
The Interaction of DMRTA2 with HSP90β Inhibits p53 Ubiquitination and Activates the p53 Pathway to Suppress the Malignant Progression of Non-Small-Cell Lung Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods and Materials
2.1. Bioinformatics
2.2. Cell Culture
2.3. Cell Transfection
2.4. CRISPR Genome Editing
2.5. MTS Experiment
2.6. Colony Formation Experiment
2.7. Transwell Experiment
2.8. Flow Cytometry
2.9. Western Blot Experiment
2.10. Immunoprecipitation Experiment
2.11. Immunofluorescence
2.12. qRT-PCR
2.13. Statistical Analysis
3. Results
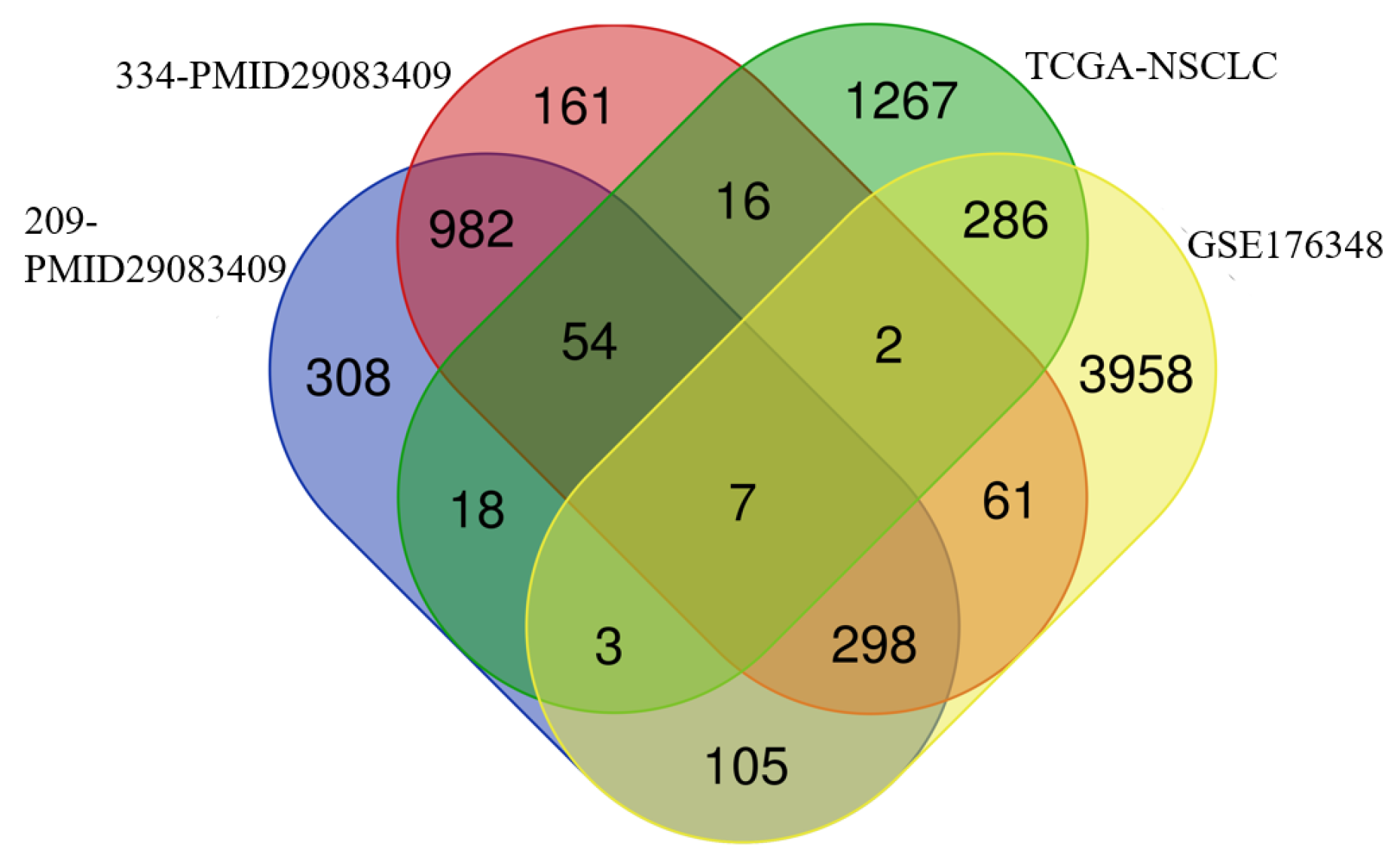
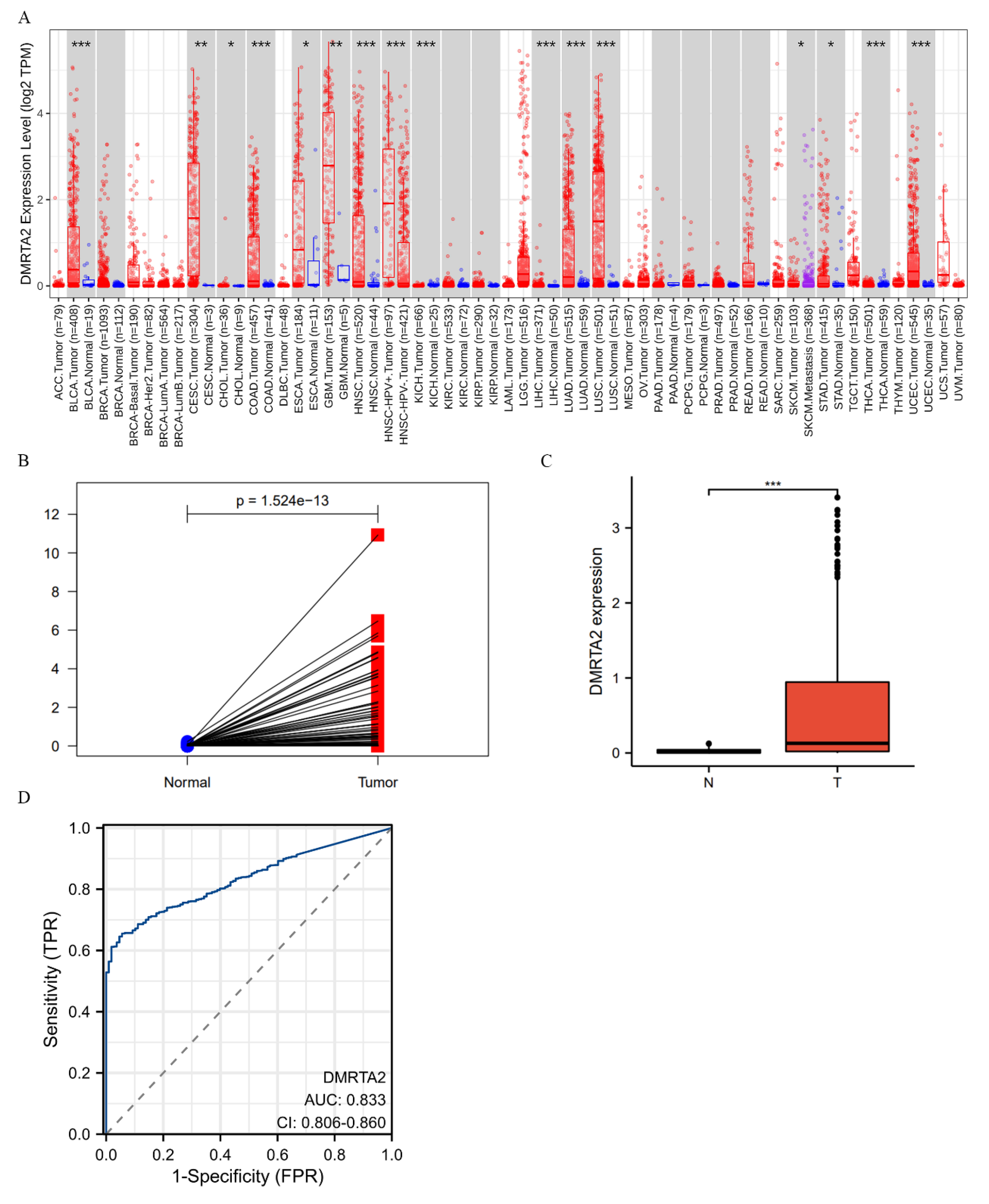
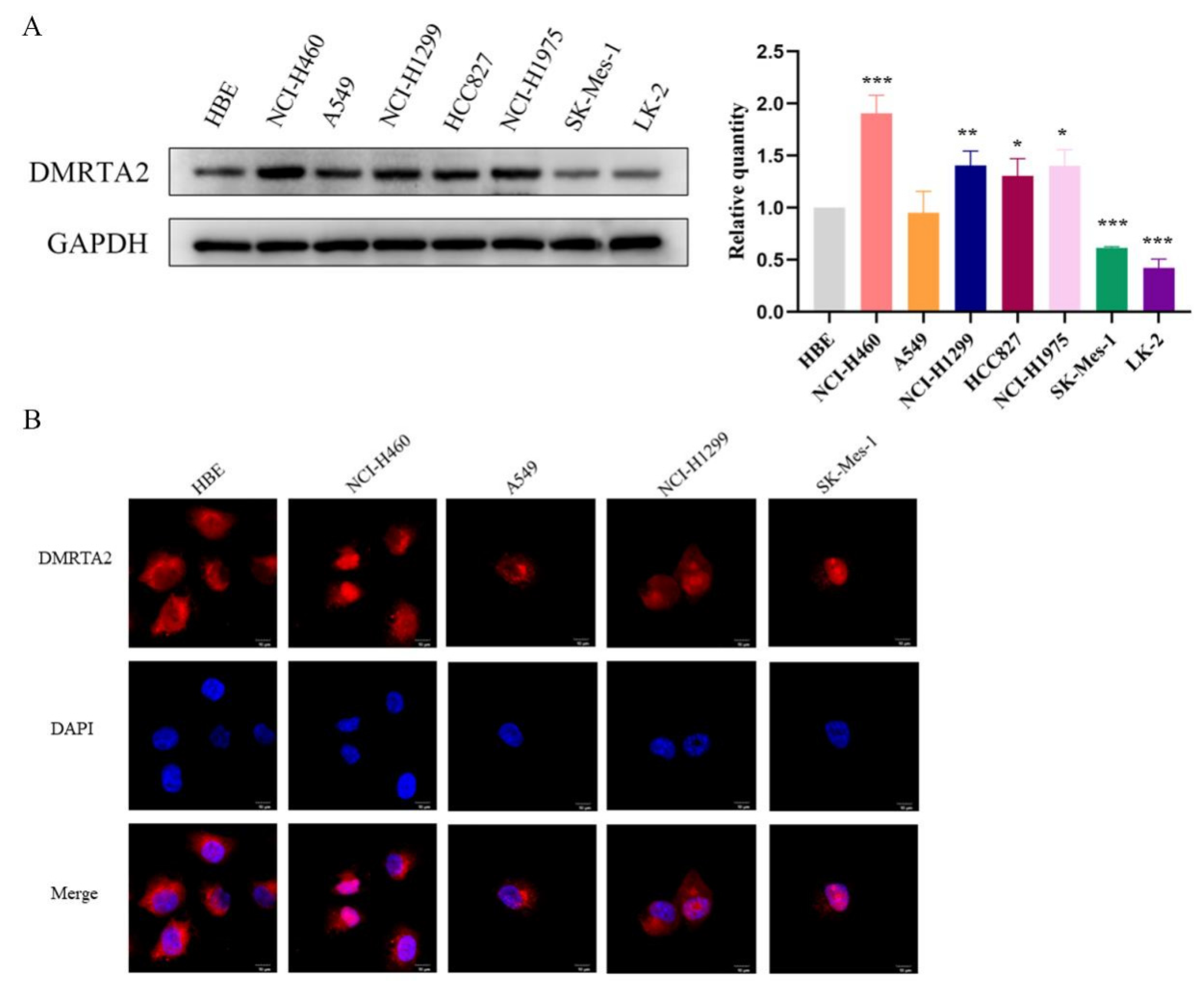
3.1. DMRTA2 Is Highly Expressed in NSCLC and Negatively Associated with Poor Prognosis
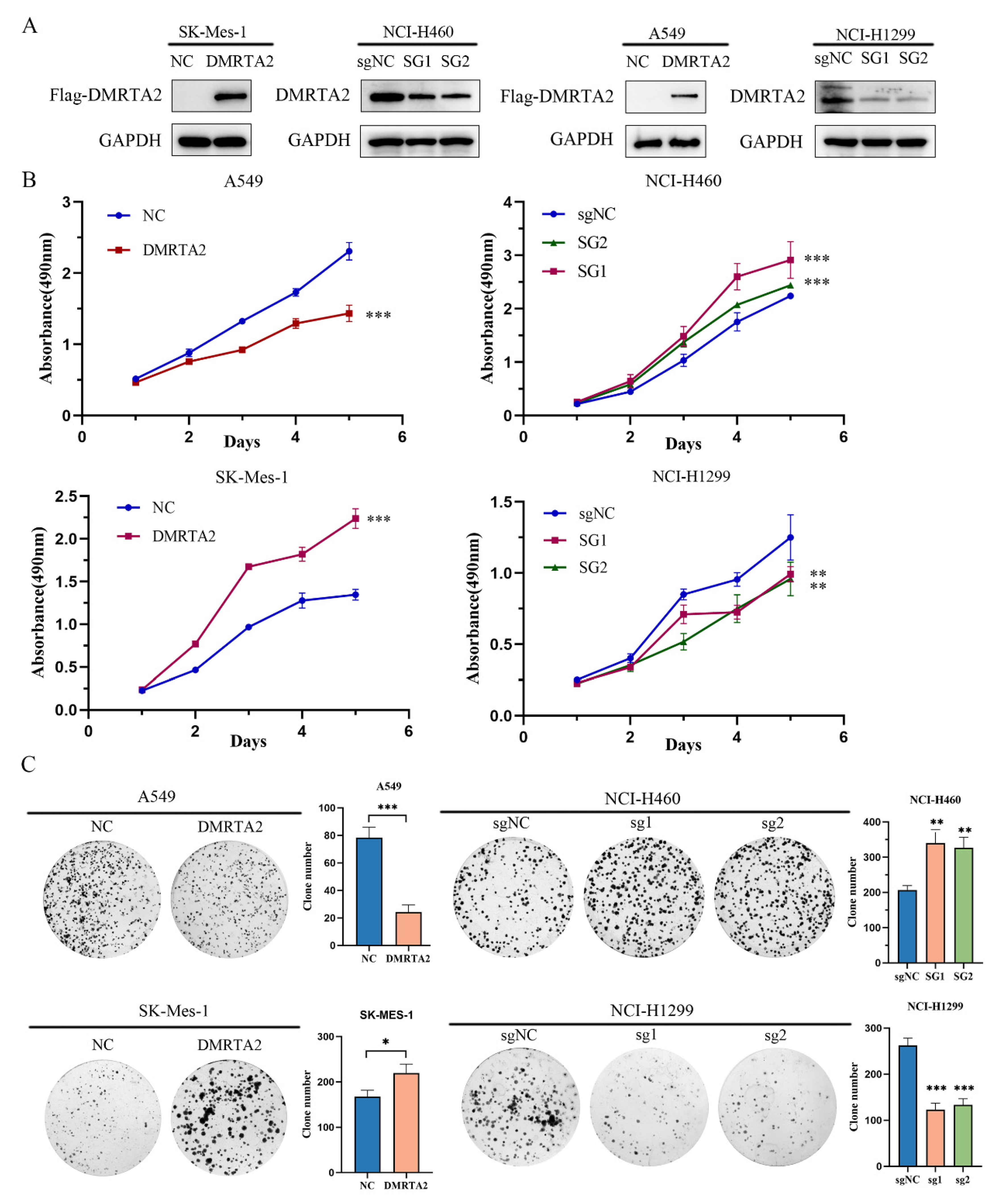
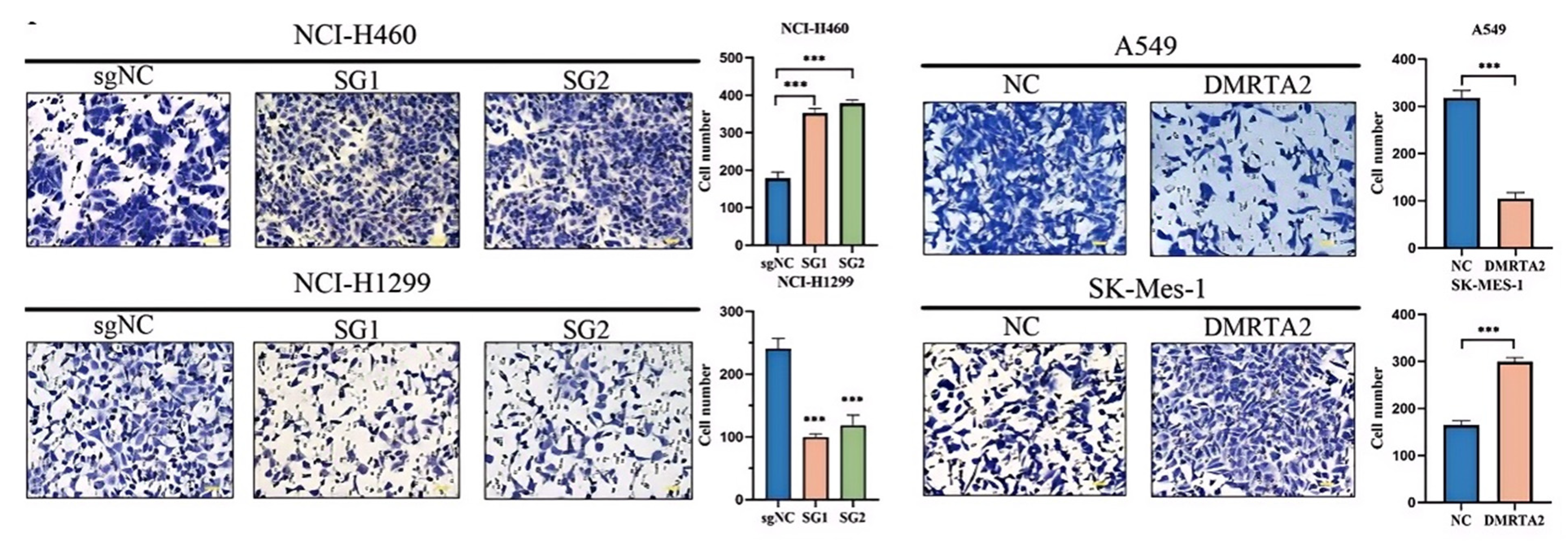
3.2. DMRTA2 Affects the Malignant Phenotype of Lung Cancer Cells
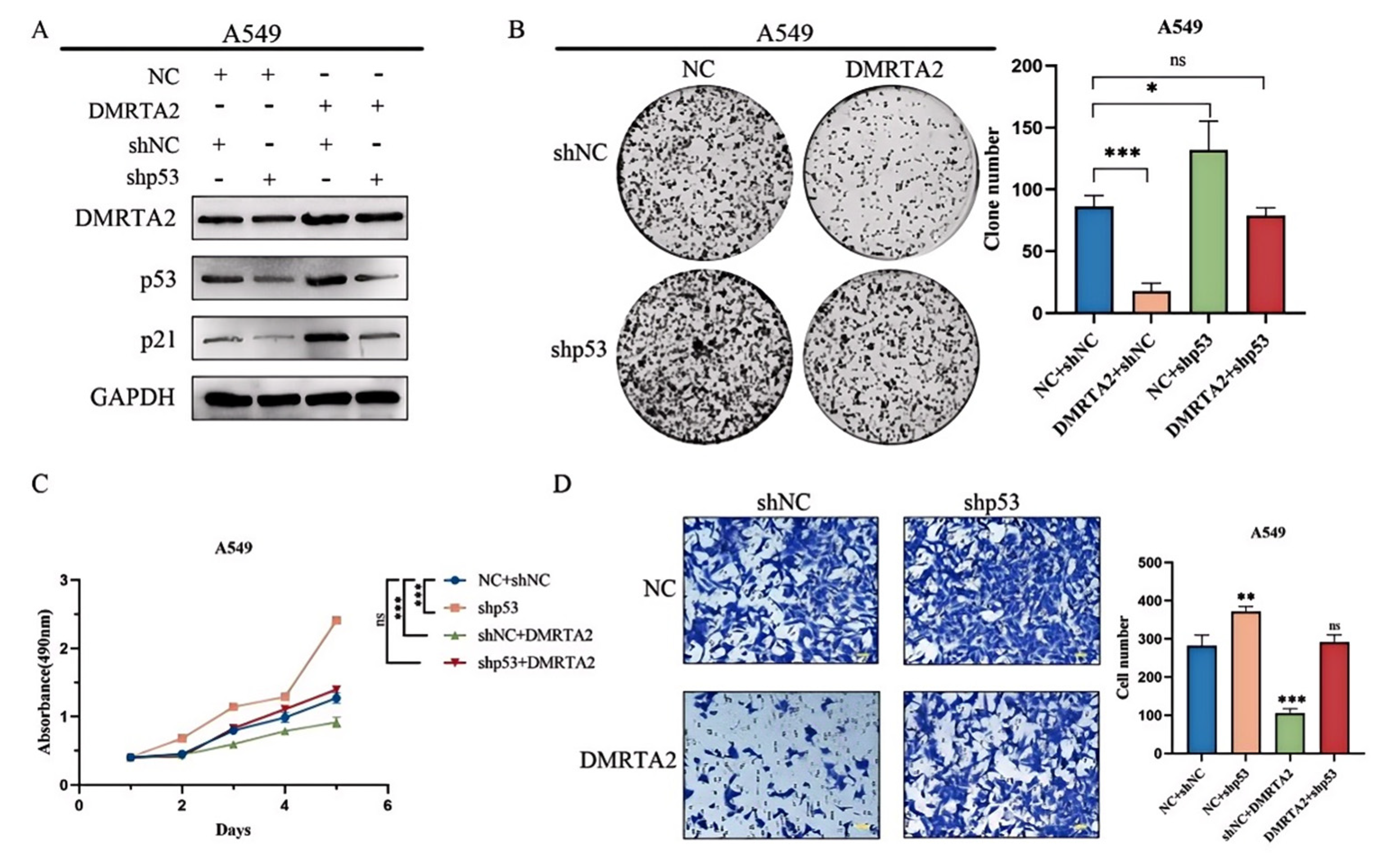
3.3. DMRTA2 Inhibits Lung Cancer Cell Proliferation and Invasion by Regulating the p53 Signaling Pathway
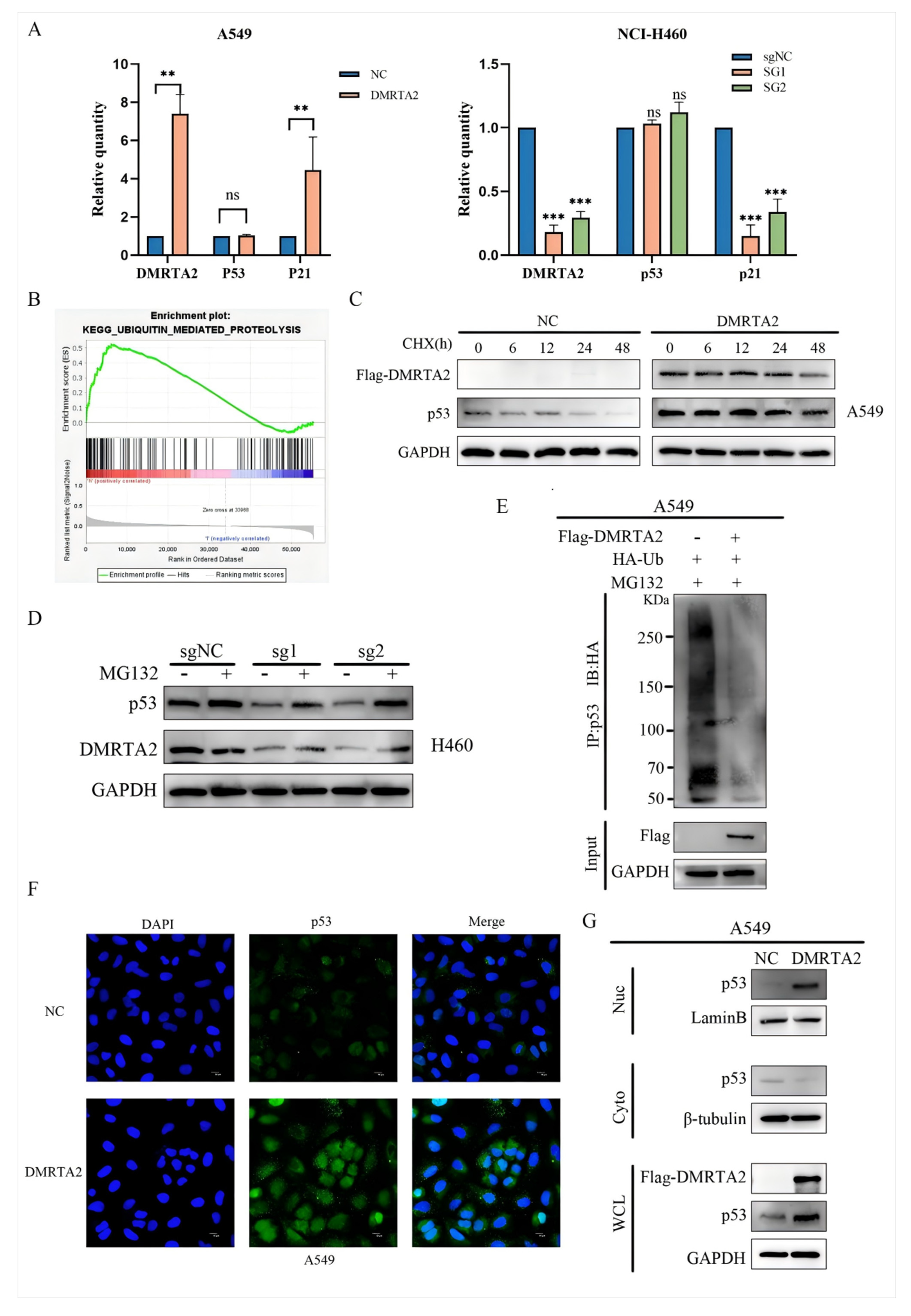
3.4. DMRTA2 Inhibits p53 Ubiquitination and Affects p53 Entry and Exit from the Nucleus
