosis in cancer cells by enhancing the activity of caspase-3, caspase-7, caspase-8, and caspase-9 [306].
A study explored the mechanism of Hericium erinaceus (Yamabushitake)-mushroom-induced apoptosis of U937 human monocytic leukemia cells. When compared to the vehicle-treated group, the HWE and MWE treatments significantly raised the activities of caspase-9 and caspase-3. There was also an increase in caspase-8 activity. However, compared to caspase-3 and -9, the induction ratio of caspase-8 activity was significantly lower [307]. In NB4 human leukemia cells, hispolon from Phellinus linteus causes G0/G1 cell cycle arrest and apoptosis. It also induces the expression of proteins linked to apoptosis, including the proapoptotic Bax protein, poly (ADP ribose) polymerase, and the cleavage form of caspase 3, caspase 8, and caspase 9 [308].
It was discovered that procaspase-6, procaspase-8, and procaspase-9 expression significantly decreased when cells were treated with Lupeol; procaspase-3 protein status remained unchanged. These findings imply that caspase-8 and caspase-9 mediate and caspase-6 carries out the death of prostate cancer cells triggered by luteol [302]. The biological activity of Thymus vulgaris (TVEO) and Thymus serpyllum (TSEO) essential oils differ. TVEO activated caspase-3 and caspase-8 to induce apoptosis in cervical cancer HeLa cells, whereas TSEO activated caspase-3 to induce apoptosis [309]. Through the activation of caspase cascades and the downregulation of the inhibitor of apoptosis proteins via JNK/p38 signaling, curcumin analog L48H37 causes apoptosis in human oral cancer cells [310]. The apoptosis of MCF7 was induced, followed by the activation of pro-apoptotic genes (p53, Bax, Parp, Caspase-3, -8, -9) and the inactivation of antiapoptotic genes, such as the Bcl2 gene, in a study involving the growth inhibition and apoptosis of cancer cells by the ethyl 4-[(4-methylbenzyl)oxy] benzoate complex in vitro and in vivo [311]. Both in vivo and in vitro, tanshinone IIA causes intrinsic apoptosis in osteosarcoma cells linked to mitochondrial malfunction. In xerograft mice and Tan IIA-treated 143B cells, apoptosis was linked to the activation of the caspase cascade through Bcl-2 family modification [312].
17. Role of RTKs and Its Inhibitors for the Development and Treatment of BRAF Mutant Lung Cancers
The BRAF gene encodes V-Raf murine sarcoma viral oncogene homolog B (BRAF) kinase, which is essential for cell signaling, growth, and survival. The onset and spread of cancer are caused by mutations in the BRAF gene. BRAF mutations are frequently found in women, never-smokers, and aggressive histological forms of non-small cell lung cancer (NSCLC). They also account for between 1% and 2% of adenocarcinomas. Patients with BRAF-mutated non-small cell lung cancer had limited response to traditional treatment. However, the way that NSCLC is treated has changed significantly with the introduction of immune checkpoint inhibitors (ICIs) and targeted therapy [313].
Based on the location of the mutation, BRAF mutations can be categorized into three types. Class II mutants, such as K601, L597, G464, and G469 alterations, are found in the activation region or P-loop and signal as RAS-independent dimers [314,315]. Class I mutants, such as V600E/K/D/R, occur in the valine residue at amino acid position 600 of exon 15 [314,316]. BRAF kinase function is compromised by class III mutants that arise in the P-loop, catalytic loop, or DFG motif; in fact, non-V600 mutations account for about half of BRAF mutations in non-small cell lung cancer (NSCLC) [317,318,319]. Furthermore, because class II and III BRAF mutations are susceptible to existing BRAF inhibitors, it is necessary to produce novel-generation BRAF inhibitors. The most prevalent BRAF mutation in lung cancer, particularly V600E, can lead to uncontrolled cell growth [313].
RTKs are characterized as key elements to go beyond BRAF inhibitor resistance and as appropriate targets for drugs, allowing for the more effective treatment of BRAF mutant tumors. RTKs are essential for the reactivation of MAPK/ERK signal transmission, the formation of bypassing signaling pathways, and the emergence of treatment resistance to BRAF inhibitors [320]. The dysregulation of RTKs, especially EGFR, HER2, and HER3, is implicated in resistance to BRAF inhibitors in BRAF mutant cancers. The inhibition of BRAFV00E often leads to the disruption of ERK-mediated negative feedback within the MAPK pathway, which is implicated in compensatory EGFR activation, the reactivation of downstream MAPK signaling, and therapeutic failure [321,322].
Therapeutic approaches with a combination of BRAF inhibitors and RTK inhibitors, such as EGFR inhibitors and pan ErbB inhibitors or monoclonal antibodies, have shown promising preclinical activity [320]. Additionally, the activation of other RTKs including MET/HGFR and AXL further contributes to adaptive resistance via alternative bypass pathways [323]. Novel dual inhibitors (targeting EGFR and RAF) and triple combinations (BRAF, MEK, and RTK inhibitors) have demonstrated promising efficacy in overcoming resistance. Therefore, RTKs represent both key mediators of resistance and promising therapeutic targets, which highlights their significance in refining treatment strategies for BRAF mutant cancers [320].
18. Conclusions
TK dysregulation results in cancer, unchecked cell division, and oncogenic conversion. Therefore, inhibiting TK can be a revolutionary advancement in cancer research, care, and treatment that clinically enhances quality of life. From this point of view, several preclinical studies are now being conducted to investigate TKIs. But, there are still a lot of unanswered questions regarding the mechanisms underlying TKIs and their selectivity. Numerous mechanisms, such as genetic mutations, changes in gene expression, and modifications to the tumor microenvironment, may result in TKI resistance. Additionally, the target gene may occasionally undergo alterations such as mutations and amplifications, allowing cells to proliferate even in the presence of an inhibitor. In such cases, strong kinase inhibitory activity from natural products looks very promising for lung cancer and, hence, NSCLC management. In this review, we discussed the fact that many plant-derived natural products including resveratrol, ginsenosides, astragalosides IV, BA, polyphyllin, and leelamines are reported to increase the anticancer activities of TKIs. However, these molecules are still not documented to inhibit TKs directly or indirectly. Rather, they function as pathway modulators or chemosensitizers affecting the potential of TKIs via complementary mechanisms. Therefore, their inclusion into TKI-based therapeutic strategies may provide a beneficial adjunct in lung cancer management. It is highly recommended to complete further research on the drug-like properties of such natural-product-derived molecules for the development of selective TKIs. Further, the research into bioactivity data and scaffolds obtained from natural sources for drug-screening important biological characteristics is necessary for the efficient treatment of lung cancer.
Author Contributions
Conceptualization, S.A. and F.A. writing—review and editing, S.A., S.V.P., V.K., A.H.R. and F.A.; visualization, S.A., A.H.R. and F.A.; supervision, S.A. and F.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No datasets were generated or analyzed during the current study.
Acknowledgments
The researchers would like to thank the Deanship of Graduate Studies and Scientific Research at Qassim University for financial support (QU-APC-2025).
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| SCLC | Small cell lung cancer |
| NSCLC | Non-small cell lung cancer |
| TKs | Tyrosine kinases |
| TKIs | Tyrosine kinase inhibitors |
| ATP | Adenosine triphosphate |
| EGFR | Epidermal growth factor receptor |
| COPD | Chronic obstructive pulmonary disease |
| EGFR | Epidermal growth factor receptor |
| ALK | Anaplastic lymphoma kinase |
| KRAS | Kirsten rat sarcoma viral oncogene homolog |
| LCLC | Large cell carcinoma |
| SCC | Squamous cell carcinoma |
| EMT | Epithelial–mesenchymal Transition |
| TSGs | Tumor suppressor genes |
| PI3K | Phosphoinositide 3-kinase |
| Akt | Serine-threonine kinase |
| TGF-β | Transforming growth factor β |
| GPNMB | Glycoprotein nonmetastatic melanoma protein B |
| HGF | Hepatocyte growth factor |
| IL-10 | Interleukin-10 |
| IL-6 | Interleukin-6 |
| EGFR-TKI | EGFR-tyrosine kinase inhibitor |
| MAPK | Mitogen-activated protein kinase |
| JAK-STAT | Janus kinase/Signal transducer and activator of transcription |
| DDRs | Discoidin domain receptor |
| Eph | Erythropoietin-producing human hepatocellular receptor |
| SRC | Proto-oncogene c-Src |
| SYK | Spleen tyrosine kinase |
| FLT3 | Fms-like tyrosine kinase 3 |
| TKIR | TKI-resistant |
| VEGFR | Vascular endothelial growth factor receptor |
| VEGF | Vascular endothelial growth factor |
| HER2 | Human epidermal growth factor receptor 2 |
| MMP-9 | Matrix metalloproteinase-9 |
| MMP-2 | Matrix metalloproteinase-2 |
| Fak | Focal adhesion kinase |
| Bcl-2 | B-cell lymphoma/leukemia 2 |
| Bcl-xL | B-cell lymphoma ex-tra-large |
| NF-κB | Nuclear factor-kappa b |
| COX-2 | Cyclooxygenase-2 |
| STAT-3 | Signal transducer and activator of transcription 3 |
| TNF-α | Tumor necrosis factor-alpha |
| HIF-1α | Hypoxia-inducible factor-1 alpha |
| PDK | Pyruvate dehydrogenase kinase |
| SAP | Signaling lymphocyte activation molecule-associated protein |
References
- Alrumaihi, F.; Raut, R.; Yahia, E.A.; Kumar, V.; Anwar, S. A Review on Risk Factors, Diagnostic Innovations, and Plant Based Therapies for the Management of Erectile Dysfunction. Uro 2024, 4, 60–88. [Google Scholar] [CrossRef]
- Almatroodi, S.A.; Alsahli, M.A.; Almatroudi, A.; Anwar, S.; Verma, A.K.; Dev, K.; Rahmani, A.H. Cinnamon and its active compounds: A potential candidate in disease and tumour management through modulating various genes activity. Gene Rep. 2020, 21, 100966. [Google Scholar] [CrossRef]
- Najmi, A.; Javed, S.A.; Al Bratty, M.; Alhazmi, H.A. Modern approaches in the discovery and development of plant-based natural products and their analogues as potential therapeutic agents. Molecules 2022, 27, 349. [Google Scholar] [CrossRef] [PubMed]
- Anwar, S.; Raut, R.; Alhumaydhi, F.A. A comprehensive investigation on alleviating oxidative stress and inflammation in hyperglycaemic conditions through in vitro experiments and computational analysis. Saudi J. Biol. Sci. 2024, 31, 104003. [Google Scholar] [CrossRef]
- Anwar, S.; Almatroudi, A.; Alsahli, M.A.; Khan, M.A.; Khan, A.A.; Rahmani, A.H. Natural products: Implication in cancer prevention and treatment through modulating various biological activities. Anti-Cancer Agents Med. Chem. 2020, 20, 2025–2040. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Canli, Ö.; Nicolas, A.M.; Gupta, J.; Finkelmeier, F.; Goncharova, O.; Pesic, M.; Neumann, T.; Horst, D.; Löwer, M.; Sahin, U.; et al. Myeloid Cell-Derived Reactive Oxygen Species Induce Epithelial Mutagenesis. Cancer Cell 2017, 32, 869–883.e5. [Google Scholar] [CrossRef] [PubMed]
- Denk, D.; Greten, F.R. Inflammation: The incubator of the tumor microenvironment. Trends Cancer 2022, 8, 901–914. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Setton, J.; Lee, N.Y.; Riaz, N.; Powell, S.N. The therapeutic significance of mutational signatures from DNA repair deficiency in cancer. Nat. Commun. 2018, 9, 3292. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Huang, R.; Zhou, P.K. DNA damage repair: Historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Signal Transduct. Target Ther. 2021, 6, 254. [Google Scholar] [CrossRef]
- Holley, R. Control of growth of mammalian cells in cell culture. Nature 1975, 258, 487–490. [Google Scholar] [CrossRef]
- Raufi, A.G.; May, M.S.; Hadfield, M.J.; Seyhan, A.A.; El-Deiry, W.S. Advances in Liquid Biopsy Technology and Implications for Pancreatic Cancer. Int. J. Mol. Sci. 2023, 24, 4238. [Google Scholar] [CrossRef] [PubMed]
- Forder, A.; Zhuang, R.; Souza, V.G.P.; Brockley, L.J.; Pewarchuk, M.E.; Telkar, N.; Stewart, G.L.; Benard, K.; Marshall, E.A.; Reis, P.P.; et al. Mechanisms Contributing to the Comorbidity of COPD and Lung Cancer. Int. J. Mol. Sci. 2023, 24, 2859. [Google Scholar] [CrossRef]
- Jiao, Q.; Bi, L.; Ren, Y.; Song, S.; Wang, Q.; Wang, Y.S. Advances in studies of tyrosine kinase inhibitors and their acquired resistance. Mol. Cancer 2018, 17, 36. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef]
- Park, W.; Han, J.H.; Wei, S.; Yang, E.-S.; Cheon, S.-Y.; Bae, S.-J.; Ryu, D.; Chung, H.-S.; Ha, K.-T. Natural Product-Based Glycolysis Inhibitors as a Therapeutic Strategy for Epidermal Growth Factor Receptor–Tyrosine Kinase Inhibitor-Resistant Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2024, 25, 807. [Google Scholar] [CrossRef]
- Ibodeng, G.O.; Uche, I.N.; Mokua, R.; Galo, M.; Odigwe, B.; Galeas, J.N.; Dasgupta, S. A snapshot of lung cancer: Where are we now?—A narrative review. Ann. Transl. Med. 2023, 11, 261. [Google Scholar] [CrossRef]
- Clark, S.B.; Alsubait, S. Non–Small Cell Lung Cancer. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK562307/ (accessed on 10 April 2025).
- Xie, S.; Wu, Z.; Qi, Y.; Wu, B.; Zhu, X. The metastasizing mechanisms of lung cancer: Recent advances and therapeutic challenges. Biomed. Pharmacother. 2021, 138, 111450. [Google Scholar] [CrossRef]
- Davidson, M.R.; Gazdar, A.F.; Clarke, B.E. The pivotal role of pathology in the management of lung cancer. J. Thorac. Dis. 2013, 5, S463–S478. [Google Scholar] [CrossRef]
- Zappa, C.; Mousa, S.A. Non-small cell lung cancer: Current treatment and future advances. Transl. Lung Cancer Res. 2016, 5, 288–300. [Google Scholar] [CrossRef]
- Gómez-López, S.; Whiteman, Z.E.; Janes, S.M. Mapping lung squamous cell carcinoma pathogenesis through in vitro and in vivo models. Commun. Biol. 2021, 4, 937. [Google Scholar] [CrossRef] [PubMed]
- Sabbula, B.R.; Gasalberti, D.P.; Mukkamalla, S.K.R.; Anjum, F. Squamous Cell Lung Cancer. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK564510/ (accessed on 10 April 2025).
