Epigallocatechin-3-gallate Synergistically Enhanced Arecoline-Induced Cytotoxicity by Redirecting Cycle Arrest to Apoptosis

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Cell Culture
2.2. Cell Treatment and Viability Assay
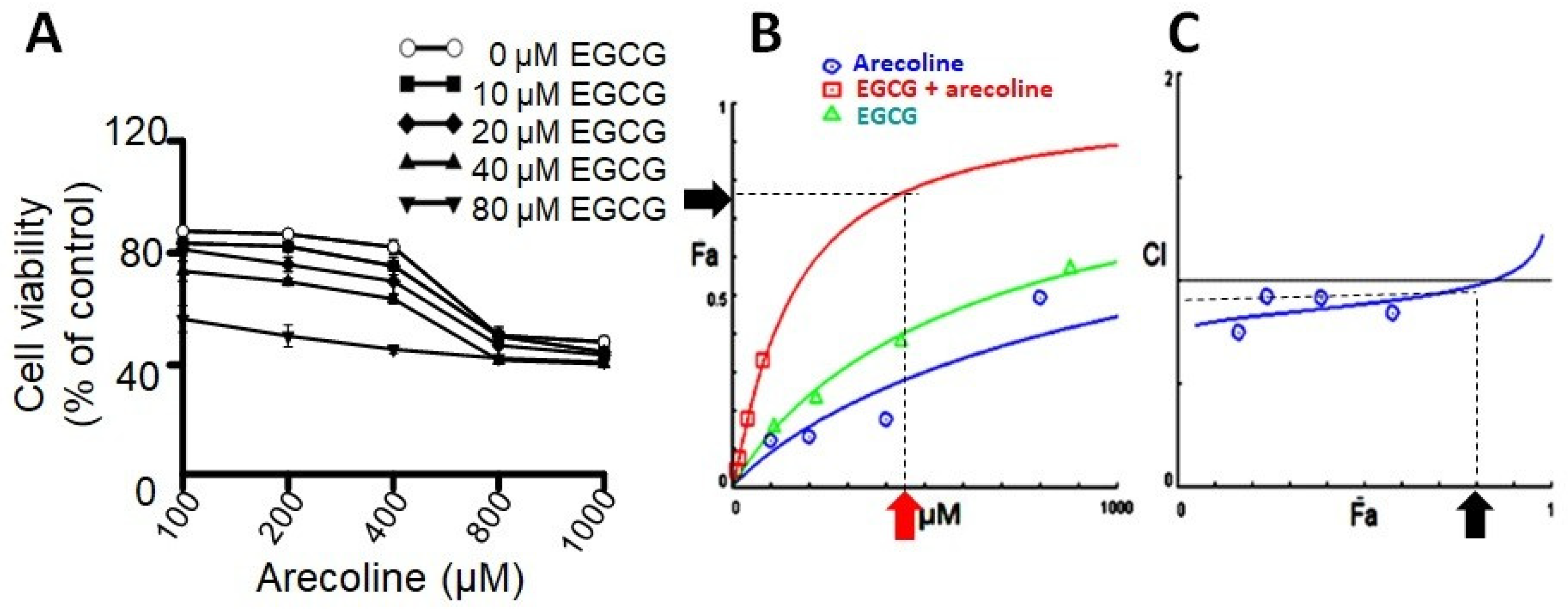
2.3. Determination of Synergistic Effect and Choice of Treatment Concentrations
2.4. Reactive Oxygen Species Analysis
2.5. Flow Cytometric Analysis of Cell Cycle and Apoptosis Phases
2.6. Annexin V-Binding Assay
2.7. Western Blot Analysis
2.8. Statistical Analysis
3. Results
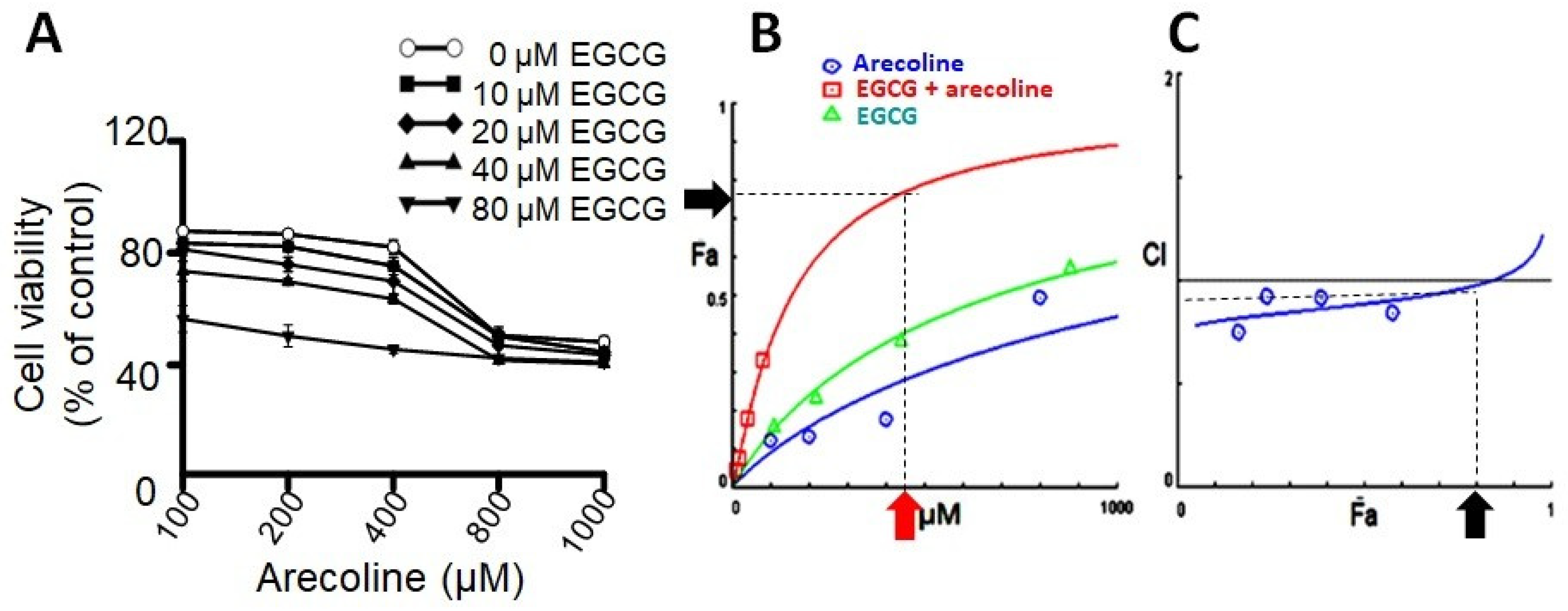
3.1. EGCG Synergistically Enhanced Cytotoxic Effects of Arecoline
3.2. EGCG and NAC Inhibited Arecoline-Generated ROS but Had Different Effects on PC Cell Viability
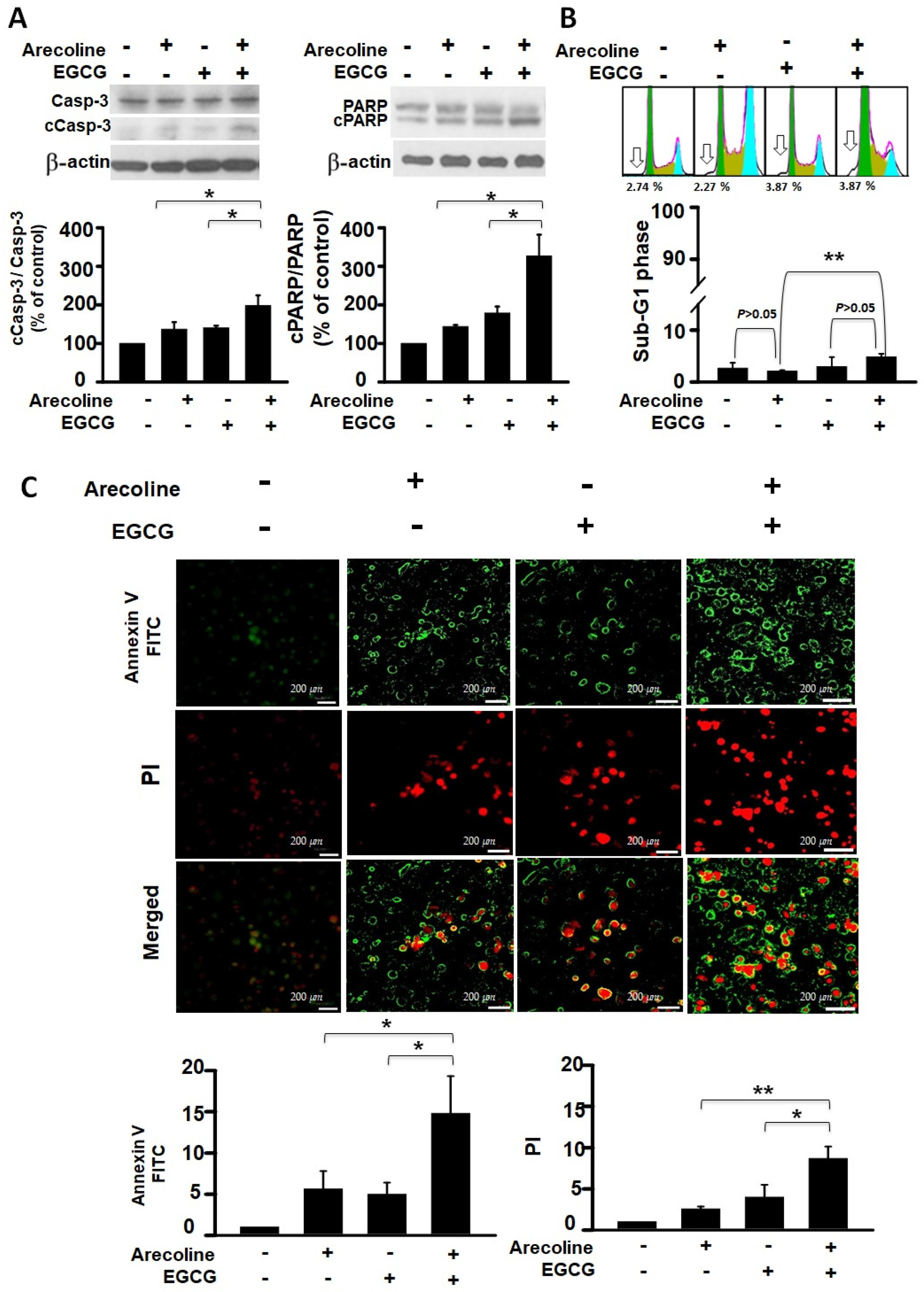
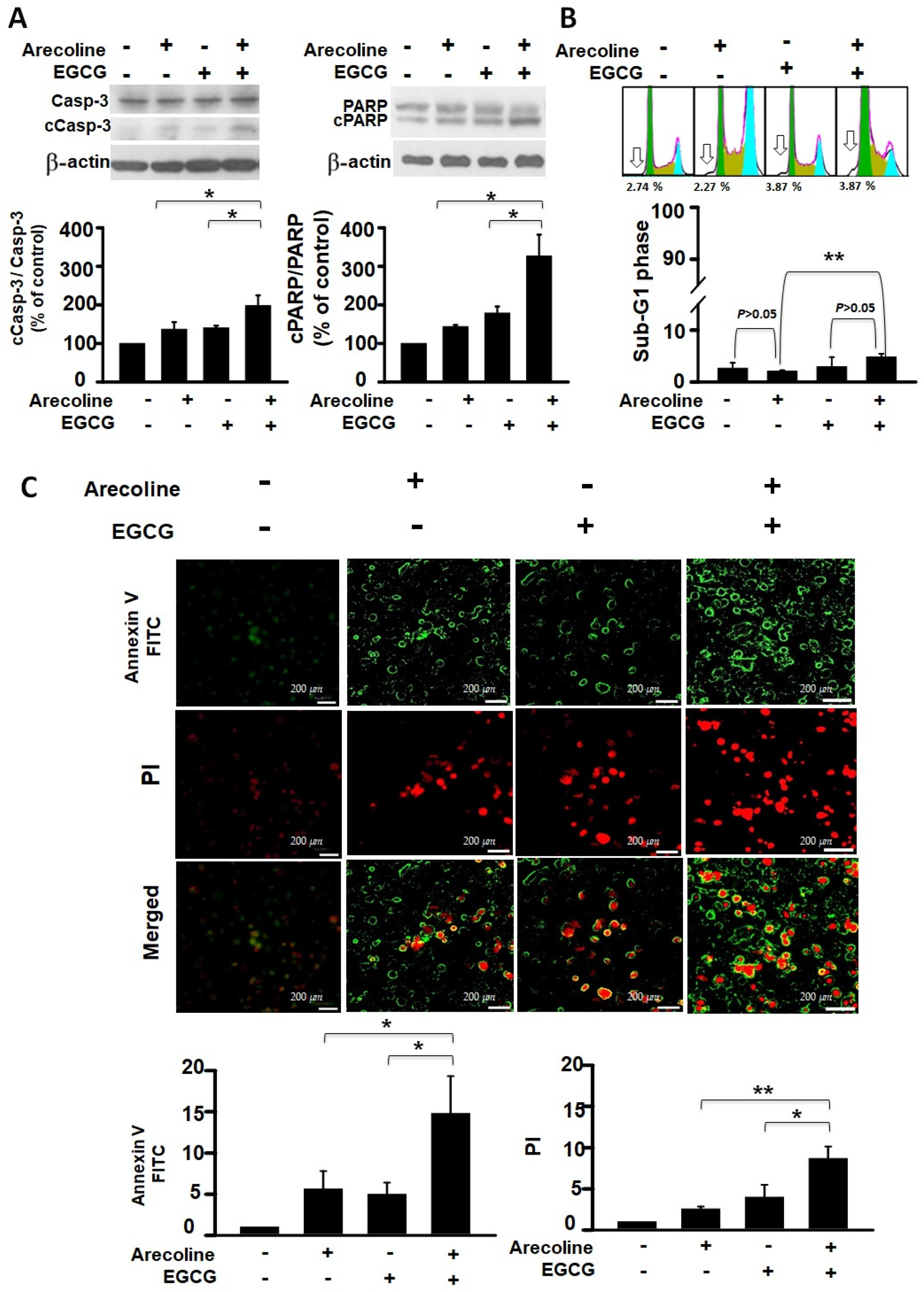
3.3. EGCG Synergistically Increased Apoptotic Events in Arecoline-Treated PC Cells
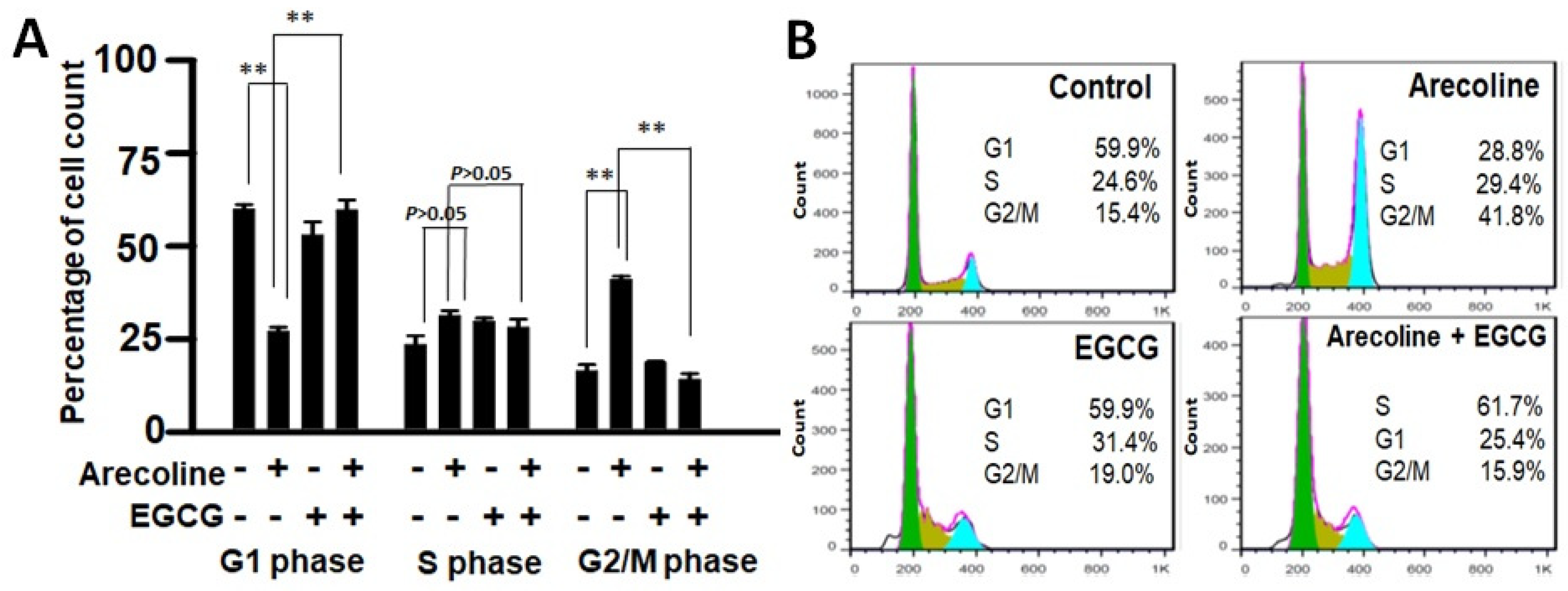
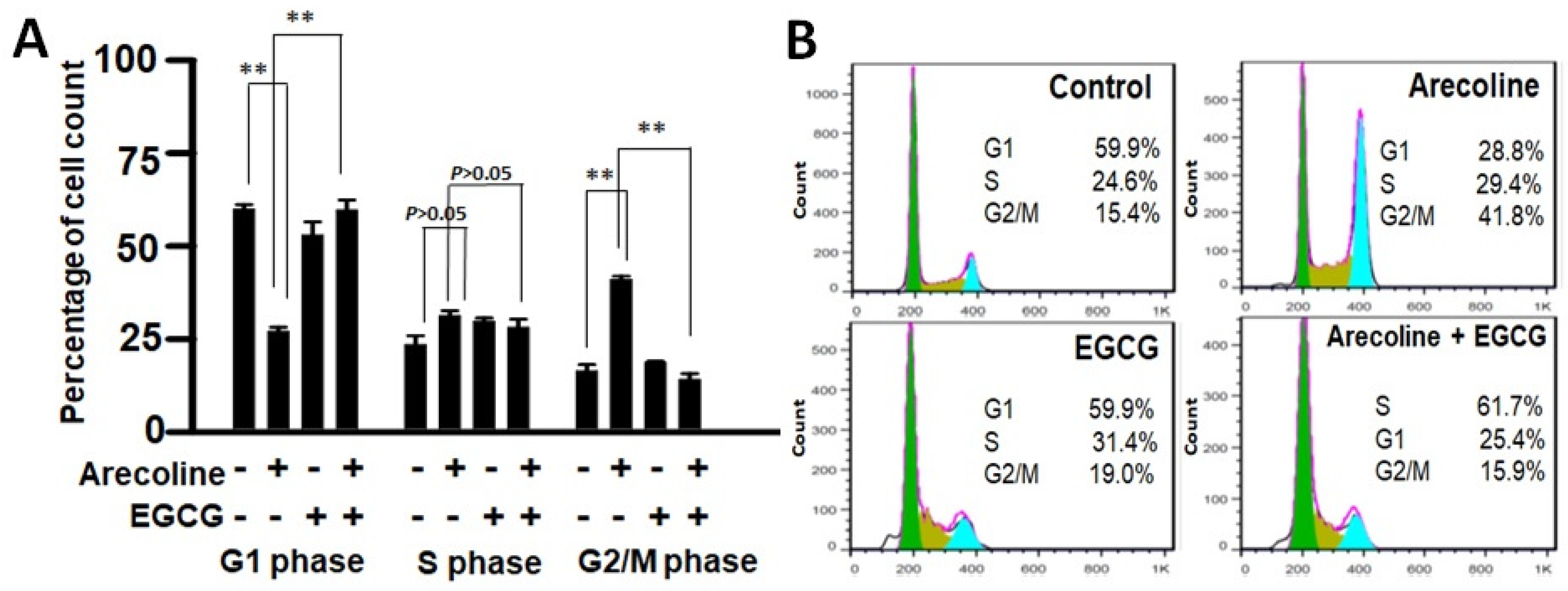
3.4. EGCG Restores Cell Cycle Progression by Redirecting Cells from Arecoline-Induced Cycle Arrest towards Apoptosis
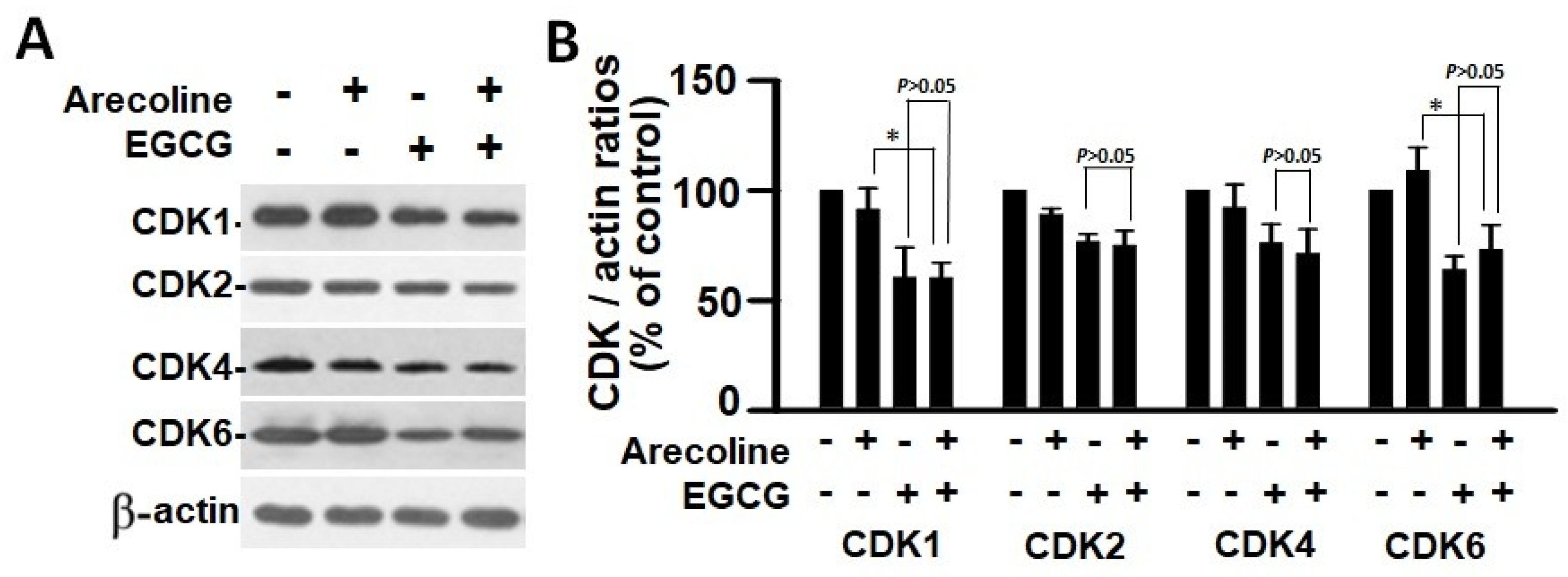
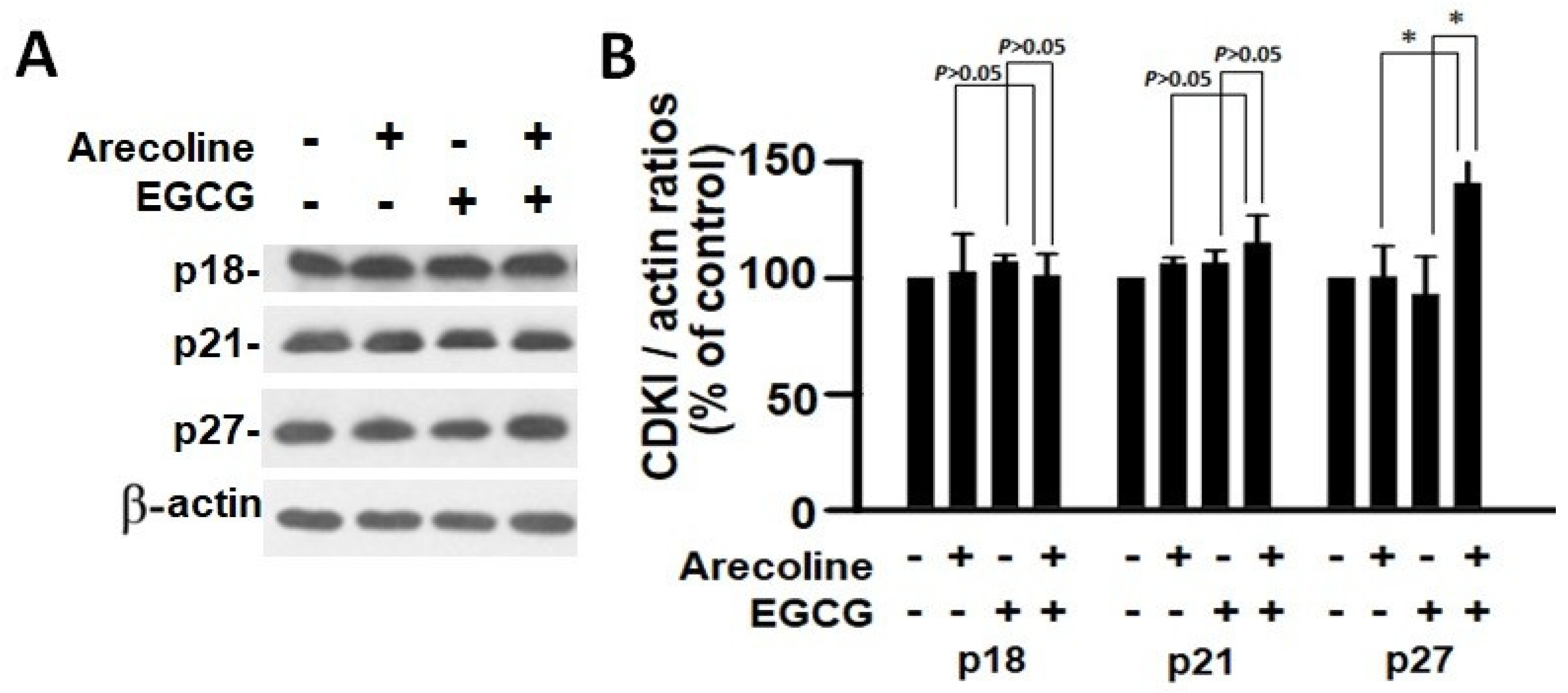