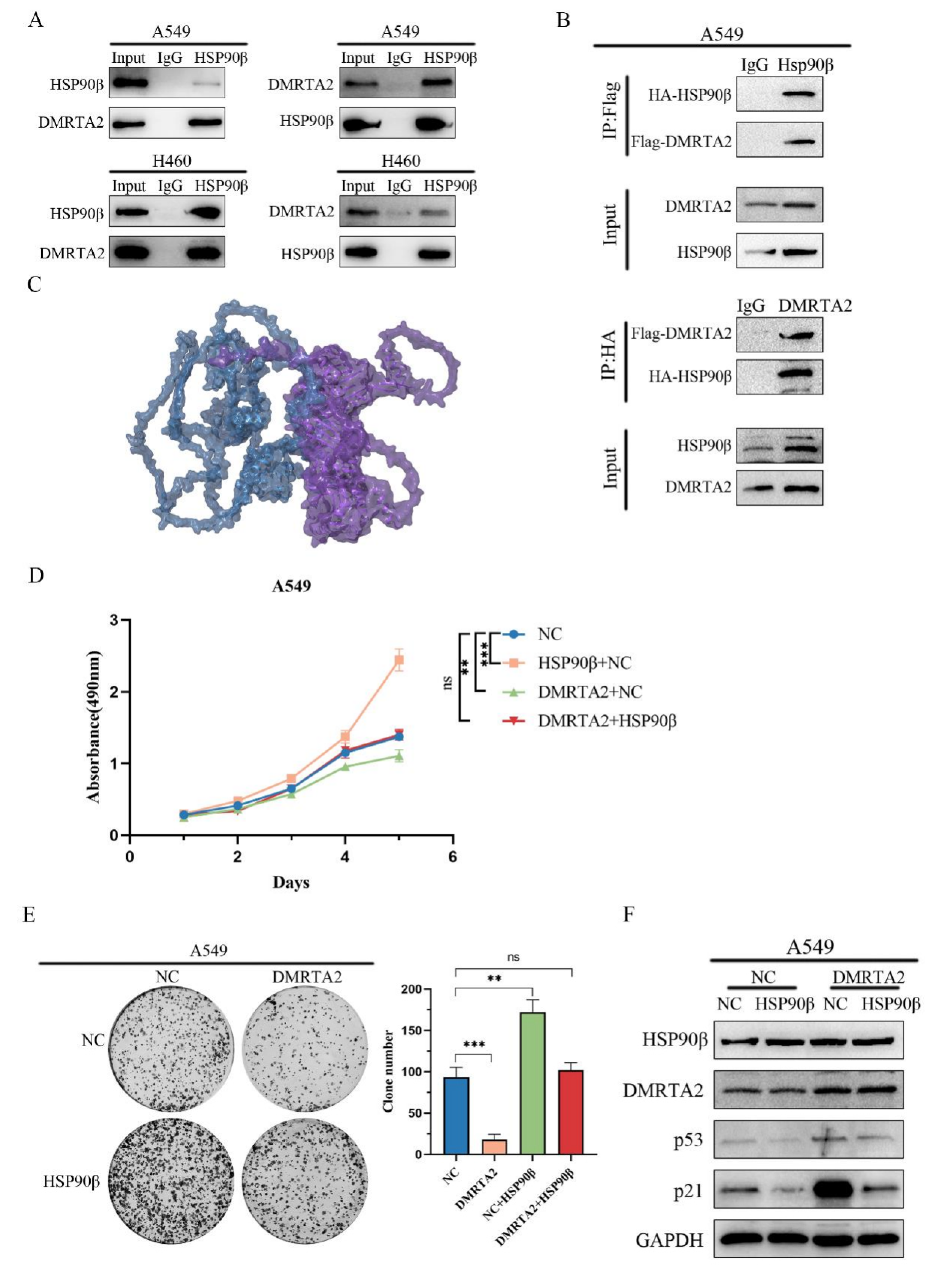
3.5. DMRTA2 Binding to HSP90β Inhibits p53 Degradation
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Cao, M.; Chen, W. Epidemiology of Lung Cancer in China. Thorac. Cancer 2019, 10, 3–7. [Google Scholar] [CrossRef]
- Zeng, H.; Chen, W.; Zheng, R.; Zhang, S.; Ji, J.S.; Zou, X.; Xia, C.; Sun, K.; Yang, Z.; Li, H.; et al. Changing Cancer Survival in China during 2003–15: A Pooled Analysis of 17 Population-Based Cancer Registries. Lancet Glob. Health. 2018, 6, e555–e567. [Google Scholar] [CrossRef]
- Ottolenghi, C.; Fellous, M.; Barbieri, M.; McElreavey, K. Novel Paralogy Relations among Human Chromosomes Support a Link between the Phylogeny of Doublesex-Related Genes and the Evolution of Sex Determination. Genomics 2002, 79, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Allemani, C.; Matsuda, T.; Di Carlo, V.; Harewood, R.; Matz, M.; Nikšić, M.; Bonaventure, A.; Valkov, M.; Johnson, C.J.; Estève, J.; et al. Global Surveillance of Trends in Cancer Survival 2000–14 (CONCORD-3): Analysis of Individual Records for 37 513 025 Patients Diagnosed with One of 18 Cancers from 322 Population-Based Registries in 71 Countries. Lancet 2018, 391, 1023–1075. [Google Scholar] [CrossRef]