- Tai, Q.; Zhang, L.; Hu, X. Clinical characteristics and treatments of large cell lung carcinoma: A retrospective study using SEER data. Transl. Cancer Res. 2020, 9, 1455–1464. [Google Scholar] [CrossRef]
- Rudin, C.M.; Brambilla, E.; Faivre-Finn, C.; Sage, J. Small-cell lung cancer. Nat. Rev. Dis. Primers 2021, 7, 3. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fischer, B.; Marinov, M.; Arcaro, A. Targeting receptor tyrosine kinase signalling in small cell lung cancer (SCLC): What have we learned so far? Cancer Treat. Rev. 2007, 33, 391–406. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Cui, Y.; Zheng, X.; Zhao, Y.; Sun, G. Small-cell lung cancer brain metastasis: From molecular mechanisms to diagnosis and treatment. Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166557. [Google Scholar] [CrossRef]
- Wistuba, I.I.; Gazdar, A.F.; Minna, J.D. Molecular genetics of small cell lung carcinoma. Semin. Oncol. 2001, 28, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Hilal, T. Current understanding and approach to well differentiated lung neuroendocrine tumors: An update on classification and management. Ther. Adv. Med. Oncol. 2017, 9, 189–199. [Google Scholar] [CrossRef]
- Petty, W.J.; Paz-Ares, L. Emerging Strategies for the Treatment of Small Cell Lung Cancer: A Review. JAMA Oncol. 2023, 9, 419–429. [Google Scholar] [CrossRef]
- Lahiri, A.; Maji, A.; Potdar, P.D.; Singh, N.; Parikh, P.; Bisht, B.; Mukherjee, A.; Paul, M.K. Lung cancer immunotherapy: Progress, pitfalls, and promises. Mol. Cancer 2023, 22, 40. [Google Scholar] [CrossRef]
- de Visser, K.E.; Joyce, J.A. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef] [PubMed]
- Wyckoff, J.; Wang, W.; Lin, E.Y.; Wang, Y.; Pixley, F.; Stanley, E.R.; Graf, T.; Pollard, J.W.; Segall, J.; Condeelis, J. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 2004, 64, 7022–7029. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Hagiwara, K. Epidermal growth factor receptor (EGFR) mutation and personalized therapy in advanced nonsmall cell lung cancer (NSCLC). Target. Oncol. 2013, 8, 27–33. [Google Scholar] [CrossRef]
- Shin, S.H.; Koh, Y.G.; Lee, W.G.; Seok, J.; Park, K.Y. The use of epidermal growth factor in dermatological practice. Int. Wound J. 2023, 20, 2414–2423. [Google Scholar] [CrossRef] [PubMed]
- Fu, K.; Xie, F.; Wang, F.; Fu, L. Therapeutic strategies for EGFR-mutated non-small cell lung cancer patients with osimertinib resistance. J. Hematol. Oncol. 2022, 15, 173. [Google Scholar] [CrossRef]
- Popat, S.; Ahn, M.J.; Ekman, S.; Leighl, N.B.; Ramalingam, S.S.; Reungwetwattana, T.; Siva, S.; Tsuboi, M.; Wu, Y.L.; Yang, J.C. Osimertinib for EGFR-Mutant Non-Small-Cell Lung Cancer Central Nervous System Metastases: Current Evidence and Future Perspectives on Therapeutic Strategies. Target. Oncol. 2023, 18, 9–24. [Google Scholar] [CrossRef] [PubMed]
- Alix-Panabières, C.; Pantel, K. Liquid Biopsy: From Discovery to Clinical Application. Cancer Discov. 2021, 11, 858–873. [Google Scholar] [CrossRef] [PubMed]
- Cicenas, J.; Zalyte, E.; Bairoch, A.; Gaudet, P. Kinases and Cancer. Cancers 2018, 10, 63. [Google Scholar] [CrossRef]
- Bhanumathy, K.K.; Balagopal, A.; Vizeacoumar, F.S.; Vizeacoumar, F.J.; Freywald, A.; Giambra, V. Protein Tyrosine Kinases: Their Roles and Their Targeting in Leukemia. Cancers 2021, 13, 184. [Google Scholar] [CrossRef]
- Paul, M.K.; Mukhopadhyay, A.K. Tyrosine kinase—Role and significance in Cancer. Int. J. Med. Sci. 2004, 1, 101–115. [Google Scholar] [CrossRef]
- Hunter, T.; Cooper, J.A. Protein-tyrosine kinases. Annu. Rev. Biochem. 1985, 54, 897–930. [Google Scholar] [CrossRef]
- Sengupta, P.; Das, R.; Majumder, P.; Mukhopadhyay, D. Connecting the ends: Signaling via receptor tyrosine kinases and cytoskeletal degradation in neurodegeneration. Explor. Neurosci. 2024, 3, 1–26. [Google Scholar] [CrossRef]
- Hunter, T. Protein kinases and phosphatases: The yin and yang of protein phosphorylation and signalling. Cell 1995, 80, 225–236. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2000, 103, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- Pawson, T.; Gish, G.D.; Nash, P. SH2 domains, interaction modules and cellular wiring. Trends Cell Biol. 2001, 11, 504–511. [Google Scholar] [CrossRef]
- Zhou, Z.; Liu, Z.; Ou, Q.; Wu, X.; Wang, X.; Shao, Y.; Liu, H.; Yang, Y. Targeting FGFR in non-small cell lung cancer: Implications from the landscape of clinically actionable aberrations of FGFR kinases. Cancer Biol. Med. 2021, 18, 490–501. [Google Scholar] [CrossRef]
- Martellucci, S.; Clementi, L.; Sabetta, S.; Mattei, V.; Botta, L.; Angelucci, A. Src Family Kinases as Therapeutic Targets in Advanced Solid Tumors: What We Have Learned so Far. Cancers 2020, 12, 1448. [Google Scholar] [CrossRef]
- Huang, L.; Fu, L. Mechanisms of resistance to EGFR tyrosine kinase inhibitors. Acta Pharm. Sin. B 2015, 5, 390–401. [Google Scholar] [CrossRef]
- Cheng, Z.; Cui, H.; Wang, Y.; Yang, J.; Lin, C.; Shi, X.; Zou, Y.; Chen, J.; Jia, X.; Su, L. The advance of the third-generation EGFR-TKI in the treatment of non-small cell lung cancer (Review). Oncol. Rep. 2024, 51, 16. [Google Scholar] [CrossRef]
- Zhu, X.; Chen, L.; Liu, L.; Niu, X. EMT-Mediated Acquired EGFR-TKI Resistance in NSCLC: Mechanisms and Strategies. Front. Oncol. 2019, 9, 1044. [Google Scholar] [CrossRef]
- Laudadio, E.; Mangano, L.; Minnelli, C. Chemical Scaffolds for the Clinical Development of Mutant-Selective and Reversible Fourth-Generation EGFR-TKIs in NSCLC. ACS Chem. Biol. 2024, 19, 839–854. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Li, Y. Receptor tyrosine kinases: Biological functions and anticancer targeted therapy. MedComm 2023, 4, e446. [Google Scholar] [CrossRef]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Sig. Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef] [PubMed]
- Bethune, G.; Bethune, D.; Ridgway, N.; Xu, Z. Epidermal growth factor receptor (EGFR) in lung cancer: An overview and update. J. Thorac. Dis. 2010, 2, 48–51. [Google Scholar] [PubMed]
- Pophali, P.A.; Patnaik, M.M. The Role of New Tyrosine Kinase Inhibitors in Chronic Myeloid Leukemia. Cancer J. 2016, 22, 40–50. [Google Scholar] [CrossRef]
- Xu, L.; Xie, Y.; Gou, Q.; Cai, R.; Bao, R.; Huang, Y.; Tang, R. HER2-targeted therapies for HER2-positive early-stage breast cancer: Present and future. Front. Pharmacol. 2024, 15, 1446414. [Google Scholar] [CrossRef]
- Tomuleasa, C.; Tigu, A.B.; Munteanu, R.; Moldovan, C.-S.; Kegyes, D.; Onaciu, A.; Gulei, D.; Ghiaur, G.; Einsele, H.; Croce, C.M. Therapeutic advances of targeting receptor tyrosine kinases in cancer. Sig. Transduct. Target. Ther. 2024, 9, 201. [Google Scholar] [CrossRef]
- Wang, X.S.; Bai, Y.F.; Verma, V.; Yu, R.L.; Tian, W.; Ao, R.; Deng, Y.; Zhu, X.Q.; Liu, H.; Pan, H.X.; et al. Randomized Trial of First-Line Tyrosine Kinase Inhibitor With or Without Radiotherapy for Synchronous Oligometastatic EGFR-Mutated Non-Small Cell Lung Cancer. J. Natl. Cancer Inst. 2023, 115, 742–748. [Google Scholar] [CrossRef]
- Zhou, Q.; Yang, J.J.; Chen, Z.H.; Zhang, X.C.; Yan, H.H.; Xu, C.R.; Su, J.; Chen, H.J.; Tu, H.Y.; Zhong, W.Z.; et al. Serial cfDNA assessment of response and resistance to EGFR-TKI for patients with EGFR-L858R mutant lung cancer from a prospective clinical trial. J. Hematol. Oncol. 2016, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Jänne, P.A.; Yang, J.C.; Kim, D.W.; Planchard, D.; Ohe, Y.; Ramalingam, S.S.; Ahn, M.J.; Kim, S.W.; Su, W.C.; Horn, L.; et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 1689–1699. [Google Scholar] [CrossRef]
- Yang, J.C.; Ahn, M.J.; Kim, D.W.; Ramalingam, S.S.; Sequist, L.V.; Su, W.C.; Kim, S.W.; Kim, J.H.; Planchard, D.; Felip, E.; et al. Osimertinib in Pretreated T790M-Positive Advanced Non-Small-Cell Lung Cancer: AURA Study Phase II Extension Component. J. Clin. Oncol. 2017, 35, 1288–1296. [Google Scholar] [CrossRef] [PubMed]
- Christopoulos, P.; Bozorgmehr, F.; Brückner, L.; Chung, I.; Krisam, J.; Schneider, M.A.; Stenzinger, A.; Eickhoff, R.; Mueller, D.W.; Thomas, M. Brigatinib versus other second-generation ALK inhibitors as initial treatment of anaplastic lymphoma kinase positive non-small cell lung cancer with deep phenotyping: Study protocol of the ABP trial. BMC Cancer 2021, 21, 743. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, P.L.; Suri, Y.; Basu, A.; Koshkin, V.S.; Desai, A. Mechanisms of tyrosine kinase inhibitor resistance in renal cell carcinoma. Cancer Drug Resist. 2023, 6, 858–873. [Google Scholar] [CrossRef]
- Khaddour, K.; Jonna, S.; Deneka, A.; Patel, J.D.; Abazeed, M.E.; Golemis, E.; Borghaei, H.; Boumber, Y. Targeting the Epidermal Growth Factor Receptor in EGFR-Mutated Lung Cancer: Current and Emerging Therapies. Cancers 2021, 13, 3164. [Google Scholar] [CrossRef]
- Zhou, C. Multi-targeted tyrosine kinase inhibitors for the treatment of non-small cell lung cancer: An era of individualized therapy. Transl. Lung Cancer Res. 2012, 1, 72–77. [Google Scholar] [CrossRef]
- Gazdar, A.F. Activating and resistance mutations of EGFR in non-small-cell lung cancer: Role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene 2009, 28 (Suppl. S1), S24–S31. [Google Scholar] [CrossRef]
- Morgillo, F.; Della Corte, C.M.; Fasano, M.; Ciardiello, F. Mechanisms of resistance to EGFR-targeted drugs: Lung cancer. ESMO Open 2016, 1, e000060. [Google Scholar] [CrossRef]
- Wu, S.G.; Shih, J.Y. Management of acquired resistance to EGFR TKI-targeted therapy in advanced non-small cell lung cancer. Mol. Cancer 2018, 17, 38. [Google Scholar] [CrossRef]
- Poudel, G.; Tolland, M.G.; Hughes, T.P.; Pagani, I.S. Mechanisms of Resistance and Implications for Treatment Strategies in Chronic Myeloid Leukaemia. Cancers 2022, 14, 3300. [Google Scholar] [CrossRef] [PubMed]
- Birnboim-Perach, R.; Benhar, I. Using Combination therapy to overcome diverse challenges of Immune Checkpoint Inhibitors treatment. Int. J. Biol. Sci. 2024, 20, 3911–3922. [Google Scholar] [CrossRef]
- Cui, J.J. A New Challenging and Promising Era of Tyrosine Kinase Inhibitors. ACS Med. Chem. Lett. 2014, 5, 272–274. [Google Scholar] [CrossRef] [PubMed]
- Klaeger, S.; Heinzlmeir, S.; Wilhelm, M.; Polzer, H.; Vick, B.; Koenig, P.A.; Reinecke, M.; Ruprecht, B.; Petzoldt, S.; Meng, C.; et al. The target landscape of clinical kinase drugs. Science 2017, 358, eaan4368. [Google Scholar] [CrossRef]
- Codex, Y. Advancements in Targeted Kinase Inhibitors: Revolutionizing Personalized Medicine in Oncology. Yubetsu Codex Medicine & Physiology. Available online: https://codex.yubetsu.com/article/c020b487e78046d89a0ed3afaac73ec5 (accessed on 10 April 2025).