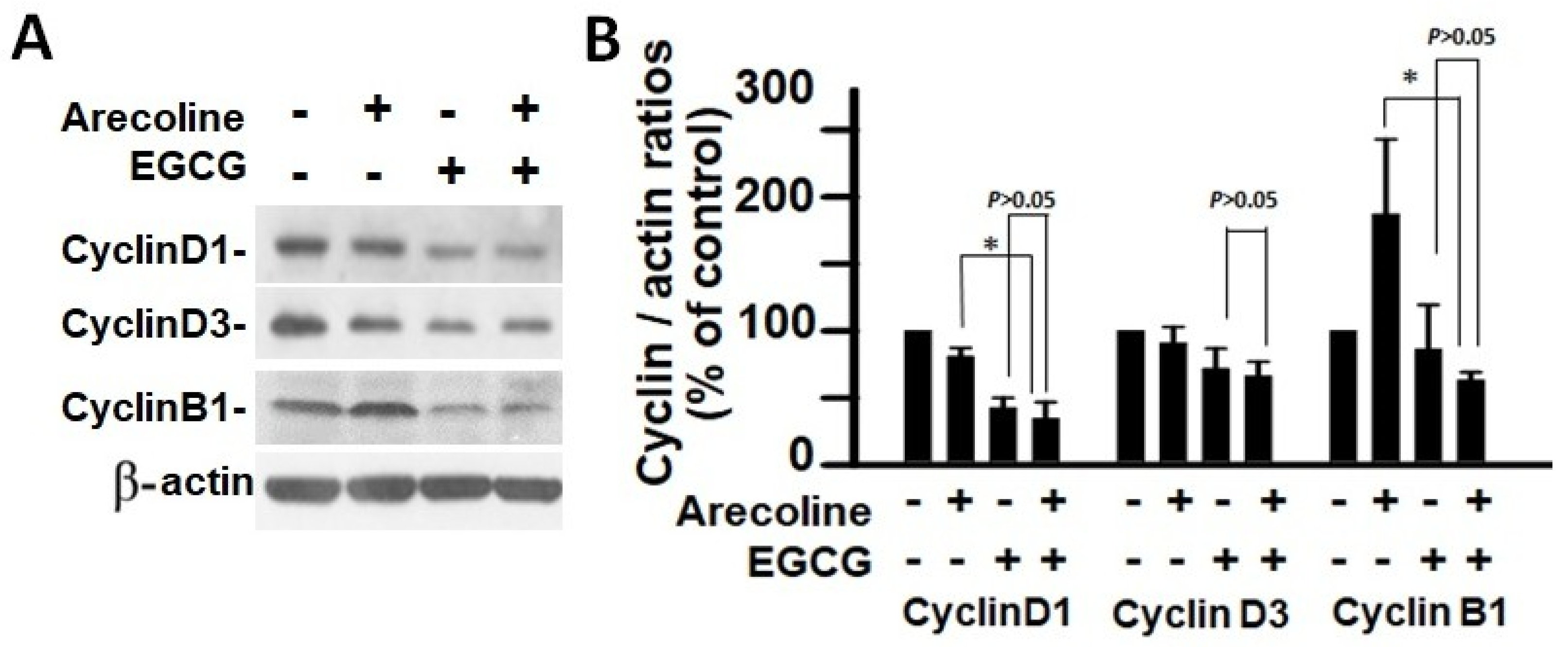
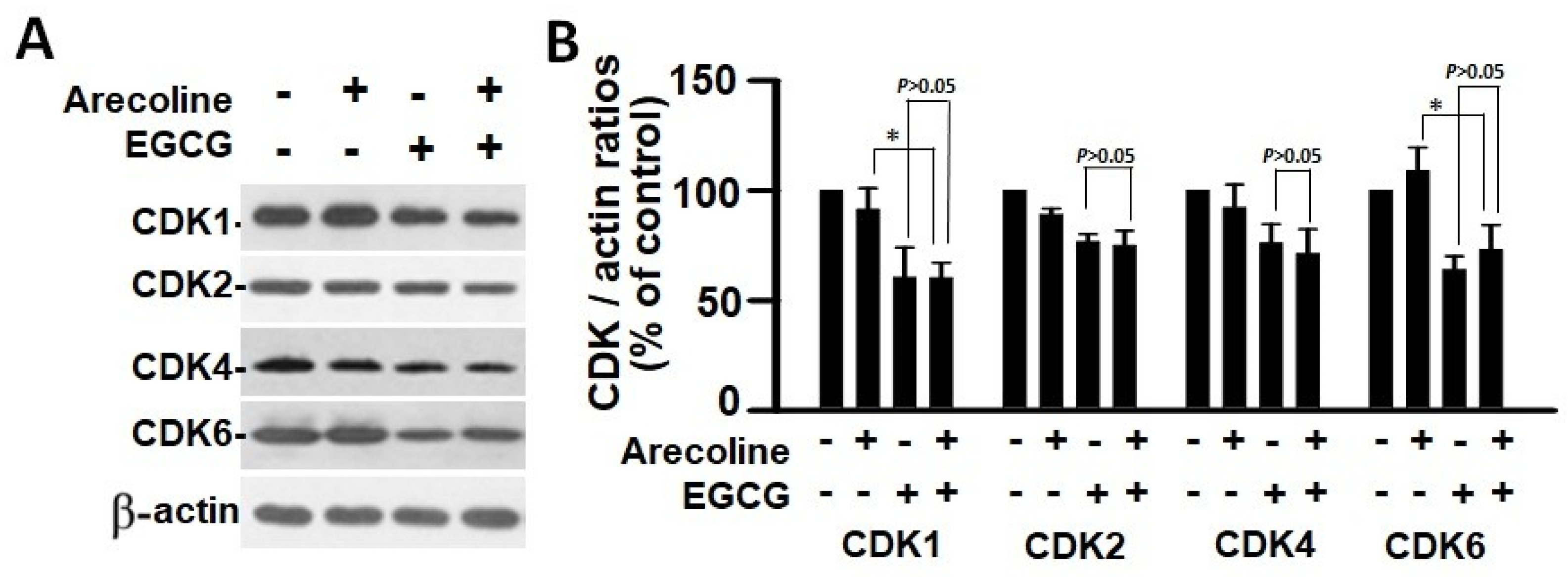
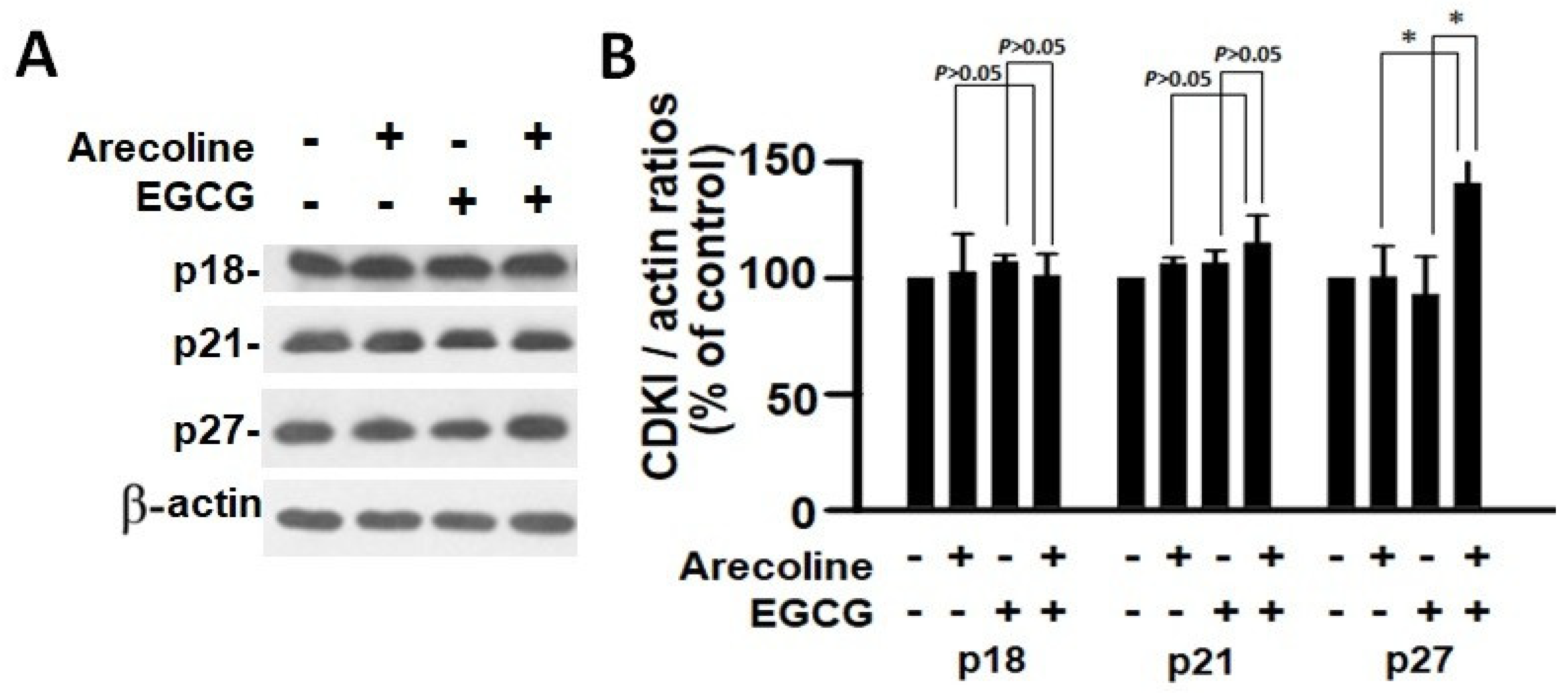
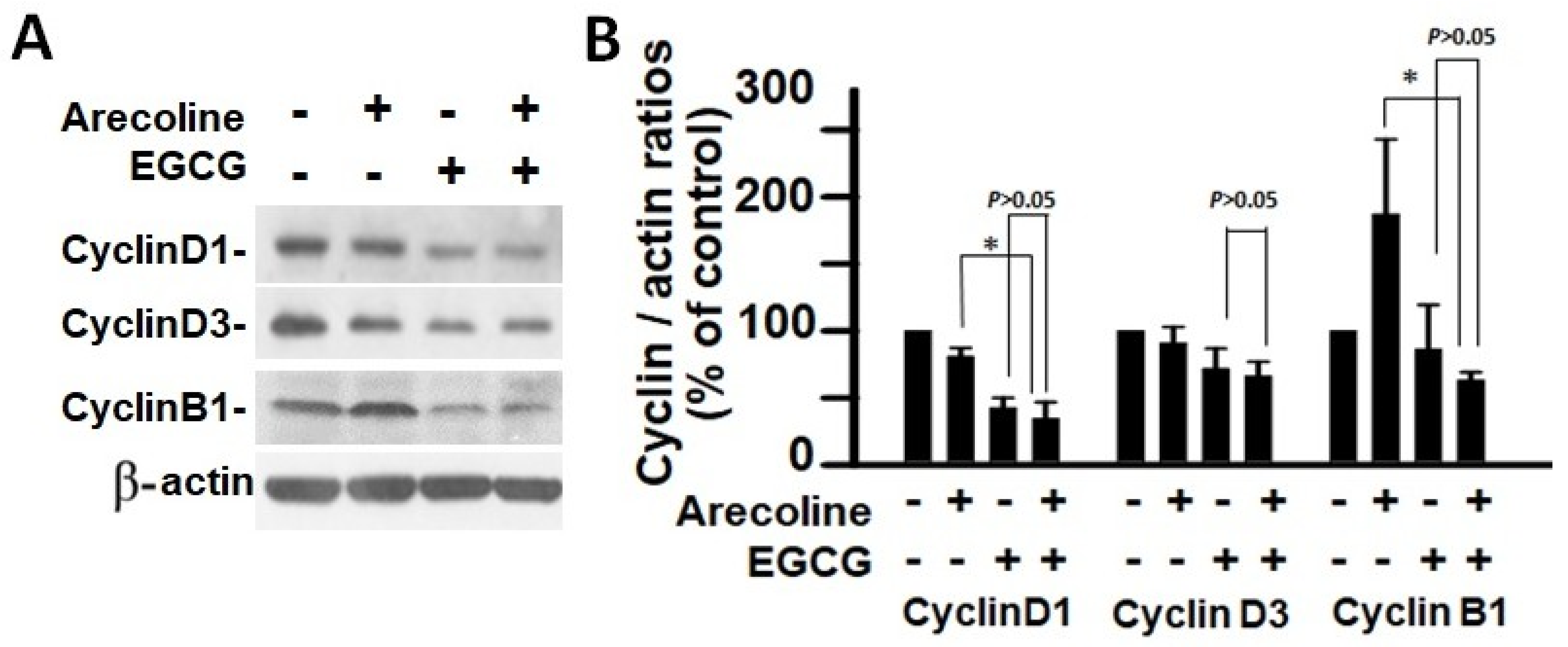
3.5. EGCG and Arecoline Synergistically Impacted the Abundance of Cyclins, Cyclin-Dependent Kinase (CDK), and CDK Inhibitors
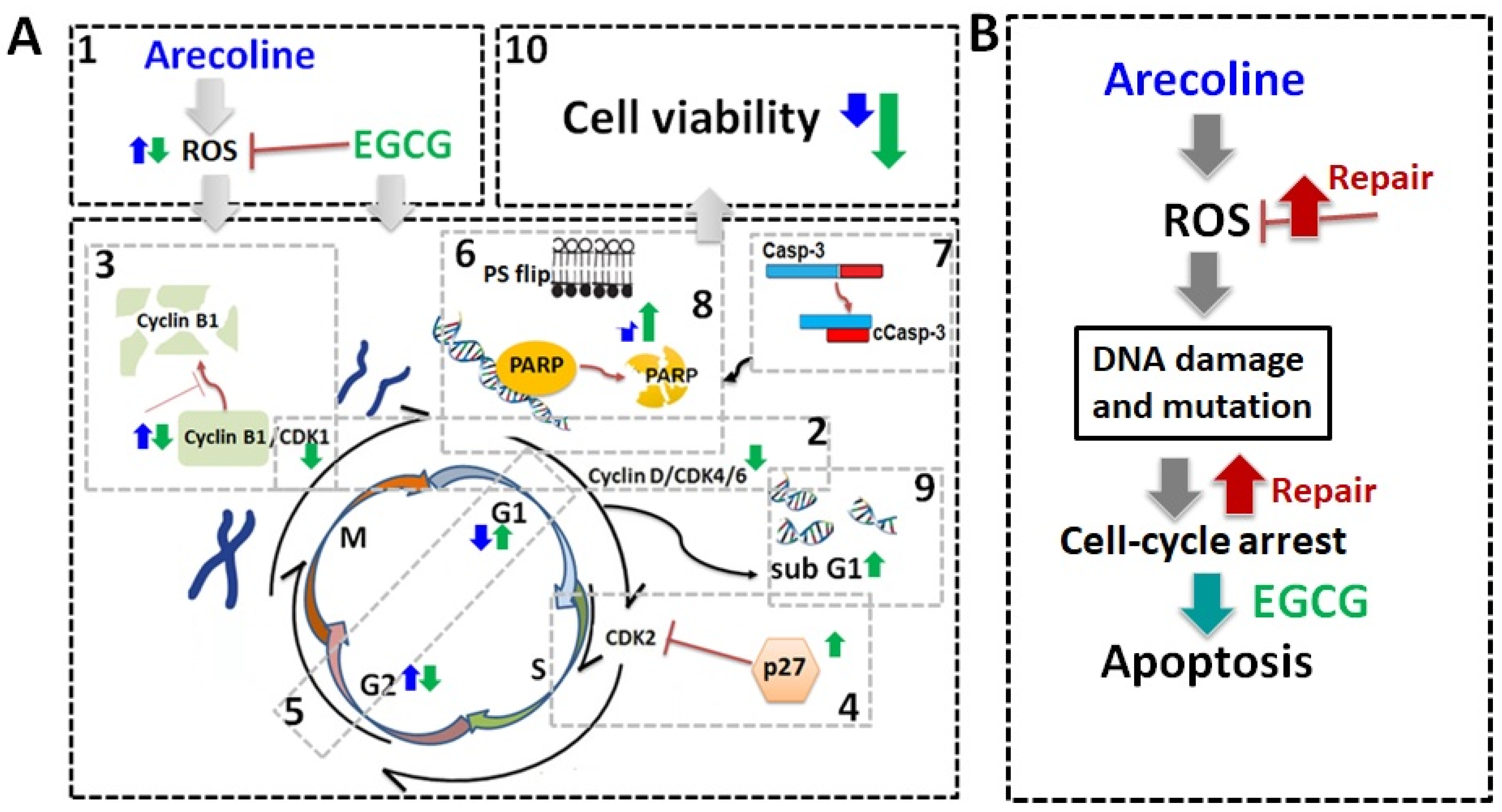
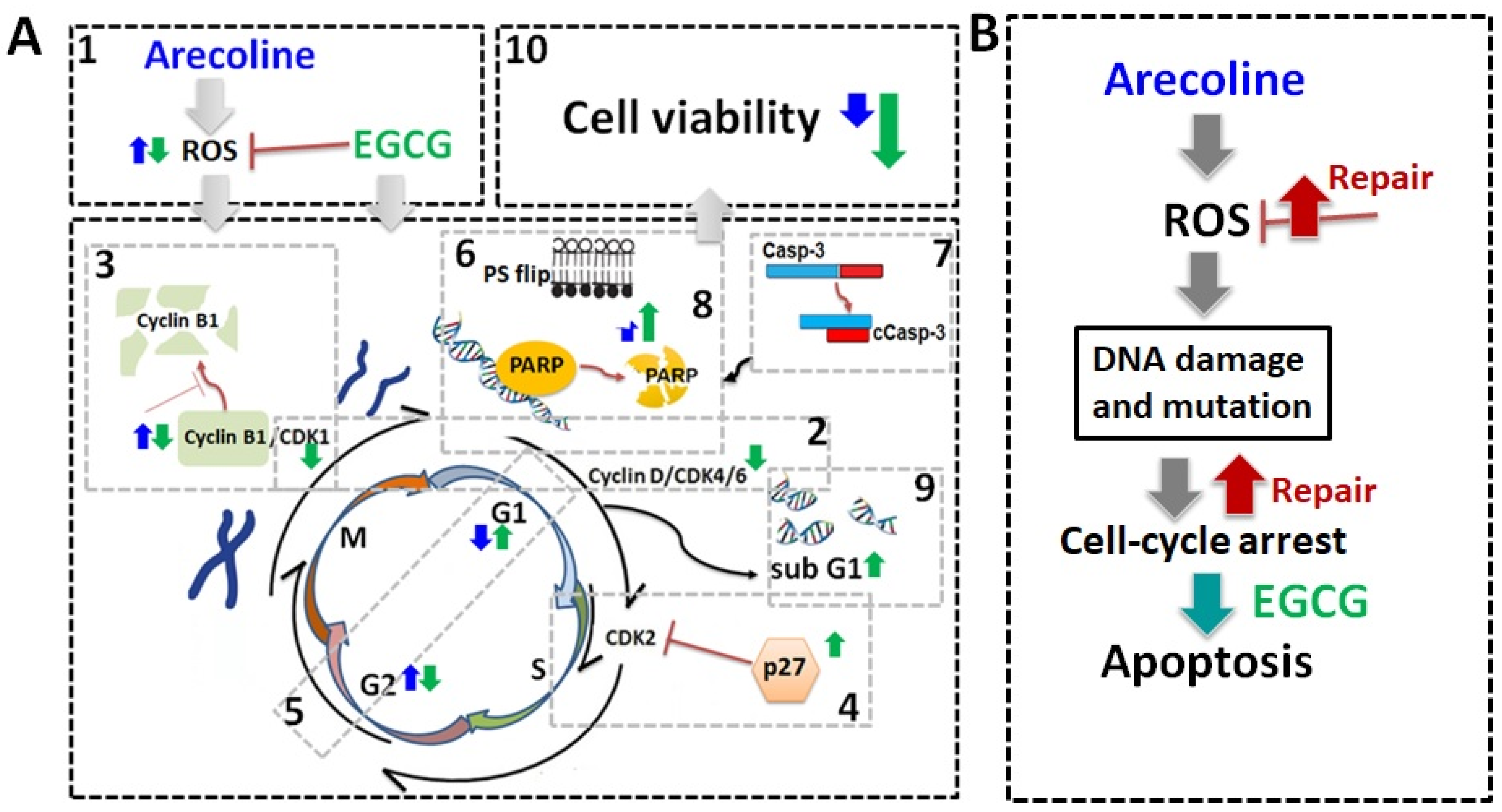
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Martins, S.G.; Zilhao, R.; Thorsteinsdottir, S.; Carlos, A.R. Linking Oxidative Stress and DNA Damage to Changes in the Expression of Extracellular Matrix Components. Front. Genet. 2021, 12, 673002. [Google Scholar] [CrossRef]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Chen, H.; Liang, J.; Li, Y.; Yang, J.; Luo, C.; Tang, Y.; Ding, Y.; Liu, X.; Yuan, Q.; et al. Dual Role of Reactive Oxygen Species and their Application in Cancer Therapy. J. Cancer 2021, 12, 5543–5561. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J.; Peng, W.; Hu, M.B.; Xu, M.; Wu, C.J. The pharmacology, toxicology and potential applications of arecoline: A review. Pharm. Biol. 2016, 54, 2753–2760. [Google Scholar] [CrossRef] [PubMed]