- Wang, F.; Yu, Y.; Ji, D.; Li, H. The DMRT Gene Family in Amphioxus. J. Biomol. Struct. Dyn. 2012, 30, 191–200. [Google Scholar] [CrossRef]
- Matson, C.K.; Zarkower, D. Sex and the Singular DM Domain: Insights into Sexual Regulation, Evolution and Plasticity. Nat. Rev. Genet. 2012, 13, 163–174. [Google Scholar] [CrossRef]
- Erdman, S.E.; Burtis, K.C. The Drosophila Doublesex Proteins Share a Novel Zinc Finger Related DNA Binding Domain. EMBO J. 1993, 12, 527–535. [Google Scholar] [CrossRef]
- Zhu, L.; Wilken, J.; Phillips, N.B.; Narendra, U.; Chan, G.; Stratton, S.M.; Kent, S.B.; Weiss, M.A. Sexual Dimorphism in Diverse Metazoans Is Regulated by a Novel Class of Intertwined Zinc Fingers. Genes Dev. 2000, 14, 1750–1764. [Google Scholar] [CrossRef] [PubMed]
- Raymond, C.S.; Shamu, C.E.; Shen, M.M.; Seifert, K.J.; Hirsch, B.; Hodgkin, J.; Zarkower, D. Evidence for Evolutionary Conservation of Sex-Determining Genes. Nature 1998, 391, 691–695. [Google Scholar] [CrossRef]