- Fukuda, S.; Suda, K.; Hamada, A.; Oiki, H.; Ohara, S.; Ito, M.; Soh, J.; Mitsudomi, T.; Tsutani, Y. Potential Utility of a 4th-Generation EGFR-TKI and Exploration of Resistance Mechanisms-An In Vitro Study. Biomedicines 2024, 12, 1412. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Springob, K.; Kutchan, T.M. Introduction to the Different Classes of Natural Products. In Plant-Derived Natural Products; Osbourn, A., Lanzotti, V., Eds.; Springer: New York, NY, USA, 2009. [Google Scholar] [CrossRef]
- Anwar, S.; Kumar, V.; Kanwal, B.; Yahia, E.A. A review on utilization of ashoka plant in oxidative stress induced women reproductive health complications. IJCRT 2023, 11, f249–f258. [Google Scholar]
- Younus, H.; Anwar, S. Prevention of non-enzymatic glycosylation (glycation): Implication in the treatment of diabetic complication. Int. J. Health Sci. 2016, 10, 261–277. [Google Scholar] [CrossRef]
- Alsahli, M.A.; Almatroodi, S.A.; Almatroudi, A.; Khan, A.A.; Anwar, S.; Almutary, A.G.; Alrumaihi, F.; Rahmani, A.H. 6-Gingerol, a major ingredient of ginger attenuates diethylnitrosamine-induced liver injury in rats through the modulation of oxidative stress and anti-inflammatory activity. Mediat. Inflamm. 2021, 2021, 6661937. [Google Scholar] [CrossRef]
- Anwar, S.; Almatroudi, A.; Allemailem, K.S.; Jacob Joseph, R.; Khan, A.A.; Rahmani, A.H. Protective Effects of Ginger Extract against Glycation and Oxidative Stress-Induced Health Complications: An In Vitro Study. Processes 2020, 8, 468. [Google Scholar] [CrossRef]
- Rahmani, A.H.; Alsahli, M.A.; Almatroudi, A.; Almogbel, M.A.; Khan, A.A.; Anwar, S.; Almatroodi, S.A. The Potential Role of Apigenin in Cancer Prevention and Treatment. Molecules 2022, 27, 6051. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, A.H.; Babiker, A.Y.; Anwar, S. Hesperidin, a Bioflavonoid in Cancer Therapy: A Review for a Mechanism of Action through the Modulation of Cell Signaling Pathways. Molecules 2023, 28, 5152. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Zhu, G.; Peng, Q.; Li, X.; Zou, X.; Zhang, W.; Zhao, L.; Li, X.; Wu, P.; Luo, A.; et al. Natural products combating EGFR-TKIs resistance in cancer. Eur. J. Med. Chem. Rep. 2025, 13, 100251. [Google Scholar] [CrossRef]
- Paller, C.J.; Denmeade, S.R.; Carducci, M.A. Challenges of conducting clinical trials of natural products to combat cancer. Clin. Adv. Hematol. Oncol. 2016, 14, 447–455. [Google Scholar]
- Al-Yozbaki, M.; Wilkin, P.J.; Gupta, G.K.; Wilson, C.M. Therapeutic potential of natural compounds in lung cancer. Curr. Med. Chem. 2021, 28, 7988–8002. [Google Scholar] [CrossRef]
- Jeong, H.; Phan, A.N.H.; Choi, J.W. Anti-cancer Effects of Polyphenolic Compounds in Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor-resistant Non-small Cell Lung Cancer. Pharmacogn. Mag. 2017, 13, 595–599. [Google Scholar] [CrossRef]
- Thoennissen, N.H.; O’Kelly, J.; Lu, D.; Iwanski, G.B.; La, D.T.; Abbassi, S.; Leiter, A.; Karlan, B.; Mehta, R.; Koeffler, H.P. Capsaicin causes cell-cycle arrest and apoptosis in ER-positive and -negative breast cancer cells by modulating the EGFR/HER-2 pathway. Oncogene 2010, 29, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.P.; Yun, H.J.; Choi, J.H.; Han, E.H.; Kim, H.G.; Song, G.Y.; Kwon, K.I.; Jeong, T.C.; Jeong, H.G. Suppression of EGF-induced tumor cell migration and matrix metalloproteinase-9 expression by capsaicin via the inhibition of EGFR-mediated FAK/Akt, PKC/Raf/ERK, p38 MAPK, and AP-1 signaling. Mol. Nutr. Food Res. 2011, 55, 594–605. [Google Scholar] [CrossRef]
- Tang, Y.; Sun, A.; Liu, R.; Zhang, Y. Simultaneous determination of fangchinoline and tetrandrine in Stephania tetrandra S. Moore by using 1-alkyl-3-methylimidazolium-based ionic liquids as the RP-HPLC mobile phase additives. Anal. Chim. Acta 2013, 767, 148–154. [Google Scholar] [CrossRef]
- Guo, B.; Zhang, T.; Su, J.; Wang, K.; Li, X. Oxymatrine targets EGFR(p-Tyr845) and inhibits EGFR-related signaling pathways to suppress the proliferation and invasion of gastric cancer cells. Cancer Chemother. Pharmacol. 2015, 75, 353–363. [Google Scholar] [CrossRef]
- Liu, T.; Liu, X.; Li, W. Tetrandrine, a Chinese plant-derived alkaloid, is a potential candidate for cancer chemotherapy. Oncotarget 2016, 7, 40800–40815. [Google Scholar] [CrossRef]
- Suh, Y.A.; Jo, S.Y.; Lee, H.Y.; Lee, C. Inhibition of IL-6/STAT3 axis and targeting Axl and Tyro3 receptor tyrosine kinases by apigenin circumvent taxol resistance in ovarian cancer cells. Int. J. Oncol. 2014, 46, 1405–1411. [Google Scholar] [CrossRef]
- Huang, S.; Yu, M.; Shi, N.; Zhou, Y.; Li, F.; Li, X.; Huang, X.; Jin, J. Apigenin and Abivertinib, a novel BTK inhibitor synergize to inhibit diffuse large B-cell lymphoma in vivo and vitro. J. Cancer 2020, 11, 2123–2132. [Google Scholar] [CrossRef]
- Leung, H.W.; Yang, W.H.; Lai, M.Y.; Lin, C.J.; Lee, H.Z. Inhibition of 12-lipoxygenase during baicalein-induced human lung nonsmall carcinoma H460 cell apoptosis. Food Chem. Toxicol. 2007, 45, 403–411. [Google Scholar] [CrossRef]
- Hong, R.L.; Spohn, W.H.; Hung, M.C. Curcumin inhibits tyrosine kinase activity of p185 neu and also depletes p185 neu. Clin. Cancer Res. 1999, 5, 1884–1891. [Google Scholar] [PubMed]
- Kim, K.C.; Baek, S.H.; Lee, C. Curcumin-induced downregulation of Axl receptor tyrosine kinase inhibits cell proliferation and circumvents chemoresistance in non-small lung cancer cells. Int. J. Oncol. 2015, 47, 2296–2303. [Google Scholar] [CrossRef] [PubMed]
- Hong, Z.; Cao, X.; Li, N.; Zhang, Y.; Lan, L.; Zhou, Y.; Pan, X.; Shen, L.; Yin, Z.; Luo, L. Luteolin is effective in the non-small cell lung cancer model with L858R/T790M EGF receptor mutation and erlotinib resistance. Br. J. Pharmacol. 2014, 171, 2842–2853. [Google Scholar] [CrossRef]
- Yu, X.; Gao, F.; Li, W.; Zhou, L.; Liu, W.; Li, M. Formononetin inhibits tumor growth by suppression of EGFR-Akt-Mcl-1 axis in non-small cell lung cancer. J. Exp. Clin. Cancer Res. 2020, 39, 62. [Google Scholar] [CrossRef]
- Wang, L.; Chen, N.; Cheng, H. Fisetin inhibits vascular endothelial growth factor-induced angiogenesis in retinoblastoma cells. Oncol. Lett. 2020, 20, 1239–1244. [Google Scholar] [CrossRef]
- Ferry, D.R.; Smith, A.; Malkhandi, J.; Fyfe, D.W.; de Takats, P.; Anderson, D.; Baker, J.; Kerr, D.J. Phase I clinical trial of the flavonoid quercetin: Pharmacokinetics and evidence for in vivo tyrosine kinase inhibition. Clin. Cancer Res. 1996, 2, 659–668. [Google Scholar]
- Twum, Y.; Marshall, K.; Gao, W. Caffeic acid phenethyl ester surmounts acquired resistance of AZD9291 in non-small cell lung cancer cells. BioFactors 2023, 49, 1143–1157. [Google Scholar] [CrossRef]
- Yang, C.; Wang, X.; Lu, G.; Picinich, S.C. Cancer prevention by tea: Animal studies, molecular mechanisms and human relevance. Nat. Rev. Cancer 2009, 9, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Farabegoli, F.; Govoni, M.; Spisni, E.; Papi, A. EGFR inhibition by (-)-epigallocatechin-3-gallate and IIF treatments reduces breast cancer cell invasion. Biosci. Rep. 2017, 37, BSR20170168. [Google Scholar] [CrossRef]
- Shimizu, M.; Adachi, S.; Masuda, M.; Kozawa, O.; Moriwaki, H. Cancer chemoprevention with green tea catechins by targeting receptor tyrosine kinases. Mol. Nutr. Food Res. 2011, 55, 832–843. [Google Scholar] [CrossRef]
- Milligan, S.A.; Burke, P.; Coleman, D.T.; Bigelow, R.L.; Steffan, J.J.; Carroll, J.L.; Williams, B.J.; Cardelli, J.A. The green tea polyphenol EGCG potentiates the antiproliferative activity of c-Met and epidermal growth factor receptor inhibitors in non-small cell lung cancer cells. Clin. Cancer Res. 2009, 15, 4885–4894. [Google Scholar] [CrossRef] [PubMed]
- Minnelli, C.; Cianfruglia, L.; Laudadio, E.; Mobbili, G.; Galeazzi, R.; Armeni, T. Effect of Epigallocatechin-3-Gallate on EGFR Signaling and Migration in Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2021, 22, 11833. [Google Scholar] [CrossRef]
- Wang, J.; Sun, P.; Wang, Q.; Zhang, P.; Wang, Y.; Zi, C.; Wang, X.; Sheng, J. (−)-Epigallocatechin-3-gallate derivatives combined with cisplatin exhibit synergistic inhibitory effects on non-small-cell lung cancer cells. Cancer Cell Int. 2019, 19, 266. [Google Scholar] [CrossRef] [PubMed]
- Labrecque, L.; Lamy, S.; Chapus, A.; Mihoubi, S.; Durocher, Y.; Cass, B.; Bojanowski, M.W.; Gingras, D.; Béliveau, R. Combined inhibition of PDGF and VEGF receptors by ellagic acid, a dietary-derived phenolic compound. Carcinogenesis 2005, 26, 821–826. [Google Scholar] [CrossRef]
- Zeng, Y.; Ma, J.; Xu, L.; Wu, D. Natural product gossypol and its derivatives in precision cancer medicine. Curr. Med. Chem. 2019, 26, 1849–1873. [Google Scholar] [CrossRef]
- Yu, Y.; Fan, S.M.; Ye, Y.C.; Tashiro, S.i.; Onodera, S.; Ikejima, T. The tyrphostin AG1478 augments oridonin-induced A431 cell apoptosis by blockage of JNK MAPK and enhancement of oxidative stress. Free Radic. Res. 2012, 46, 1393–1405. [Google Scholar] [CrossRef]
- Pan, J.; Lee, Y.; Zhang, Q.; Xiong, D.; Wan, T.C.; Wang, Y.; You, M. Honokiol Decreases Lung Cancer Metastasis through Inhibition of the STAT3 Signaling Pathway. Cancer Prev. Res. 2017, 10, 133–141. [Google Scholar] [CrossRef]
- Wang, X.; Liu, Q.; Fu, Y.; Ding, R.B.; Qi, X.; Zhou, X.; Sun, Z.; Bao, J. Magnolol as a Potential Anticancer Agent: A Proposed Mechanistic Insight. Molecules 2022, 27, 6441. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Notas, G.; Nifli, A.P.; Kampa, M.; Vercauteren, J.; Kouroumalis, E.; Castanas, E. Resveratrol exerts its antiproliferative effect on HepG2 hepatocellular carcinoma cells, by inducing cell cycle arrest, and NOS activation. Biochim. Biophys. Acta 2006, 1760, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Zhang, T.; Jing, S.; Zuo, P.; Li, T.; Wang, Y.; Xing, S.; Zhang, J.; Wei, Z. 20 (S)-Ginsenoside Rg3 inhibits lung cancer cell proliferation by targeting EGFR-mediated Ras/Raf/MEK/ERK pathway. Am. J. Chin. Med. 2021, 49, 753–765. [Google Scholar] [CrossRef]
- Tan, Q.; Lin, S.; Zeng, Y.; Yao, M.; Liu, K.; Yuan, H.; Liu, C.; Jiang, G. Ginsenoside Rg3 attenuates the osimertinib resistance by reducing the stemness of non-small cell lung cancer cells. Environ. Toxicol. 2020, 35, 643–651. [Google Scholar] [CrossRef]
- Liu, T.; Zuo, L.; Guo, D.; Chai, X.; Xu, J.; Cui, Z.; Wang, Z.; Hou, C. Ginsenoside Rg3 regulates DNA damage in non-small cell lung cancer cells by activating VRK1/P53BP1 pathway. Biomed. Pharmacother. 2019, 120, 109483. [Google Scholar] [CrossRef]
- Xu, F.; Cui, W.Q.; Wei, Y.; Cui, J.; Qiu, J.; Hu, L.L.; Gong, W.-Y.; Dong, J.-C.; Liu, B.-J. Astragaloside IV inhibits lung cancer progression and metastasis by modulating macrophage polarization through AMPK signaling. J. Exp. Clin. Cancer Res. 2018, 37, 207. [Google Scholar] [CrossRef]