- Volgin, A.D.; Bashirzade, A.; Amstislavskaya, T.G.; Yakovlev, O.A.; Demin, K.A.; Ho, Y.J.; Wang, D.; Shevyrin, V.A.; Yan, D.; Tang, Z.; et al. DARK Classics in Chemical Neuroscience: Arecoline. ACS Chem. Neurosci. 2019, 10, 2176–2185. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.M.; Tu, H.P.; Ko, Y.C. Systematic Review of Roles of Arecoline and Arecoline N-Oxide in Oral Cancer and Strategies to Block Carcinogenesis. Cells 2023, 12, 1208. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; He, G.; Lu, Z.; He, Q.; Li, S.; Huang, Z.; Chen, Z.; Cao, C.; Wang, A. Arecoline induces epithelial-mesenchymal transformation and promotes metastasis of oral cancer by SAA1 expression. Cancer Sci. 2021, 112, 2173–2184. [Google Scholar] [CrossRef]
- Wang, Y.C.; Huang, J.L.; Lee, K.W.; Lu, H.H.; Lin, Y.J.; Chen, L.F.; Wang, C.S.; Cheng, Y.C.; Zeng, Z.T.; Chu, P.Y.; et al. Downregulation of the DNA Repair Gene DDB2 by Arecoline Is through p53’s DNA-Binding Domain and Is Correlated with Poor Outcome of Head and Neck Cancer Patients with Betel Quid Consumption. Cancers 2020, 12, 2053. [Google Scholar] [CrossRef]