- Bellefroid, E.J.; Leclère, L.; Saulnier, A.; Keruzore, M.; Sirakov, M.; Vervoort, M.; De Clercq, S. Expanding Roles for the Evolutionarily Conserved Dmrt Sex Transcriptional Regulators during Embryogenesis. Cell. Mol. Life Sci. 2013, 70, 3829–3845. [Google Scholar] [CrossRef]
- Murphy, M.W.; Lee, J.K.; Rojo, S.; Gearhart, M.D.; Kurahashi, K.; Banerjee, S.; Loeuille, G.-A.; Bashamboo, A.; McElreavey, K.; Zarkower, D.; et al. An Ancient Protein-DNA Interaction Underlying Metazoan Sex Determination. Nat. Struct. Mol. Biol. 2015, 22, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.W.; Sarver, A.L.; Rice, D.; Hatzi, K.; Ye, K.; Melnick, A.; Heckert, L.L.; Zarkower, D.; Bardwell, V.J. Genome-Wide Analysis of DNA Binding and Transcriptional Regulation by the Mammalian Doublesex Homolog DMRT1 in the Juvenile Testis. Proc. Natl. Acad. Sci. USA 2010, 107, 13360–13365. [Google Scholar] [CrossRef]
- Murphy, M.W.; Zarkower, D.; Bardwell, V.J. Vertebrate DM Domain Proteins Bind Similar DNA Sequences and Can Heterodimerize on DNA. BMC Mol. Biol. 2007, 8, 58. [Google Scholar] [CrossRef] [PubMed]
- Shirak, A.; Seroussi, E.; Cnaani, A.; E Howe, A.; Domokhovsky, R.; Zilberman, N.; Kocher, T.D.; Hulata, G.; Ron, M. Amh and Dmrta2 Genes Map to Tilapia (Oreochromis spp.) Linkage Group 23 within Quantitative Trait Locus Regions for Sex Determination. Genetics 2006, 174, 1573–1581. [Google Scholar] [CrossRef]