- Jiang, K.; Lu, Q.; Li, Q.; Ji, Y.; Chen, W.; Xue, X. Astragaloside IV inhibits breast cancer cell invasion by suppressing Vav3 mediated Rac1/MAPK signaling. Int. Immunopharmacol. 2017, 42, 195–202. [Google Scholar] [CrossRef]
- Li, L.; Wu, J.; Zheng, F.; Tang, Q.; Wu, W.; Hann, S.S. Inhibition of EZH2 via activation of SAPK/JNK and reduction of p65 and DNMT1 as a novel mechanism in inhibition of human lung cancer cells by polyphyllin I. J. Exp. Clin. Cancer Res. 2016, 35, 112. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Chen, W.; Xu, Y.; Lv, X.; Zhang, M.; Jiang, H. Polyphyllin I modulates MALAT1/STAT3 signaling to induce apoptosis in gefitinib-resistant non-small cell lung cancer. Toxicol. Appl. Pharmacol. 2018, 356, 1–7. [Google Scholar] [CrossRef]
- Jing, S.-Y.; Wu, Z.-D.; Zhang, T.H.; Zhang, J.; Wei, Z.-Y. In vitro antitumor effect of cucurbitacin E on human lung cancer cell line and its molecular mechanism. Chin. J. Nat. Med. 2020, 18, 483–490. [Google Scholar]
- Xie, Y.L.; Tao, W.H.; Yang, T.X.; Qiao, J.G. Anticancer effect of cucurbitacin B on MKN-45 cells via inhibition of the JAK2/STAT3 signaling pathway. Exp. Ther. Med. 2016, 12, 2709–2715. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.Y.; Cao, D.; Ren, Q.N.; Zhang, S.S.; Zhou, N.N.; Mai, S.J.; Feng, B.; Wang, H.Y. Combination treatment with inhibitors of erk and autophagy enhances antitumor activity of betulinic acid in non–small-cell lung cancer in vivo and in vitro. Front. Pharmacol. 2021, 12, 684243. [Google Scholar] [CrossRef] [PubMed]
- Ahmadu, A.A.; Delehouzé, C.; Haruna, A.; Mustapha, L.; Lawal, B.A.; Udobre, A.; Baratte, B.; Triscornia, C.; Autret, A.; Robert, T.; et al. Betulin, a Newly Characterized Compound in Acacia auriculiformis Bark, Is a Multi-Target Protein Kinase Inhibitor. Molecules 2021, 26, 4599. [Google Scholar] [CrossRef]
- Jung, Y.Y.; Um, J.-Y.; Sethi, G.; Ahn, K.S. Potential Application of Leelamine as a Novel Regulator of Chemokine-Induced Epithelial-to-Mesenchymal Transition in Breast Cancer Cells. Int. J. Mol. Sci. 2022, 23, 9848. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, D.; Huang, Y.; Gao, Y.; Qian, S. Biopharmaceutics classification and intestinal absorption study of apigenin. Int. J. Pharm. 2012, 436, 311–317. [Google Scholar] [CrossRef]
- DL McKay, J.B. Blumberg a review of the bioactivity and potential health benefits of chamomile tea (Matricaria recutita L.). Phytother. Res. 2006, 20, 519–530. [Google Scholar] [CrossRef]
- Papay, Z.E.; Kosa, A.; Boddi, B.; Merchant, Z.; Saleem, I.Y.; Zariwala, M.G.; Klebovich, I.; Somavarapu, S.; Antal, I. Study on the pulmonary delivery system of apigenin-loaded albumin nanocarriers with antioxidant activity. J. Aerosol Med. Pulm. Drug Deliv. 2017, 30, 274–288. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Huang, K.M. In vitro anti-inflammatory effect of apigenin in the Helicobacter pylori-infected gastric adenocarcinoma cells. Food Chem. Toxicol. 2013, 53, 376–383. [Google Scholar] [CrossRef]
- Zhu, Z.Y.; Gao, T.; Huang, Y.; Xue, J.; Xie, M.L. Apigenin ameliorates hypertension-induced cardiac hypertrophy and down-regulates cardiac hypoxia inducible factor-lalpha in rats. Food Funct. 2016, 7, 1992–1998. [Google Scholar] [CrossRef]
- Ozcelik, B.; Kartal, M.; Orhan, I. Cytotoxicity, antiviral and antimicrobial activities of alkaloids, flavonoids, and phenolic acids. Pharm. Biol. 2011, 49, 396. [Google Scholar] [CrossRef]
- Hu, W.J.; Liu, J.; Zhong, L.K.; Wang, J. Apigenin enhances the antitumor effects of cetuximab in nasopharyngeal carcinoma by inhibiting EGFR signaling. Biomed. Pharmacother. 2018, 102, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Tian, D.; Liao, X.; Zhang, Y.; Xiao, J.; Chen, W.; Liu, Q.; Chen, Y.; Li, D.; Zhu, L.; et al. Apigenin Combined with Gefitinib Blocks Autophagy Flux and Induces Apoptotic Cell Death Through Inhibition of HIF-1α, c-Myc, p-EGFR, and Glucose Metabolism in EGFR L858R+T790M-Mutated H1975 Cells. Front. Pharmacol. 2019, 10, 260. [Google Scholar] [CrossRef] [PubMed]
- Maher, H.M.; Alzoman, N.Z.; Shehata, S.M.; Abahussain, A.O. Comparative pharmacokinetic profiles of selected irreversible tyrosine kinase inhibitors, neratinib and pelitinib, with apigenin in rat plasma by UPLC–MS/MS. J. Pharm. Biomed. Anal. 2017, 137, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Sinha, S.; Rathaur, P.; Vora, J.; Jha, P.C.; Johar, K.; Rawal, R.M.; Shrivastava, N. Reckoning apigenin and kaempferol as a potential multi-targeted inhibitor of EGFR/HER2-MEK pathway of metastatic colorectal cancer identified using rigorous computational workflow. Mol. Divers. 2022, 26, 3337–3356. [Google Scholar] [CrossRef]
- Kim, K.-C.; Choi, E.-H.; Lee, C. Axl receptor tyrosine kinase is a novel target of apigenin for the inhibition of cell proliferation. Int. J. Mol. Med. 2014, 34, 592–598. [Google Scholar] [CrossRef]
- Chang, J.H.; Cheng, C.W.; Yang, Y.C.; Chen, W.S.; Hung, W.Y.; Chow, J.M.; Chen, P.S.; Hsiao, M.; Lee, W.J.; Chien, M.H. Downregulating CD26/DPPIV by apigenin modulates the interplay between Akt and Snail/Slug signaling to restrain metastasis of lung cancer with multiple EGFR statuses. J. Exp. Clin. Cancer Res. 2018, 37, 199. [Google Scholar] [CrossRef]
- Osada, M.; Imaoka, S.; Funae, Y. Apigenin suppresses the expression of VEGF, an important factor for angiogenesis, in endothelial cells via degradation of HIF-1α protein. FEBS Lett. 2004, 575, 59–63. [Google Scholar] [CrossRef]
- Ansó, E.; Zuazo, A.; Irigoyen, M.; Urdaci, M.C.; Rouzaut, A.; Martínez-Irujo, J.J. Flavonoids inhibit hypoxia-induced vascular endothelial growth factor expression by a HIF-1 independent mechanism. Biochem. Pharmacol. 2010, 79, 1600–1609. [Google Scholar] [CrossRef]
- Ortiz, M.A.; Mikhailova, T.; Li, X.; Porter, B.A.; Bah, A.; Kotula, L. Src family kinases, adaptor proteins and the actin cytoskeleton in epithelial-to-mesenchymal transition. Cell Commun. Signal 2021, 19, 67. [Google Scholar] [CrossRef]
- Cathcart, M.C.; Useckaite, Z.; Drakeford, C.; Semik, V.; Lysaght, J.; Gately, K.; O’Byrne, K.J.; Pidgeon, G.P. Anti-cancer effects of baicalein in non-small cell lung cancer in-vitro and in-vivo. BMC Cancer 2016, 16, 707. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jelić, D.; Lower-Nedza, A.D.; Brantner, A.H.; Blažeković, B.; Bian, B.; Yang, J.; Brajša, K.; Vladimir-Knežević, S. Baicalin and Baicalein Inhibit Src Tyrosine Kinase and Production of IL-6. J. Chem. 2016, 2016, 2510621. [Google Scholar] [CrossRef]
- Park, H.-J.; Park, S.-H.; Choi, Y.-H.; Chi, G.-Y. The Root Extract of Scutellaria baicalensis Induces Apoptosis in EGFR TKI-Resistant Human Lung Cancer Cells by Inactivation of STAT3. Int. J. Mol. Sci. 2021, 22, 5181. [Google Scholar] [CrossRef]
- Jin, H.; Qiao, F.; Wang, Y.; Xu, Y.; Shang, Y. Curcumin inhibits cell proliferation and induces apoptosis of human non-small cell lung cancer cells through the up regulation of miR-192-5p and suppression of PI3K/Akt signaling pathway. Oncol. Rep. 2015, 34, 2782–2789. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wei, C.; Xi, Z. Curcumin suppresses proliferation and invasion in non-small cell lung cancer by modulation of MTA1-mediated Wnt/β-catenin pathway. Vitr. Cell. Dev. Biol. Anim. 2014, 50, 840–850. [Google Scholar] [CrossRef]
- Li, Y.; Chao, Y.; Fang, Y.; Wang, J.; Wang, M.; Zhang, H.; Ying, M.; Zhu, X.; Wang, H. MTA1 promotes the invasion and migration of non-small cell lung cancer cells by down regulating miR-125b. J. Exp. Clin. Cancer Res. 2013, 32, 33. [Google Scholar] [CrossRef]
- Xiao, K.; Jiang, J.; Guan, C.; Dong, C.; Wang, G.; Bai, L.; Sun, J.; Hu, C.; Bai, C. Curcumin induces autophagy via activating the AMPK signaling pathway in lung adenocarcinoma cells. J. Pharmacol. Sci. 2013, 123, 102–109. [Google Scholar] [CrossRef]
- Lee, J.; Lee, Y.; Chang, G.; Yu, S.L.; Hsieh, W.Y.; Chen, J.J.; Chen, H.W.; Yang, P.C. Curcumin induces EGFR degradation in lung adenocarcinoma modulates p38 activation in intestine: The versatile adjuvant for gefitinib therapy. PLoS ONE 2011, 6, e23756. [Google Scholar] [CrossRef]
- Chen, P.; Huang, H.; Wang, Y.; Jin, J.; Long, W.G.; Chen, K.; Zhao, X.H.; Chen, C.G.; Li, J. Curcumin overcome primary gefitinib resistance in non-small-cell lung cancer cells through inducing autophagy-related cell death. J. Exp. Clin. Cancer Res. 2019, 38, 254. [Google Scholar] [CrossRef]
- Yamauchi, Y.; Izumi, Y.; Yamamoto, J.; Nomori, H. Coadministration of erlotinib and curcumin augmentatively reduces cell viability in lung cancer cells. Phytother. Res. 2014, 28, 728–735. [Google Scholar] [CrossRef]
- Li, S.; Liu, Z.; Zhu, F.; Fan, X.; Wu, X.; Zhao, H.; Jiang, L. Curcumin lowers erlotinib resistance in non-small cell lung carcinoma cells with mutated EGF receptor. Oncol. Res. 2014, 21, 137–144. [Google Scholar] [CrossRef]
- Ye, M.X.; Zhao, Y.L.; Li, Y.; Miao, Q.; Li, Z.K.; Ren, X.L.; Song, L.Q.; Yin, H.; Zhang, J. Curcumin reverses cis-platin resistance and promotes human lung adenocarcinoma A549/DDP cell apoptosis through HIF-1α and caspase-3 mechanisms. Phytomedicine 2012, 19, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Chanvorachote, P.; Pongrakhananon, V.; Wannachaiyasit, S.; Luanpitpong, S.; Rojanasakul, Y.; Nimmannit, U. Curcumin sensitizes lung cancer cells to cisplatin-induced apoptosis through superoxide anion-mediated Bcl-2 degradation. Cancer Investig. 2009, 27, 624–635. [Google Scholar] [CrossRef] [PubMed]
- Dorai, T.; Gehani, N.; Katz, A. Therapeutic potential of curcumin in human prostate cancer. II. Curcumin inhibits tyrosine kinase activity of epidermal growth factor receptor and depletes the protein [Internet]. Mol. Urol. 2000, 4, 1–6. [Google Scholar] [PubMed]
- Kim, J.H.; Xu, C.; Keum, Y.S.; Reddy, B.; Conney, A.; Kong, A.N.T. Inhibition of EGFR signaling in human prostate cancer PC-3 cells by combination treatment with β-phenylethyl isothiocyanate and curcumin [Internet]. Carcinogenesis 2006, 27, 475–482. [Google Scholar] [CrossRef]
- A Open-label Prospective Cohort Trial of Curcumin Plus Tyrosine Kinase Inhibitors (TKI) for EGFR-Mutant Advanced NSCLC (CURCUMIN). Available online: https://clinicaltrials.gov/study/NCT02321293?term=Tyrosine%20Kinase%20Inhibitor&intr=Curcumin&rank=1 (accessed on 16 June 2025).
- Phase II Trial to Modulate Intermediate Endpoint Biomarkers in Former and Current Smokers. Available online: https://clinicaltrials.gov/study/NCT03598309?intr=Curcumin%20C3%20complex%C2%AE.%20Drug:%20Lovaza%C2%AE.&rank=1 (accessed on 16 June 2025).
- The Thoracic Peri-Operative Integrative Surgical Care Evaluation Trial—Stage III (POISE). Available online: https://clinicaltrials.gov/study/NCT04871412?intr=Curcumin&term=Thoracic%20Peri-Operative%20Integrative%20Surgical%20Care%20&rank=1 (accessed on 16 June 2025).