- Salehi, B.; Martorell, M.; Arbiser, J.L.; Sureda, A.; Martins, N.; Maurya, P.K.; Sharifi-Rad, M.; Kumar, P.; Sharifi-Rad, J. Antioxidants: Positive or Negative Actors? Biomolecules. Biomolecules 2018, 8, 124. [Google Scholar] [CrossRef]
- Gal, K.L.; Ibrahim, M.X.; Wiel, C.; Sayin, V.I.; Akula, M.K.; Karlsson, C.; Dalin, M.G.; Akyurek, L.M.; Lindahl, P.; Nilsson, J.; et al. Antioxidants can increase melanoma metastasis in mice. Sci. Transl. Med. 2015, 7, 308re8. [Google Scholar]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants accelerate lung cancer progression in mice. Sci. Transl. Med. 2014, 6, 221ra15. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.V.; Gal, K.L.; Zowalaty, A.E.; Pehlivanoglu, L.E.; Garellick, V.; Gul, N.; Ibrahim, M.X.; Bergh, P.O.; Henricsson, M.; Wiel, C.; et al. Antioxidants Promote Intestinal Tumor Progression in Mice. Antioxidants 2021, 10, 241. [Google Scholar] [CrossRef] [PubMed]
- Shih, Y.H.; Chiu, K.C.; Wang, T.H.; Lan, W.C.; Tsai, B.H.; Wu, L.J.; Hsia, S.M.; Shieh, T.M. Effects of melatonin to arecoline-induced reactive oxygen species production and DNA damage in oral squamous cell carcinoma. J. Formos. Med. Assoc. 2021, 120, 668–678. [Google Scholar] [CrossRef] [PubMed]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxid. Med. Cell Longev. 2019, 2019, 6175804. [Google Scholar] [CrossRef]