- Parlier, D.; Moers, V.; Van Campenhout, C.; Preillon, J.; Leclère, L.; Saulnier, A.; Sirakov, M.; Busengdal, H.; Kricha, S.; Marine, J.-C.; et al. The Xenopus Doublesex-Related Gene Dmrt5 Is Required for Olfactory Placode Neurogenesis. Dev. Biol. 2013, 373, 39–52. [Google Scholar] [CrossRef]
- Yoshizawa, A.; Nakahara, Y.; Izawa, T.; Ishitani, T.; Tsutsumi, M.; Kuroiwa, A.; Itoh, M.; Kikuchi, Y. Zebrafish Dmrta2 Regulates Neurogenesis in the Telencephalon. Genes Cells 2011, 16, 1097–1109. [Google Scholar] [CrossRef]
- Xu, S.; Xia, W.; Zohar, Y.; Gui, J.-F. Zebrafish Dmrta2 Regulates the Expression of Cdkn2c in Spermatogenesis in the Adult Testis. Biol. Reprod. 2013, 88, 14. [Google Scholar] [CrossRef]
- Young, F.I.; Keruzore, M.; Nan, X.; Gennet, N.; Bellefroid, E.J.; Li, M. The Doublesex-Related Dmrta2 Safeguards Neural Progenitor Maintenance Involving Transcriptional Regulation of Hes1. Proc. Natl. Acad. Sci. USA 2017, 114, E5599–E5607. [Google Scholar] [CrossRef] [PubMed]