- Dev, S.S.; Farghadani, R.; Abidin, S.A.; Othman, I.; Naidu, R. Flavonoids as receptor tyrosine kinase inhibitors in lung cancer. J. Funct. Foods 2023, 110, 105845. [Google Scholar]
- Rahmani, A.H.; Almatroudi, A.; Allemailem, K.S.; Khan, A.A.; Almatroodi, S.A. The Potential Role of Fisetin, a Flavonoid in Cancer Prevention and Treatment. Molecules 2022, 27, 9009. [Google Scholar] [CrossRef]
- Tabasum, S.; Singh, R.P. Fisetin suppresses migration, invasion and stem-cell-like phenotype of human non-small cell lung carcinoma cells via attenuation of epithelial to mesenchymal transition. Chem. Biol. Interact. 2019, 303, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Huang, Y.; Nie, S.; Zhou, S.; Gao, X.; Chen, G. Biological effects and mechanisms of fisetin in cancer: A promising anti-cancer agent. Eur. J. Med. Res. 2023, 28, 297. [Google Scholar] [CrossRef]
- Sabarwal, A.; van Rooyen, J.C.; Caburet, J.; Avgenikos, M.; Dheeraj, A.; Ali, M.; Mishra, D.; de Meester, J.S.B.; Stander, S.; van Otterlo, W.A.L.; et al. A novel 4′-brominated derivative of fisetin induces cell cycle arrest and apoptosis and inhibits EGFR/ERK1/2/STAT3 pathways in non-small-cell lung cancer without any adverse effects in mice. FASEB J. 2022, 36, e22654. [Google Scholar] [CrossRef] [PubMed]
- Almatroodi, S.A.; Almatroudi, A.; Khan, A.A.; Rahmani, A.H. Potential Therapeutic Targets of Formononetin, a Type of Methoxylated Isoflavone, and Its Role in Cancer Therapy through the Modulation of Signal Transduction Pathways. Int. J. Mol. Sci. 2023, 24, 9719. [Google Scholar] [CrossRef]
- Tay, K.C.; Tan, L.T.; Chan, C.K.; Hong, S.L.; Chan, K.G.; Yap, W.H.; Pusparajah, P.; Lee, L.H.; Goh, B.H. Formononetin: A Review of Its Anticancer Potentials and Mechanisms. Front. Pharmacol. 2019, 10, 820. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Sun, W.X.; Zheng, C.S.; Han, H.W.; Wang, X.; Zhang, Y.H.; Qiu, H.Y.; Tang, C.Y.; Qi, J.L.; Lu, G.H.; et al. Synthesis, characterization and biological evaluation of formononetin derivatives as novel EGFR inhibitors via inhibiting growth, migration and inducing apoptosis in breast cancer cell line. RSC Adv. 2017, 7, 48404–48419. [Google Scholar] [CrossRef]
- López-Lázaro, M. Distribution and biological activities of the flavonoid luteolin. Mini Rev. Med. Chem. 2009, 9, 31–59. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.T.; Hwang, J.J.; Lee, P.P.; Ke, F.C.; Huang, J.H.; Huang, C.J.; Kandaswami, C.; Middleton, E., Jr.; Lee, M.T. Effects of luteolin and quercetin, inhibitors of tyrosine kinase, on cell growth and metastasis-associated properties in A431 cells overexpressing epidermal growth factor receptor. Br. J. Pharmacol. 1999, 128, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Cai, X.; Zheng, X.; Zhu, X.; Feng, J.; Wang, X. Luteolin inhibits viability, migration, angiogenesis and invasion of non-small cell lung cancer vascular endothelial cells via miR-133a-3p/purine rich element binding protein B-mediated MAPK and PI3K/Akt signaling pathways. Tissue Cell 2022, 75, 101740. [Google Scholar] [CrossRef]
- Pratheeshkumar, P.; Son, Y.O.; Divya, S.P.; Roy, R.V.; Hitron, J.A.; Wang, L.; Kim, D.; Dai, J.; Asha, P.; Zhang, Z.; et al. Luteolin inhibits Cr(VI)-induced malignant cell transformation of human lung epithelial cells by targeting ROS mediated multiple cell signaling pathways. Toxicol. Appl. Pharmacol. 2014, 281, 230–241. [Google Scholar] [CrossRef]
- Patel, N.; Patel, M.; Patel, A.; Patel, S.; Sakariya, D.; Parmar, A.; Sarkar, R.; Patel, M.; Rohit, S.; Patel, S.; et al. Investigating the role of natural flavonoids in VEGFR inhibition: Molecular modelling and biological activity in A549 lung cancer cells. J. Mol. Struct. 2025, 1322, 140392. [Google Scholar] [CrossRef]
- Huang, G.; Liu, X.; Jiang, T.; Cao, Y.; Sang, M.; Song, X.; Zhou, B.; Qu, H.; Cai, H.; Xing, D.; et al. Luteolin overcomes acquired resistance to osimertinib in non-small cell lung cancer cells by targeting the HGF-MET-Akt pathway. Am. J. Cancer Res. 2023, 13, 4145–4162. [Google Scholar]
- Çetinkaya, M.; Baran, Y. Therapeutic Potential of Luteolin on Cancer. Vaccines 2023, 11, 554. [Google Scholar] [CrossRef]
- Sakurai, M.A.; Ozaki, Y.; Okuzaki, D.; Naito, Y.; Sasakura, T.; Okamoto, A.; Tabara, H.; Inoue, T.; Hagiyama, M.; Ito, A. Gefitinib and Luteolin Cause Growth Arrest of Human Prostate Cancer PC-3 Cells via Inhibition of Cyclin G-Associated Kinase and Induction of MiR-630. PLoS ONE 2014, 9, e100124. [Google Scholar] [CrossRef]
- Almatroodi, S.A.; Alsahli, M.A.; Almatroudi, A.; Verma, A.K.; Aloliqi, A.; Allemailem, K.S.; Khan, A.A.; Rahmani, A.H. Potential Therapeutic Targets of Quercetin, a Plant Flavonol, and Its Role in the Therapy of Various Types of Cancer through the Modulation of Various Cell Signaling Pathways. Molecules 2021, 26, 1315. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, S.; Malik, M.Y.; Rashid, M.; Singh, S.; Tiwari, V.; Gupta, P.; Shukla, S.; Singh, S.; Wahajuddin, M. Mechanistic exploration of quercetin against metronidazole induced neurotoxicity in rats: Possible role of nitric oxide isoforms and inflammatory cytokines. Neurotoxicology 2020, 79, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yao, J.; Han, C.; Yang, J.; Chaudhry, M.T.; Wang, S.; Liu, H.; Yin, Y. Quercetin, Inflammation and Immunity. Nutrients 2016, 8, 167. [Google Scholar] [CrossRef]
- Tang, S.-M.; Deng, X.-T.; Zhou, J.; Li, Q.-P.; Ge, X.-X.; Miao, L. Pharmacological basis and new insights of quercetin action in respect to its anti-cancer effects. Biomed. Pharm. Ther. 2020, 121, 109604. [Google Scholar] [CrossRef]
- Elumalai, P.; Ezhilarasan, D.; Raghunandhakumar, S. Quercetin inhibits the epithelial to mesenchymal transition through suppressing Akt mediated nuclear translocation of β-catenin in lung cancer cell line. Nutr. Cancer 2022, 74, 1894–1906. [Google Scholar] [CrossRef]
- Baby, B.; Antony, P.; Vijayan, R. Interactions of quercetin with receptor tyrosine kinases associated with human lung carcinoma. Nat. Product Res. 2018, 32, 2928–2931. [Google Scholar] [CrossRef] [PubMed]
- Basha, S.H.; Bethapudi, P.; Majji Rambabu, F. Anti-angiogenesis property by Quercetin compound targeting VEGFR2 elucidated in a computational approach. Eur. J. Biotechnol. Biosci. 2014, 2, 30–46. [Google Scholar]
- Hou, D.X.; Kumamoto, T. Flavonoids as protein kinase inhibitors for cancer chemoprevention on: Direct binding and molecular modeling. Antioxid. Redox Signal 2010, 13, 691–719. [Google Scholar] [CrossRef]
- Walker, E.H.; Pacold, M.E.; Perisic, O.; Stephens, L.; Hawkins, P.T.; Wymann, M.P.; Williams, R.L. Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol. Cell 2000, 6, 909–919. [Google Scholar] [CrossRef]
- Navarro-Núñez, L.; Rivera, J.; Guerrero, J.A.; Martinez, C.; Vicente, V.; Lozano, M.L. Differential effects of quercetin, apigenin and genistein on signaling pathways of protease-activated receptors PAR1 and PAR4 in platelets. Br. J. Pharmacol. 2009, 158, 1548–1556. [Google Scholar] [CrossRef]
- Kim, J.K.; Park, S.U. Quercetin and its role in biological functions: An updated review. EXCLI J. 2018, 17, 856–863. [Google Scholar] [CrossRef]
- Espíndola, K.M.M.; Ferreira, R.G.; Narvaez, L.E.M.; Silva Rosario, A.C.R.; da Silva, A.H.M.; Silva, A.G.B.; Vieira, A.P.O.; Monteiro, M.C. Chemical and Pharmacological Aspects of Caffeic Acid and Its Activity in Hepatocarcinoma. Front. Oncol. 2019, 9, 541. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mirzaei, S.; Gholami, M.H.; Zabolian, A.; Saleki, H.; Farahani, M.V.; Hamzehlou, S.; Far, F.B.; Sharifzadeh, S.O.; Samarghandian, S.; Khan, H.; et al. Caffeic acid and its derivatives as potential modulators of oncogenic molecular pathways: New hope in the fight against cancer. Pharmacol. Res. 2021, 171, 105759. [Google Scholar] [CrossRef] [PubMed]
- Ning, X.; Ren, X.; Xie, X.; Yan, P.; Wang, D.; Huang, X. A caffeic acid phenethyl ester analog inhibits the proliferation of nasopharyngeal carcinoma cells via targeting epidermal growth factor receptor. J. Biochem. Mol. Toxicol. 2020, 34, e22491. [Google Scholar] [CrossRef]
- Kuo, L.K.; Fu, Y.K.; Yeh, C.C.; Lee, C.Y.; Chung, C.J.; Shih, L.J.; Lu, H.Y.; Chuu, C.P. Combined Treatment of Caffeic Acid Phenethyl Ester with Docetaxel Inhibits Survival of Non-small-cell Lung Cancer Cells via Suppression of c-MYC. Anticancer Res. 2024, 44, 4915–4928. [Google Scholar] [CrossRef] [PubMed]
- Almatroodi, S.A.; Almatroudi, A.; Khan, A.A.; Alhumaydhi, F.A.; Alsahli, M.A.; Rahmani, A.H. Potential Therapeutic Targets of Epigallocatechin Gallate (EGCG), the Most Abundant Catechin in Green Tea, and Its Role in the Therapy of Various Types of Cancer. Molecules 2020, 25, 3146. [Google Scholar] [CrossRef]
- Min, K.; Kwon, T.K. Anticancer effects and molecular mechanisms of epigallocatechin-3-gallate. Integr. Med. Res. 2014, 3, 16–24. [Google Scholar]
- Tsai, Y.J.; Chen, B.H. Preparation of catechin extracts and nanoemulsions from green tea leaf waste and their inhibition effect on prostate cancer cell PC-3. Int. J. Nanomed. 2016, 11, 1907–1926. [Google Scholar]
- Chang, H.B.; Chen, B.H. Inhibition of lung cancer cells A549 and H460 by curcuminoid extracts and nanoemulsions prepared from Curcuma Longa Linnaeus. Int. J. Nanomed. 2015, 10, 5059–5080. [Google Scholar]
- Sadava, D.; Whitlock, E.; Kane, S.E. The green tea polyphenol, epigallocatechin-3-gallate inhibits telomerase and induces apoptosis in drug-resistant lung cancer cells. Biochem. Biophys. Res. Commun. 2007, 360, 233–237. [Google Scholar] [CrossRef]
- Rahmani, A.H.; Al Shabrmi, F.M.; Allemailem, K.S.; Aly, S.M.; Khan, M.A. Implications of green tea and its constituents in the prevention of cancer via the modulation of cell signaling pathway. Biomed. Res. Int. 2015, 2015, 925640. [Google Scholar] [CrossRef]
- Chen, B.H.; Hsieh, C.H.; Tsai, S.Y.; Wang, C.Y.; Wang, C.C. Anticancer effects of epigallocatechin-3-gallate nanoemulsion on lung cancer cells through the activation of AMP-activated protein kinase signaling pathway. Sci. Rep. 2020, 10, 5163. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Suganuma, M.; Kurusu, M.; Suzuki, K.; Tasaki, E.; Fujiki, H. Green tea polyphenol stimulates cancer preventive effects of celecoxib in human lung cancer cells by upregulation of GADD153 gene. Int. J. Cancer 2006, 119, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, H.; Tighiouart, M.; Lee, J.E.; Shin, H.J.; Khuri, F.R.; Yang, C.S.; Chen, Z.; Shin, D.M. Synergistic inhibition of head and neck tumor growth by green tea (−)-epigallocatechin-3-gallate and EGFR tyrosine kinase inhibitor. Int. J. Cancer 2008, 123, 1005–1014. [Google Scholar] [CrossRef] [PubMed]