- Negri, A.; Naponelli, V.; Rizzi, F.; Bettuzzi, S. Molecular Targets of Epigallocatechin-Gallate (EGCG): A Special Focus on Signal Transduction and Cancer. Nutrients 2018, 10, 1936. [Google Scholar] [CrossRef] [PubMed]
- Mokra, D.; Adamcakova, J.; Mokry, J. Green Tea Polyphenol (-)-Epigallocatechin-3-Gallate (EGCG): A Time for a New Player in the Treatment of Respiratory Diseases? Antioxidants 2022, 11, 1566. [Google Scholar] [CrossRef] [PubMed]
- Tai, S.; Sun, Y.; Squires, J.M.; Zhang, H.; Oh, W.K.; Liang, C.Z.; Huang, J. PC3 is a cell line characteristic of prostatic small cell carcinoma. Prostate 2011, 71, 1668–1679. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef]
- Shih, L.J.; Wang, J.Y.; Jheng, J.Y.; Siao, A.C.; Lin, Y.Y.; Tsuei, Y.W.; Kuo, Y.C.; Chuu, C.P.; Kao, Y.H. Betel Nut Arecoline Induces Different Phases of Growth Arrest between Normal and Cancerous Prostate Cells through the Reactive Oxygen Species Pathway. Int. J. Mol. Sci. 2020, 21, 9219. [Google Scholar] [CrossRef]