- Urquhart, J.E.; Beaman, G.; Byers, H.; Roberts, N.A.; Chervinsky, E.; O’Sullivan, J.; Pilz, D.; Fry, A.; Williams, S.G.; Bhaskar, S.S.; et al. DMRTA2 (DMRT5) Is Mutated in a Novel Cortical Brain Malformation. Clin. Genet. 2016, 89, 724–727. [Google Scholar] [CrossRef] [PubMed]
- Desmaris, E.; Keruzore, M.; Saulnier, A.; Ratié, L.; Assimacopoulos, S.; De Clercq, S.; Nan, X.; Roychoudhury, K.; Qin, S.; Kricha, S.; et al. DMRT5, DMRT3, and EMX2 Cooperatively Repress Gsx2 at the Pallium-Subpallium Boundary to Maintain Cortical Identity in Dorsal Telencephalic Progenitors. J. Neurosci. 2018, 38, 9105–9121. [Google Scholar] [CrossRef]
- Lee, C.-M.; Zhou, L.; Liu, J.; Shi, J.; Geng, Y.; Liu, M.; Wang, J.; Su, X.; Barad, N.; Wang, J.; et al. Single-Cell RNA-Seq Analysis Revealed Long-Lasting Adverse Effects of Tamoxifen on Neurogenesis in Prenatal and Adult Brains. Proc. Natl. Acad. Sci. USA 2020, 117, 19578–19589. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Qin, X. Significance of TP53 Mutation in Treatment and Prognosis in Head and Neck Squamous Cell Carcinoma. Biomark. Med. 2021, 15, 15–28. [Google Scholar] [CrossRef]