- Anwar, S.; Younus, H. Antiglycating potential of ellagic acid against glucose and methylglyoxal induced glycation of superoxide dismutase. J. Protein Proteom. 2017, 8, 1–2. [Google Scholar]
- Xie, C.; Kong, J.; Miao, F.; Wang, X.; Sheng, J. Combination effects of ellagic acid with erlotinib in a Ba/F3 cell line expressing EGFR H773_V774 insH mutation. Thorac. Cancer 2020, 11, 2101–2111. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Paunovic, D.; Rajkovic, J.; Novakovic, R.; Grujic-Milanovic, J.; Mekky, R.H.; Popa, D.; Calina, D.; Sharifi-Rad, J. The potential roles of gossypol as anticancer agent: Advances and future directions. Chin. Med. 2023, 18, 163. [Google Scholar] [CrossRef]
- Pal, D.; Sahu, P.; Sethi, G.; Wallace, C.E.; Bishayee, A. Gossypol and its natural derivatives: Multitargeted phytochemicals as potential drug candidates for oncologic diseases. Pharmaceutics 2022, 14, 2624. [Google Scholar] [CrossRef]
- Xu, J.; Zhu, G.Y.; Cao, D.; Pan, H.; Li, Y.W. Gossypol overcomes EGFR-TKIs resistance in non-small cell lung cancer cells by targeting YAP/TAZ and EGFRL858R/T790M. Biomed. Pharmacother. 2019, 115, 108860. [Google Scholar] [CrossRef]
- Wang, Y.; Lai, H.; Fan, X.; Luo, L.; Duan, F.; Jiang, Z.; Wang, Q.; Leung, E.L.; Liu, L.; Yao, X. Gossypol inhibits non-small cell lung cancer cells proliferation by targeting EGFRL858R/T790M. Front. Pharmacol. 2018, 9, 728. [Google Scholar] [CrossRef]
- Ranaware, A.M.; Banik, K.; Deshpande, V.; Padmavathi, G.; Roy, N.K.; Sethi, G.; Fan, L.; Kumar, A.P.; Kunnumakkara, A.B. Magnolol: A Neolignan from the Magnolia Family for the Prevention and Treatment of Cancer. Int. J. Mol. Sci. 2018, 19, 2362. [Google Scholar] [CrossRef]
- Porro, C.; Cianciulli, A.; Calvello, R.; Panaro, M.A. Reviewing the role of resveratrol as a natural modulator of microglial activities. Curr. Pharm. Des. 2015, 21, 5277–5291. [Google Scholar] [CrossRef]
- Salehi, B.; Mishra, A.P.; Nigam, M.; Sener, B.; Kilic, M.; Sharifi-Rad, M.; Fokou, P.V.T.; Martins, N.; Sharifi-Rad, J. Resveratrol: A Double-Edged sword in health benefits. Biomedicines 2018, 6, 91. [Google Scholar] [CrossRef] [PubMed]
- Holthoff, J.H.; Woodling, K.A.; Doerge, D.R.; Burns, S.T.; Hinson, J.A.; Mayeux, P.R. Resveratrol, a dietary polyphenolic phytoalexin, is a functional scavenger of peroxynitrite. Biochem. Pharmacol. 2010, 80, 1260–1265. [Google Scholar] [CrossRef] [PubMed]
- Calil, O.N.; Carvalho, G.S.G.; Franco, D.C.Z.; Silva, A.D.; Raposo, N.B.R. Antioxidant activity of Resveratrol Analogs. Lett. Drug Des. Discov. 2012, 9, 8–11. [Google Scholar] [CrossRef]
- Mendes, J.B.E.; Riekes, M.R.; Oliveira, V.M.; Michel, M.D.; Stulzer, H.K.; Zawadzki, S.F.; Mainardes, R.M.; Farago, P.V. PHBV/PCL Micro particles for Controlled Release of Resveratrol: Physicochemical characterization, antioxidant potential, and effect on hemolysis of human erythrocytes. Sci. World J. 2012, 2012, 542937. [Google Scholar] [CrossRef]
- Bernard, P.; Berthon, J.Y. Resveratrol: An original mechanism on tyrosinase inhibition. Int. J. Cosmet. Sci. 2000, 22, 219–226. [Google Scholar] [CrossRef]
- Kim, Y.M.; Yun, J.; Lee, C.K.; Lee, H.; Min, K.R.; Kim, Y. Oxyresveratrol and Hydroxystilbene Compounds: Inhibitory effect on tyrosinase and mechanism of action. J. Biol. Chem. 2002, 277, 16340–16344. [Google Scholar] [CrossRef]
- Zhu, Y.; He, W.; Gao, X.; Li, B.; Mei, C.; Xu, R.; Chen, H. Resveratrol overcomes gefitinib resistance by increasing the intracellular gefitinib concentration and triggering apoptosis, autophagy and senescence in PC9/G NSCLC cells. Sci. Rep. 2015, 5, 17730. [Google Scholar] [CrossRef]
- Helms, S. Cancer prevention and therapeutics: Panax ginseng. Altern. Med. Rev. 2004, 9, 259–274. [Google Scholar] [PubMed]
- Varjas, T.; Nowrasteh, G.; Budan, F.; Nadasi, E.; Horvath, G.; Makai, S.; Gracza, T.; Cseh, J. Ember Chemo preventive effect of Panax ginseng. Phytother. Res. 2009, 23, 1399–1403. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Xin, Y.; Li, Y.; Xu, F.; Xi, X.; Guo, H.; Cui, X.; Cao, H.; Zhang, X.; Han, C. Ginsenosides: A potential neuroprotective agent. BioMed Res. Int. 2018, 2018, 8174345. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Wang, W.; Sun, Q.; Tuohayi, J. Ginsenoside Rg3 promotes the antitumor activity of gefitinib in lung cancer cell lines. Exp. Ther. Med. 2018, 17, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A. The snail superfamily of zinc-finger transcription factors. Nat. Rev. Mol. Cell Biol. 2002, 3, 155–166. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, J.; Wang, X.; Wei, N.; Liu, H.; Zhang, X. CD146-mediated acquisition of stemness phenotype enhances tumour invasion metastasis after EGFR-TKI resistance in lung cancer. Clin. Respir. J. 2019, 13, 23–33. [Google Scholar] [CrossRef]
- Xu, T.; Jin, Z.; Yuan, Y.; Wei, H.; Xu, X.; He, S.; Chen, S.; Hou, W.; Guo, Q.; Hua, B. Ginsenoside Rg3 serves as an adjuvant chemotherapeutic agent and VEGF inhibitor in the treatment of non-small cell lung cancer: A meta-analysis and systematic review. Evid. Based Complement. Altern. Med. 2016, 2016, 7826753. [Google Scholar] [CrossRef]
- Chian, S.; Zhao, Y.; Xu, M.; Yu, X.; Ke, X.; Gao, R.; Yin, L. Ginsenoside Rd reverses cisplatin resistance in non-small-cell lung cancer A549 cells by downregulating the nuclear factor erythroid 2-related factor 2 pathway. Anticancer. Drugs 2019, 30, 838–845. [Google Scholar] [CrossRef]
- Jiang, Z.; Yang, Y.; Yang, Y.; Zhang, Y.; Yue, Z.; Pan, Z.; Ren, X. Ginsenoside Rg3 attenuates cisplatin resistance in lung cancer by down regulating PD-L1 and resuming immune. Biomed. Pharmacother. 2017, 96, 378–383. [Google Scholar] [CrossRef]
- Wang, Q.N.; Yang, X.F.; Song, Y.; Sun, X.W.; Li, W.T.; Zhang, L.; Hu, X.; Wang, H.; Zhao, N.; Zhuang, R.; et al. Astragaloside IV-targeting miRNA-1 attenuates lipopolysaccharide-induced cardiac dysfunction in rats through inhibition of apoptosis and autophagy. Life Sci. 2021, 275, 119414. [Google Scholar] [CrossRef]
- Liang, X.Y.; Hong, F.F.; Yang, S.L. Astragaloside IV alleviates liver inflammation, oxidative stress and apoptosis to protect against experimental non-alcoholic fatty liver disease. Diabetes Metab. Syndr. Obes. 2021, 14, 1871–1883. [Google Scholar] [CrossRef]
- Xing, L.N.; Fang, J.; Zhu, B.B.; Wang, L.; Chen, J.L.; Wang, Y.M.; Huang, J.; Wang, H.; Yao, X. Astragaloside IV protects against podocyte apoptosis by inhibiting oxidative stress via activating PPARγ Klotho-FoxO1 axis in diabetic nephropathy. Life Sci. 2021, 269, 119068. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, G.; Chen, M.B.; Cai, R.Z. Astragaloside IV enhances the sensibility of lung adenocarcinoma cells to bevacizumab by inhibiting autophagy. Drug Dev. Res. 2021, 83, 461–469. [Google Scholar] [CrossRef]
- Ye, Q.; Su, L.; Chen, D.G.; Zheng, W.Y.; Liu, Y. Astragaloside IV induced miR-134 expression reduces EMT and increases chemotherapeutic sensitivity by suppressing CREB1 signaling in colorectal cancer cell line SW-480. Cell. Physiol. Biochem. 2017, 43, 1617–1626. [Google Scholar] [CrossRef] [PubMed]
- Min, L.; Wang, H.; Qi, H. Astragaloside IV inhibits the progression of liver cancer by modulating macrophage polarization through the TLR4/NF-κB/STAT3 signaling pathway. Am. J. Transl. Res. 2022, 14, 1551–1566. [Google Scholar]
- Zheng, Y.F.; Dai, Y.; Liu, W.P.; Wang, N.; Cai, Y.L.; Wang, S.Q.; Zhang, F.; Liu, P.; Chen, Q.; Wang, Z. Astragaloside IV enhances taxol chemo sensitivity of breast cancer via caveolin-1-targeting oxidant damage. J. Cell. Physiol. 2018, 234, 4277–4290. [Google Scholar] [CrossRef]
- Dai, P.; Liu, D.; Zhang, L.; Ye, J.; Wang, Q.; Zhang, H.W.; Lin, X.H.; Lai, G.X. Astragaloside IV sensitizes non-small cell lung cancer cells to gefitinib potentially via regulation of SIRT6. Tumor. Biol. 2017; ahead of print. [Google Scholar] [CrossRef]
- Zhou, R.; Guo, T.; Li, J. Research progress on the antitumor effects of astragaloside IV. Eur. J. Pharmacol. 2023, 938, 175449. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Gong, G.Y.; Ma, L.L.; Wang, Z.Q.; Song, D.; Fang, M.Y. Anti-cancer effects of Polyphyllin I: An update in 5 years. Chem. Biol. Interact. 2020, 316, 108936. [Google Scholar] [CrossRef]
- Lou, W.; Chen, Y.; Zhu, K.Y.; Deng, H.; Wu, T.; Wang, J. Polyphyllin I Overcomes EMT-Associated Resistance to Erlotinib in Lung Cancer Cells via IL-6/STAT3 Pathway Inhibition. Biol. Pharm. Bull. 2017, 40, 1306–1313. [Google Scholar] [CrossRef]
- Zheng, R.; Jiang, H.; Li, J.; Liu, X.; Xu, H. Polyphyllin II Restores Sensitization of the Resistance of PC-9/ZD Cells to Gefitinib by a Negative Regulation of the PI3K/Akt/mTOR Signaling Pathway. Curr. Cancer Drug Targets 2017, 17, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Alghasham, A.A. Cucurbitacin—A promising target for cancer therapy. Int. J. Health Sci. 2013, 7, 77–89. [Google Scholar] [CrossRef]
- Clericuzio, M.; Mella, M.; Vita-Finzi, P.; Zema, M.; Vidari, G. Cucurbitane triterpenoids from Leucopaxillus gentianeus. J. Nat. Prod. 2004, 67, 1823–1828. [Google Scholar] [CrossRef]
- Chen, J.C.; Chiu, M.H.; Nie, R.L.; Cordell, G.A.; Qiu, S.X. Cucurbitacins and cucurbitane glycosides: Structures and biological activities. Nat. Prod. Rep. 2005, 22, 386–399. [Google Scholar] [CrossRef] [PubMed]
- Wiart, C. The definition and significance of Cucurbitacin B a STAT3 inhibitors. Cancer Lett. 2013, 328, 188. [Google Scholar] [CrossRef] [PubMed]
- Clericuzio, M.; Tabasso, S.; Bianco, M.A.; Pratesi, G.; Beretta, G.; Tinelli, S.; Zunino, F.; Vidari, G. Cucurbitane triterpenes from the fruiting bodies and cultivated mycelia of Leucopaxillus gentianeus. J. Nat. Prod. 2006, 69, 1796–1799. [Google Scholar] [CrossRef] [PubMed]
- Abou-Khalil, R.; Jraij, A.; Magdalou, J.; Ouaini, N.; Tome, D.; Greige-Gerges, H. Interaction of cucurbitacins with human serum albumin: Thermodynamic characteristics influence on the binding of site specific ligands. J. Photochem. Photobiol. B 2009, 95, 189–195. [Google Scholar] [CrossRef]