- Saha, I.; Chatterjee, A.; Mondal, A.; Maiti, B.R.; Chatterji, U. Arecoline augments cellular proliferation in the prostate gland of male Wistar rats. Toxicol. Appl. Pharmacol. 2011, 255, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, J.; Byun, H.; Kim, S.J.; Joo, J.; Park, H.H.; Shin, H. Evaluation of the anti-oxidative and ROS scavenging properties of biomaterials coated with epigallocatechin gallate for tissue engineering. Acta Biomater. 2021, 124, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Spagnuolo, G.; D’Antò, V.; Cosentino, C.; Schmalz, G.; Schweikl, H.; Rengo, S. Effect of N-acetyl-L-cysteine on ROS production and cell death caused by HEMA in human primary gingival fibroblasts. Biomaterials 2006, 27, 1803–1809. [Google Scholar] [CrossRef] [PubMed]
- Li, G.X.; Chen, Y.K.; Hou, Z.; Xiao, H.; Jin, H.; Lu, G.; Lee, M.J.; Liu, B.; Guan, F.; Yang, Z.; et al. Pro-oxidative activities and dose-response relationship of (-)-epigallocatechin-3-gallate in the inhibition of lung cancer cell growth: A comparative study in vivo and in vitro. Carcinogenesis 2010, 31, 902–910. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Takada, K. Reactive oxygen species in cancer: Current findings and future directions. Cancer Sci. 2021, 112, 3945–3952. [Google Scholar] [CrossRef]