- Hassin, O.; Oren, M. Drugging P53 in Cancer: One Protein, Many Targets. Nat. Rev. Drug Discov. 2023, 22, 127–144. [Google Scholar] [CrossRef]
- Liebl, M.C.; Hofmann, T.G. The Role of P53 Signaling in Colorectal Cancer. Cancers 2021, 13, 2125. [Google Scholar] [CrossRef]
- Sullivan, K.D.; Galbraith, M.D.; Andrysik, Z.; Espinosa, J.M. Mechanisms of Transcriptional Regulation by P53. Cell Death Differ. 2018, 25, 133–143. [Google Scholar] [CrossRef]
- Kung, C.-P.; Weber, J.D. It’s Getting Complicated—A Fresh Look at P53-MDM2-ARF Triangle in Tumorigenesis and Cancer Therapy. Front. Cell Dev. Biol. 2022, 10, 818744. [Google Scholar] [CrossRef]
- Deng, L.; Meng, T.; Chen, L.; Wei, W.; Wang, P. The Role of Ubiquitination in Tumorigenesis and Targeted Drug Discovery. Signal Transduct. Target. Ther. 2020, 5, 11. [Google Scholar] [CrossRef]
- Cockram, P.E.; Kist, M.; Prakash, S.; Chen, S.-H.; Wertz, I.E.; Vucic, D. Ubiquitination in the Regulation of Inflammatory Cell Death and Cancer. Cell Death Differ. 2021, 28, 591–605. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ren, W.; Wang, Q.; He, Y.; Ma, D.; Cai, Z. The Regulation of Necroptosis by Ubiquitylation. Apoptosis 2022, 27, 668–684. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, S.; Li, L.; Du, J. The Interaction of DMRTA2 with HSP90β Inhibits p53 Ubiquitination and Activates the p53 Pathway to Suppress the Malignant Progression of Non-Small-Cell Lung Cancer. Curr. Issues Mol. Biol. 2025, 47, 497. https://doi.org/10.3390/cimb47070497
Deng S, Li L, Du J. The Interaction of DMRTA2 with HSP90β Inhibits p53 Ubiquitination and Activates the p53 Pathway to Suppress the Malignant Progression of Non-Small-Cell Lung Cancer. Current Issues in Molecular Biology. 2025; 47(7):497. https://doi.org/10.3390/cimb47070497
Chicago/Turabian StyleDeng, Shiyang, Ling Li, and Jiang Du. 2025. "The Interaction of DMRTA2 with HSP90β Inhibits p53 Ubiquitination and Activates the p53 Pathway to Suppress the Malignant Progression of Non-Small-Cell Lung Cancer" Current Issues in Molecular Biology 47, no. 7: 497. https://doi.org/10.3390/cimb47070497
APA StyleDeng, S., Li, L., & Du, J. (2025). The Interaction of DMRTA2 with HSP90β Inhibits p53 Ubiquitination and Activates the p53 Pathway to Suppress the Malignant Progression of Non-Small-Cell Lung Cancer. Current Issues in Molecular Biology, 47(7), 497. https://doi.org/10.3390/cimb47070497