- Wakimoto, N.; Yin, D.; O’Kelly, J.; Haritunians, T.; Karlan, B.; Said, J.; Xing, H.; Koeffler, H.P. Cucurbitacin B has a potent antiproliferative effect on breast cancer cells in vitro and in vivo. Cancer Sci. 2008, 99, 1793–1797. [Google Scholar] [CrossRef]
- Kausar, H.; Munagala, R.; Bansal, S.S.; Aqil, F.; Vadhanam, M.V.; Gupta, R.C. Cucurbitacin B potently suppresses non-small-cell lung cancer growth: Identification of intracellular thiols as critical targets. Cancer Lett. 2013, 332, 35–45. [Google Scholar] [CrossRef]
- Li, W.; Liu, Y.; Cai, S.; Yang, C.; Lin, Z.; Zhou, L.; Liu, L.; Cheng, X.; Zeng, W. Not all mutations of KRAS predict poor prognosis in patients with colorectal cancer. Int. J. Clin. Exp. Pathol. 2019, 12, 957–967. [Google Scholar]
- Wei, L.; Qu, W.; Sun, J.; Wang, X.; Lv, L.; Xie, L.; Song, X. Knockdown of cancerous inhibitor of protein phosphatase 2A may sensitize NSCLC cells to cisplatin. Cancer Gene Ther. 2014, 21, 194–199. [Google Scholar] [CrossRef]
- Sablina, A.A.; Chen, W.; Arroyo, J.D.; Corral, L.; Hector, M.; Bulmer, S.E.; DeCaprio, J.A.; Hahn, W.C. The tumor suppressor PP2A Abeta regulates the RalA GTPase. Cell 2007, 129, 969–982. [Google Scholar] [CrossRef]
- Liu, P.; Xiang, Y.; Liu, X.; Zhang, T.; Yang, R.; Chen, S.; Xu, L.; Yu, Q.; Zhao, H.; Zhang, L.; et al. Cucurbitacin B induces the lysosomal degradation of EGFR and suppresses the CIP2A/PP2A/Akt signaling axis in gefitinib-resistant non-small cell lung cancer. Molecules 2019, 24, 647. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.H.; Ku, J.M.; Lim, Y.S.; Lee, S.Y.; Kim, J.H.; Cheon, C.; Ko, S.G. Cucurbitacin D overcomes gefitinib resistance by blocking EGF binding to EGFR and inducing cell death in NSCLCs. Front. Oncol. 2020, 10, 62. [Google Scholar] [CrossRef]
- Saneja, A.; Arora, D.; Kumar, R.; Dubey, R.D.; Panda, A.K.; Gupta, P.N. Therapeutic applications of betulinic acid nanoformulation. Ann. N. Y. Acad. Sci. 2018, 1421, 5. [Google Scholar] [CrossRef]
- Zheng, Y.; Liu, P.; Wang, N.; Wang, S.; Yang, B.; Li, M.; Chen, J.; Situ, H.; Xie, M.; Lin, Y.; et al. Betulinic acid suppresses breast cancer metastasis by targeting GRP78-mediated glycolysis and ER stress apoptotic pathway Oxid. Med. Cell Longev. 2019, 2019, 8781690. [Google Scholar] [CrossRef]
- XLiu, I.; Jutooru, P.; Lei, K.; Kim, S.O.; Lee, L.K.; Brents, P.L.; Prather, S. Safe Betulinic acid targets YY1 and ErbB2 through cannabinoid receptor-dependent disruption of microRNA-27a: ZBTB10 in breast cancer Mol. Cancer Ther. 2012, 11, 1421–1431. [Google Scholar] [CrossRef]
- Cai, Y.; Zheng, Y.; Gu, J.; Wang, S.; Wang, N.; Yang, B.; Zhang, F.; Wang, D.; Fu, W.; Wang, Z. Betulinic acid chemosensitizes breast cancer by triggering ER stress-mediated apoptosis by directly targeting GRP78. Cell Death Dis. 2018, 9, 636. [Google Scholar] [CrossRef] [PubMed]
- Jäger, S.; Trojan, H.; Kopp, T.; Laszczyk, M.N.; Scheffler, A. Pentacyclic triterpene distribution in various plants—Rich sources for a new group of multi-potent plant extracts. Molecules 2009, 14, 2016–2031. [Google Scholar] [CrossRef]
- Cichewicz, R.H.; Kouzi, S.A. Chemistry, biological activity, and chemotherapeutic potential of betulinic acid for the prevention and treatment of cancer and HIV infection. Med. Res. Rev. 2004, 24, 90–114. [Google Scholar] [CrossRef]
- Silva, M.G.V.; Vieira, I.G.P.; Mendes, F.N.P.; Albuquerque, I.L.; Dos Santos, R.N.; Silva, F.O.; Morais, S.M. Variation of ursolic acid content in eight Ocimum species from northeastern Brazil. Molecules 2008, 13, 2482–2487. [Google Scholar] [CrossRef]
- Jäger, S.; Winkler, L.; Pfüller, I.; Scheffler, A. Solubility studies of oleanolic acid and betulinic acid in aqueous solutions and plant extracts of Viscum album L. Planta Med. 2007, 73, 157–162. [Google Scholar] [CrossRef]
- Laszczyk, M.; Jäger, S.; Simon-Haarhaus, B.; Scheffler, A.; Schempp, C.M. Physical, chemical and pharmacological characterization of a new oleo gel-forming triterpene extract from the outer bark of birch (betulae cortex). Planta Med. 2006, 72, 1389–1395. [Google Scholar] [CrossRef] [PubMed]
- Ressmann, A.K.; Kremsmayr, T.; Gaertner, P.; Zirbs, R.; Bica, K. Toward a benign strategy for the manufacturing of betulinic acid. Green Chem. 2017, 19, 1014–1022. [Google Scholar] [CrossRef]
- Feng, Y.; Li, M.; Liu, J.; Xu, T.Y.; Fang, R.S.; Chen, Q.H.; He, G.Q. A novel one-step microbial transformation of betulin to betulinic acid catalysed by Cunninghamella blakesleeana. Food Chem. 2013, 136, 73–79. [Google Scholar] [CrossRef]
- Madsen, K.M.; Udatha, G.D.; Semba, S.; Otero, J.M.; Koetter, P.; Nielsen, J.; Ebizuka, Y.; Kushiro, T.; Panagiotou, G. Linking genotype and phenotype of Saccharomyces cerevisiae strains reveals metabolic engineering targets and leads to triterpene hyper-producers. PLoS ONE 2011, 6, 14763. [Google Scholar] [CrossRef]
- Thimmappa, R.; Geisler, K.; Louveau, T.; O’Maille, P.; Osbourn, A. Triterpene biosynthesis in plants. Annu. Rev. Plant Biol. 2014, 65, 225–257. [Google Scholar] [CrossRef]
- Zhang, X.; Hu, J.Y.; Chen, Y. Betulinic acid and the pharmacological effects of tumour suppression. Mol. Med. Rep. 2016, 14, 4489–4495. [Google Scholar] [CrossRef]
- Lin, C.K.; Tseng, C.K.; Chen, K.H.; Wu, S.H.; Liaw, C.C.; Lee, J.C. Betulinic acid exerts anti-hepatitis C virus activity via the suppression of NF-κB and MAPK-ERK1/2-mediated COX-2 expression. Br. J. Pharm. 2015, 172, 4481–4492. [Google Scholar] [CrossRef] [PubMed]
- Alakurtti; Mäkelä, T.; Koskimies, S.; Yli-Kauhaluoma, J. Pharmacological properties of the ubiquitous natural product botuli Eur. J. Pharm. Sci. 2006, 29, 113. [Google Scholar] [CrossRef]
- Lingaraju, M.C.; Pathak, N.N.; Begum, J.; Balaganur, V.; Bhat, R.A.; Ramachandra, H.D.; Ayanur, A.; Ram, M.; Singh, V.; Kumar, D.; et al. Betulinic acid attenuates lung injury by modulation of inflammatory cytokine response in experimentally-induced polymicrobial sepsis in mice. Cytokine 2015, 71, 101–108. [Google Scholar] [CrossRef]
- Ko, J.L.; Lin, C.H.; Chen, H.C.; Hung, W.H.; Chien, P.J.; Chang, H.Y.; Wang, B.Y. Effects and mechanisms of betulinic acid on improving EGFR TKI-resistance of lung cancer cells. Environ. Toxicol. 2018, 33, 1153–1159. [Google Scholar] [CrossRef]
- Shi, Y.; Au, J.S.; Thongprasert, S.; Srinivasan, S.; Tsai, C.M.; Khoa, M.T.; Heeroma, K.; Cornelio, G.; Yang, P.C. A prospective, molecular epidemiology study of EGFR mutations in Asian patients with advanced non-small-cell lung cancer of adenocarcinoma histology (PIONEER). J. Thorac. Oncol. 2014, 9, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Cao, J.; Chen, K.; Cheng, L.; Zhou, C.; Yan, B.; Qian, W.; Li, J.; Duan, W.; Ma, J.; et al. Betulinic acid inhibits stemness and EMT of pancreatic cancer cells via activation of AMPK signaling. Int. J. Oncol. 2019, 54, 98–110. [Google Scholar] [CrossRef]
- Yin, B.; Fang, D.M.; Zhou, X.L.; Gao, F. Natural products as important tyrosine kinase inhibitors. Eur. J. Med. Chem. 2019, 182, 111664. [Google Scholar] [CrossRef] [PubMed]
- Gowda, R.; Inamdar, G.S.; Kuzu, O.; Dinavahi, S.S.; Krzeminski, J.; Battu, M.B.; Voleti, S.R.; Amin, S.; Robertson, G.P. Identifying the structure-activity relationship of leelamine necessary for inhibiting intracellular cholesterol transport. Oncotarget 2017, 8, 28260–28277. [Google Scholar] [CrossRef]
- Jin, L.; Cho, M.; Kim, B.-S.; Han, J.H.; Park, S.; Lee, I.-K.; Ryu, D.; Kim, J.H.; Bae, S.-J.; Ha, K.-T. Drug evaluation based on phosphomimetic PDHA1 reveals the complexity of activity-related cell death in A549 non-small cell lung cancer cells. BMB Rep. 2021, 54, 563. [Google Scholar] [CrossRef]
- Wu, J.; Li, Y.; He, Q.; Yang, X. Exploration of the Use of Natural Compounds in Combination with Chemotherapy Drugs for Tumor Treatment. Molecules 2023, 28, 1022. [Google Scholar] [CrossRef]
- Yang, Z.; Tam, K.Y. Combination Strategies Using EGFR-TKi in NSCLC Therapy: Learning from the Gap between Pre-Clinical Results and Clinical Outcomes. Int. J. Biol. Sci. 2018, 14, 204–216. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Foucquier, J.; Guedj, M. Analysis of Drug Combinations: Current Methodological Landscape. Pharmacol. Res. Perspect. 2015, 3, e00149. [Google Scholar] [CrossRef]
- Zhong, W.; Qin, Y.; Chen, S.; Sun, T. Antitumor Effect of Natural Product Molecules against Lung Cancer. In A Global Scientific Vision-Prevention, Diagnosis, and Treatment of Lung Cancer; InTech: Rijeka, Croatia, 2017. [Google Scholar]
- Bayat Mokhtari, R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Castañeda, A.M.; Meléndez, C.M.; Uribe, D.; Pedroza-Díaz, J. Synergistic effects of natural compounds and conventional chemotherapeutic agents: Recent insights for the development of cancer treatment strategies. Heliyon 2022, 8, e09519. [Google Scholar] [CrossRef]
- Boța, M.; Vlaia, L.; Jîjie, A.-R.; Marcovici, I.; Crişan, F.; Oancea, C.; Dehelean, C.A.; Mateescu, T.; Moacă, E.-A. Exploring Synergistic Interactions between Natural Compounds and Conventional Chemotherapeutic Drugs in Preclinical Models of Lung Cancer. Pharmaceuticals 2024, 17, 598. [Google Scholar] [CrossRef] [PubMed]
- Gahtori, R.; Tripathi, A.H.; Kumari, A.; Negi, N.; Paliwal, A.; Tripathi, P.; Joshi, P.; Rai, R.C.; Upadhyay, S.K. Anticancer Plant-Derivatives: Deciphering Their Oncopreventive and Therapeutic Potential in Molecular Terms. Future J. Pharm. Sci. 2023, 9, 14. [Google Scholar] [CrossRef]
- Huang, T.H.; Wu, T.H.; Guo, Y.H.; Li, T.L.; Chan, Y.L.; Wu, C.J. The Concurrent Treatment of Scutellaria baicalensis Georgi Enhances the Therapeutic Efficacy of Cisplatin but Also Attenuates Chemotherapy-Induced Cachexia and Acute Kidney Injury. J. Ethnopharmacol. 2019, 243, 112075. [Google Scholar] [CrossRef]
- Han, S.Y.; Zhao, M.B.; Zhuang, G.B.; Li, P.P. Marsdenia Tenacissima Extract Restored Gefitinib Sensitivity in Resistant Non-Small Cell Lung Cancer Cells. Lung Cancer 2012, 75, 30–37. [Google Scholar] [CrossRef]
- Wang, F.; Wang, W.; Li, J.; Zhang, J.; Wang, X.; Wang, M. Sulforaphane reverses gefitinib tolerance in human lung cancer cells via modulation of sonic hedgehog signaling. Oncol. Lett. 2018, 15, 109–114. [Google Scholar] [CrossRef]
- Han, Y.; Shi, J.; Xu, Z.; Zhang, Y.; Cao, X.; Yu, J.; Li, J.; Xu, S. Identification of solamargine as a cisplatin sensitizer through phenotypical screening in cisplatin-resistant NSCLC organoids. Front. Pharmacol. 2022, 13, 802168. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Liu, X.; Liu, Q.; Ho, I.H.; Wei, X.; Yin, T.; Zhan, Y.; Zhang, W.; Zhang, W.; Chen, B.; et al. Hederagenin potentiated cisplatin and paclitaxel-mediated cytotoxicity by impairing autophagy in lung cancer cells. Cell Death Dis. 2020, 11, 611. [Google Scholar] [CrossRef]