- Wang, Y.; Qi, H.; Liu, Y.; Duan, C.; Liu, X.; Xia, T.; Chen, D.; Piao, H.L.; Liu, H.X. The double-edged roles of ROS in cancer prevention and therapy. Theranostics 2021, 11, 4839–4857. [Google Scholar] [CrossRef]
- Kaufmann, S.H.; Earnshaw, W.C. Induction of apoptosis by cancer chemotherapy. Exp. Cell Res. 2000, 256, 42–49. [Google Scholar] [CrossRef]
- Chen, F.; Vallyathan, V.; Castranova, V.; Shi, X. Cell apoptosis induced by carcinogenic metals. Mol. Cell Biochem. 2001, 222, 183–188. [Google Scholar] [CrossRef]
- Robertson, J.D.; Orrenius, S. Molecular mechanisms of apoptosis induced by cytotoxic chemicals. Crit. Rev. Toxicol. 2000, 30, 609–627. [Google Scholar] [CrossRef]
- Shapiro, G.I.; Harper, J.W. Anticancer drug targets: Cell cycle and checkpoint control. J. Clin. Investig. 1999, 104, 1645–1653. [Google Scholar] [CrossRef]
- DiPaola, R.S. To arrest or not to G(2)-M Cell-cycle arrest. Clin. Cancer Res. 2002, 8, 3311–3314. [Google Scholar] [PubMed]
- Tyagi, A.K.; Singh, R.P.; Agarwal, C.; Chan, D.C.F.; Agarwal, R. Silibinin strongly synergizes human prostate carcinoma DU145 cells to doxorubicin-induced growth inhibition, G(2)-M arrest, and apoptosis. Clin. Cancer Res. 2002, 8, 3512–3519. [Google Scholar] [PubMed]