- Shi, S.; Bai, X.; Ji, Q.; Wan, H.; An, H.; Kang, X.; Guo, S. Molecular mechanism of ion channel protein TMEM16A regulated by natural product of narirutin for lung cancer adjuvant treatment. Int. J. Biol. Macromol. 2022, 223, 1145–1157. [Google Scholar] [CrossRef]
- Zhang, W.; Shi, H.; Chen, C.; Ren, K.; Xu, Y.; Liu, X.; He, L. Curcumin enhances cisplatin sensitivity of human NSCLC cell lines through influencing Cu-Sp1-CTR1 regulatory loop. Phytomedicine 2018, 48, 51–61. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, Y.M.; Chen, Y.; Chen, J.T.; Liu, Y. Targeted polysaccharide nanoparticle for adamplatin prodrug delivery. J. Med. Chem. 2013, 56, 9725–9736. [Google Scholar] [CrossRef]
- Pinmai, K.; Chunlaratthanabhorn, S.; Ngamkitidechakul, C.; Soonthornchareon, N.; Hahnvajanawong, C. Synergistic growth inhibitory effects of Phyllanthus emblica and Terminalia bellerica extracts with conventional cytotoxic agents: Doxorubicin and cisplatin against human hepatocellular carcinoma and lung cancer cells. World J. Gastroenterol. 2008, 14, 1491–1497. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tan, K.T.; Li, S.; Li, Y.R.; Cheng, S.-L.; Lin, S.-H.; Tung, Y.-T. Synergistic Anticancer Effect of a Combination of Paclitaxel and 5-Demethylnobiletin Against Lung Cancer Cell Line In Vitro and In Vivo. Appl. Biochem. Biotechnol. 2019, 187, 1328–1343. [Google Scholar] [CrossRef]
- Peng, M.; Zheng, Z.; Chen, S.; Fang, L.; Feng, R.; Zhang, L.; Tang, Q.; Liu, X. Sensitization of Non-Small Cell Lung Cancer Cells to Gefitinib and Reversal of Epithelial-Mesenchymal Transition by Aloe-Emodin Via PI3K/Akt/TWIS1 Signal Blockage. Front. Oncol. 2022, 12, 908031. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jiao, L.; Xu, J.; Sun, J.; Chen, Z.; Gong, Y.; Bi, L.; Lu, Y.; Yao, J.; Zhu, W.; Hou, A.; et al. Chinese Herbal Medicine Combined With EGFR-TKI in EGFR Mutation-Positive Advanced Pulmonary Adenocarcinoma (CATLA): A Multicenter, Randomized, Double-Blind, Placebo-Controlled Trial. Front. Pharmacol. 2019, 10, 732. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, J.; Sun, L.; Cui, J.; Wang, J.; Liu, X.; Aung, T.N.; Qu, Z.; Chen, Z.; Adelson, D.L.; Lin, L. Yiqi Chutan Tang Reduces Gefitinib-Induced Drug Resistance in Non-Small-Cell Lung Cancer by Targeting Apoptosis and Autophagy. Cytom. A 2020, 97, 70–77. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Abd El-Hafeez, A.A.; Fujimura, T.; Kamei, R.; Hirakawa, N.; Baba, K.; Ono, K.; Kawamoto, S. Synergistic tumor suppression by a Perilla frutescens-derived methoxyflavanone and anti-cancer tyrosine kinase inhibitors in A549 human lung adenocarcinoma. Cytotechnology 2018, 70, 913–919. [Google Scholar] [CrossRef]
- Chen, Z.; Vallega, K.A.; Chen, H.; Zhou, J.; Ramalingam, S.S.; Sun, S.Y. The natural product berberine synergizes with osimertinib preferentially against MET-amplified osimertinib-resistant lung cancer via direct MET inhibition. Pharmacol. Res. 2022, 175, 105998. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Corvaja, C.; Passaro, A.; Attili, I.; Aliaga, P.T.; Spitaleri, G.; Signore, E.D.; de Marinis, F. Advancements in fourth-generation EGFR TKIs in EGFR-mutant NSCLC: Bridging biological insights and therapeutic development. Cancer Treat. Rev. 2024, 130, 102824. [Google Scholar] [CrossRef] [PubMed]
- Eno, M.S.; Brubaker, J.D.; Campbell, J.E.; De Savi, C.; Guzi, T.J.; Williams, B.D.; Wilson, D.; Wilson, K.; Brooijmans, N.; Kim, J.; et al. Discovery of BLU-945, a Reversible, Potent, and Wild-Type-Sparing Next-Generation EGFR Mutant Inhibitor for Treatment-Resistant Non-Small-Cell Lung Cancer. J. Med. Chem. 2022, 65, 9662–9677. [Google Scholar] [CrossRef]
- Su, C.; Sun, S.Y. Fourth-generation epidermal growth factor receptor-tyrosine kinases inhibitors: Hope and challenges. Transl. Cancer Res. 2024, 13, 3929–3934. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jan, R.; Chaudhry, G.E. Understanding Apoptosis and Apoptotic Pathways Targeted Cancer Therapeutics. Adv. Pharm. Bull. 2019, 9, 205–218. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Saleem, M.; Kweon, M.H.; Yun, J.M.; Adhami, V.M.; Khan, N.; Syed, D.N.; Mukhtar, H. A novel dietary triterpene Lupeol induces fas-mediated apoptotic death of androgen-sensitive prostate cancer cells and inhibits tumor growth in a xenograft model. Cancer Res. 2005, 65, 11203–11213. [Google Scholar] [CrossRef]
- Olsson, M.; Zhivotovsky, B. Caspases and cancer. Cell Death Differ. 2011, 18, 1441–1449. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ekonomopoulou, M.T.; Babas, E.; Mioglou-Kalouptsi, E.; Malandri, M.; Iakovidou-Kritsi, Z. Changes in activities of caspase-8 and caspase-9 in human cervical malignancy. Int. J. Gynecol. Cancer 2011, 21, 435–438. [Google Scholar] [CrossRef] [PubMed]
- Anto, R.J.; Mukhopadhyay, A.; Denning, K.; Aggarwal, B.B. Curcumin (diferuloylmethane) induces apoptosis through activation of caspase-8, BID cleavage and cytochrome c release: Its suppression by ectopic expression of Bcl-2 and Bcl-xl. Carcinogenesis 2002, 23, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Hajiaghaalipour, F.; Kanthimathi, M.S.; Sanusi, J.; Rajarajeswaran, J. White tea (Camellia sinensis) inhibits proliferation of the colon cancer cell line, HT-29, activates caspases and protects DNA of normal cells against oxidative damage. Food Chem. 2015, 169, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.P.; Kang, M.Y.; Choi, Y.H.; Kim, J.H.; Nam, S.H.; Friedman, M. Mechanism of Hericium erinaceus (Yamabushitake) mushroom-induced apoptosis of U937 human monocytic leukemia cells. Food Funct. 2011, 2, 348–356. [Google Scholar] [CrossRef]
- Chen, Y.C.; Chang, H.Y.; Deng, J.S.; Chen, J.J.; Huang, S.S.; Lin, I.H.; Kuo, W.L.; Chao, W.; Huang, G.J. Hispolon from Phellinus linteus induces G0/G1 cell cycle arrest and apoptosis in NB4 human leukaemia cells. Am. J. Chin. Med. 2013, 41, 1439–1457. [Google Scholar] [CrossRef]
- Preljević, K.; Pašić, I.; Vlaović, M.; Matić, I.Z.; Krivokapić, S.; Petrović, N.; Stanojković, T.; Živković, V.; Perović, S. Comparative analysis of chemical profiles, antioxidant, antibacterial, and anticancer effects of essential oils of two Thymus species from Montenegro. Fitoterapia 2024, 174, 105871. [Google Scholar] [CrossRef]
- Su, C.W.; Kao, S.H.; Chen, Y.T.; Hsieh, Y.H.; Yang, W.E.; Tsai, M.Y.; Lin, C.W.; Yang, S.F. Curcumin analog L48H37 induces apoptosis in human Oral cancer cells by activating caspase cascades and downregulating the inhibitor of apoptosis proteins through JNK/p38 signaling. Am. J. Chin. Med. 2024, 52, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Auwal, A.; Al Banna, M.H.; Pronoy, T.U.; Hossain, M.M.; Rashel, K.M.; Kabir, S.R.; Ansary, M.R.; Islam, F. In vitro and In vivo Growth Inhibition and apoptosis of cancer cells by ethyl 4-[(4-methylbenzyl) oxy] benzoate complex. Anti-Cancer Agents Med. Chem. 2025; ahead of print. [Google Scholar]
- Huang, S.T.; Huang, C.C.; Huang, W.L.; Lin, T.K.; Liao, P.L.; Wang, P.W.; Liou, C.W.; Chuang, J.H. Tanshinone IIA induces intrinsic apoptosis in osteosarcoma cells both in vivo and in vitro associated with mitochondrial dysfunction. Sci. Rep. 2017, 7, 40382. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Guo, S.; Zhang, H.; Zhang, Z.; Shen, S.; Li, X. BRAF-Mutated Non-Small Cell Lung Cancer: Current Treatment Status and Future Perspective. Front. Oncol. 2022, 12, 863043. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yao, Z.; Torres, N.M.; Tao, A.; Gao, Y.J.; Luo, L.S.; Li, Ł.Q.; de Stanchina, E.; Abdel-Wahab, O.; Solit, D.B.; Poulikakos, P.I.; et al. BRAF Mutants Evade ERK-Dependent Feedback by Different Mechanisms That Determine Their Sensitivity to Pharmacologic Inhibition. Cancer Cell 2015, 28, 370–383. [Google Scholar] [CrossRef]
- Yao, Z.; Yaeger, R.; Rodrik-Outmezguine, V.S.; Tao, A.; Torres, N.M.; Chang, M.T.; Drosten, M.; Zhao, H.; Cecchi, F.; Hembrough, T.; et al. Tumours with Class 3 BRAF Mutants are Sensitive to the Inhibition of Activated RAS. Nature 2017, 548, 234–238. [Google Scholar] [CrossRef]
- Dankner, M.; Rose, A.A.N.; Rajkumar, S.; Siegel, P.M.; Watson, I.R. Classifying BRAF Alterations in Cancer: New Rational Therapeutic Strategies for Actionable Mutations. Oncogene 2018, 37, 3183–3199. [Google Scholar] [CrossRef]
- Paik, P.K.; Arcila, M.E.; Fara, M.; Sima, C.S.; Miller, V.A.; Kris, M.G.; Ladanyi, M.; Riely, G.J. Clinical Characteristics of Patients with Lung Adenocarcinomas Harboring BRAF Mutations. J. Clin. Oncol. 2011, 29, 2046–2051. [Google Scholar] [CrossRef]
- Litvak, A.M.; Paik, P.K.; Woo, K.M.; Sima, C.S.; Hellmann, M.D.; Arcila, M.E.; Ladanyi, M.; Rudin, C.M.; Kris, M.G.; Riely, G.J. Clinical Characteristics and Course of 63 Patients with BRAF Mutant Lung Cancers. J. Thorac. Oncol. 2014, 9, 1669–1674. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive Molecular Profiling of Lung Adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Biersack, B.; Tahtamouni, L.; Höpfner, M. Role and Function of Receptor Tyrosine Kinases in BRAF Mutant Cancers. Receptors 2024, 3, 58–106. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Ebi, H.; Turke, A.B.; Coffee, E.M.; Nishino, M.; Cogdill, A.P.; Brown, R.D.; Della Pelle, P.; Dias-Santagata, D.; Hung, K.E.; et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012, 2, 227–235. [Google Scholar] [CrossRef]
- Held, M.A.; Langdon, C.G.; Platt, J.T.; Graham-Steed, T.; Liu, Z.; Chakraborty, A.; Bacchiocchi, A.; Koo, A.; Haskins, J.W.; Bosenberg, M.W.; et al. Genotype-selective combination therapies for melanoma identified by high-throughput drug screening. Cancer Discov. 2013, 3, 52–67. [Google Scholar] [CrossRef]
- Baur, F.; Nietzer, S.L.; Kunz, M.; Saal, F.; Jeromin, J.; Matschos, S.; Linnebacher, M.; Walles, H.; Dandekar, T.; Dandekar, G. Connecting cancer pathways to tumor engines: A stratification tool for colorectal cancer combining human in vitro tissue models with Boolean in silico models. Cancers 2020, 12, 28. [Google Scholar] [CrossRef]
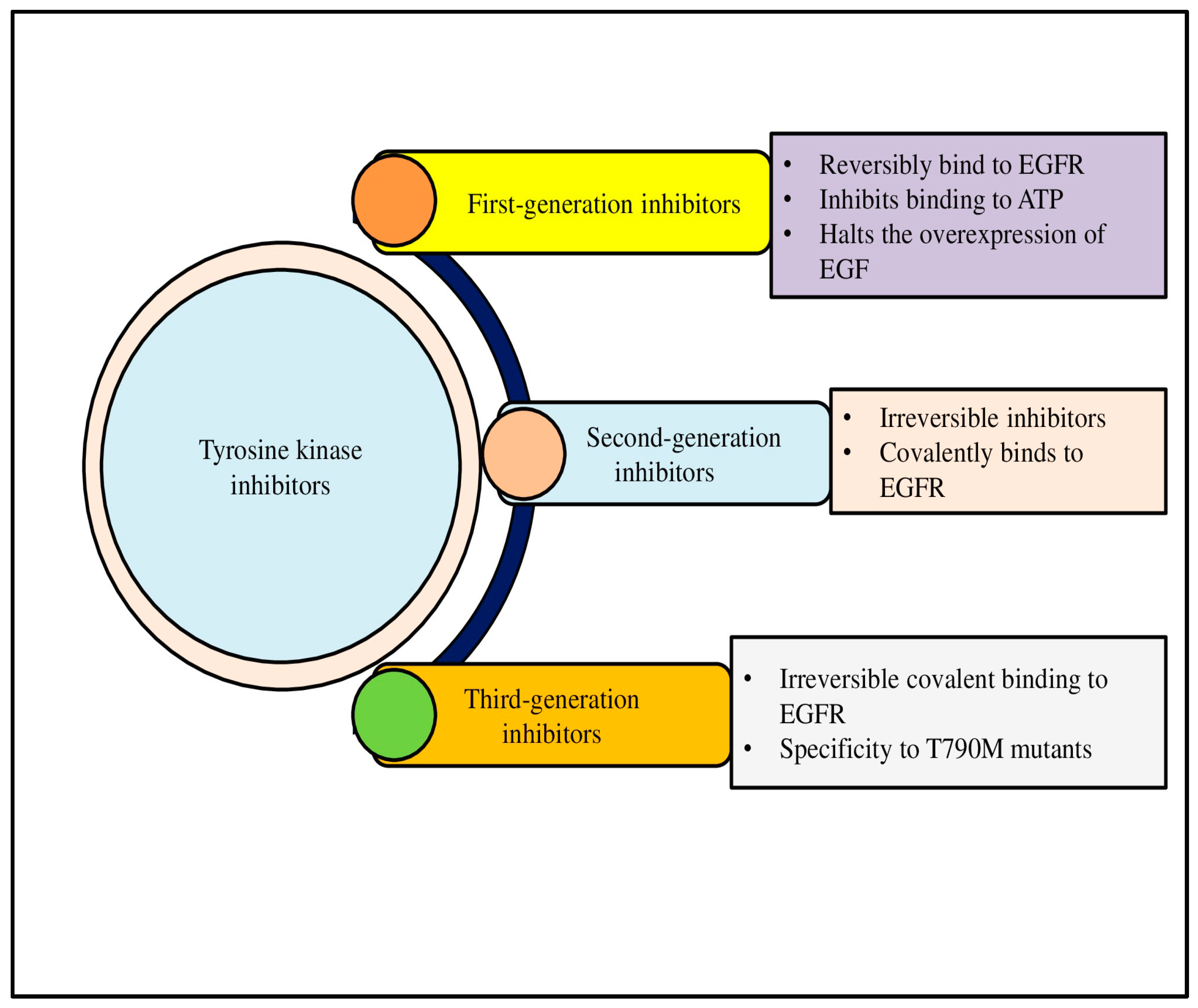
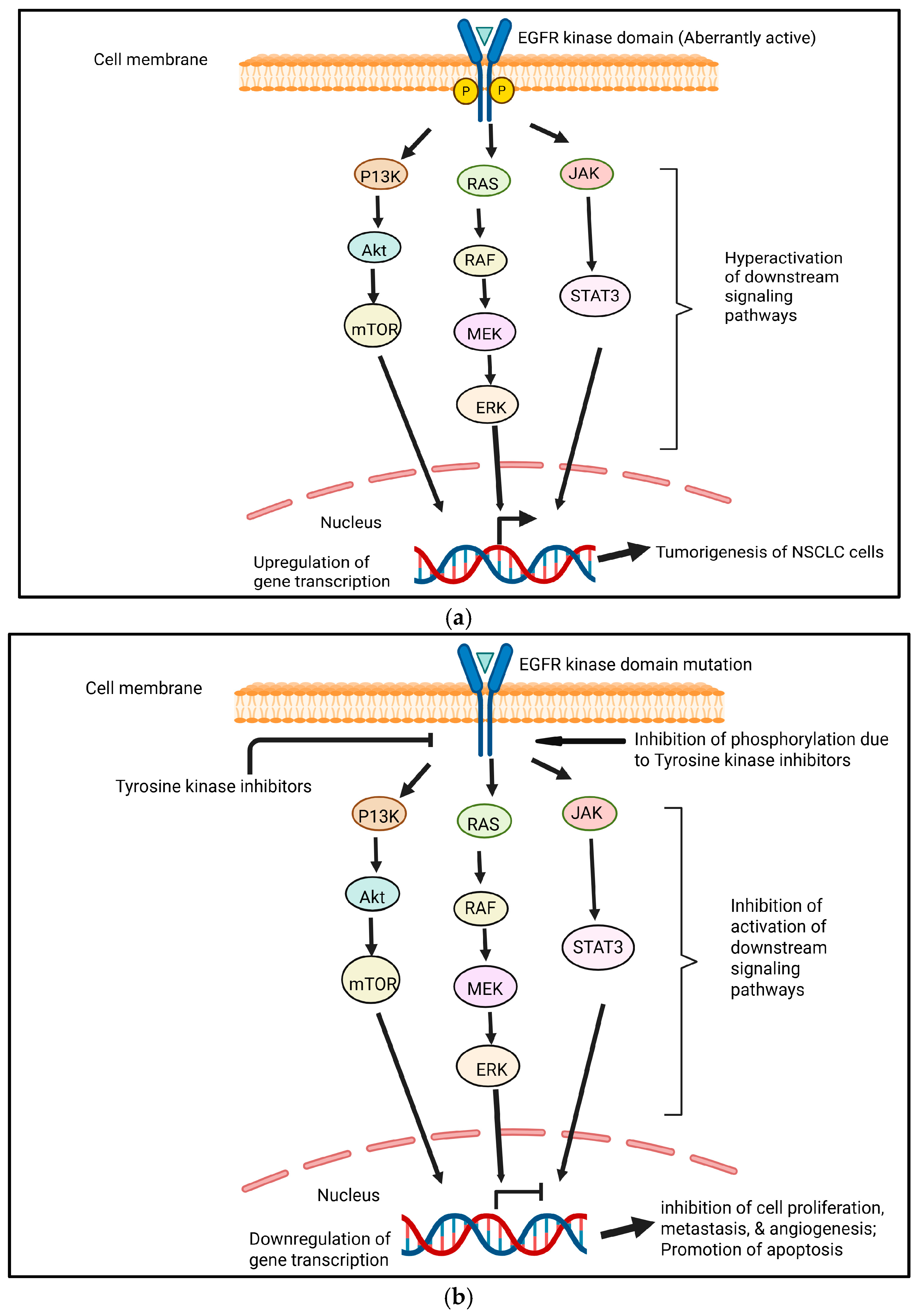
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).