- Nair, U.J.; Floyd, R.A.; Nair, J.; Bussachini, V.; Friesen, M.; Bartsch, H. Formation of reactive oxygen species and of 8-hydroxydeoxyguanosine in DNA in vitro with betel quid ingredients. Chem. Biol. Interact. 1987, 63, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.H.; Hsu, H.H.; Shibu, M.A.; Day, C.H.; Bau, D.T.; Ho, C.C.; Lin, Y.M.; Chen, M.C.; Wang, S.H.; Huang, C.Y. Down- regulation of beta-catenin and the associated migration ability by Taiwanin C in arecoline and 4-NQO-induced oral cancer cells via GSK-3beta activation. Mol. Carcinog. 2017, 56, 1055–1067. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.R.; Cheng, J.T.; Shih, C.S. Gastric mucosal damage induced by arecoline seizure in rats. Life Sci. 2000, 66, 2337–2349. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.M.; Wang, L.; Gu, H.F.; Wu, K.; Xiao, F.; Chen, Y.; Guo, R.M.; Tang, X.Q. Arecoline Induces Neurotoxicity to PC12 Cells: Involvement in ER Stress and Disturbance of Endogenous H2S Generation. Neurochem. Res. 2016, 41, 2140–2148. [Google Scholar] [CrossRef] [PubMed]
- Shih, Y.T.; Chen, P.S.; Wu, C.H.; Tseng, Y.T.; Wu, Y.C.; Lo, Y.C. Arecoline, a major alkaloid of the areca nut, causes neurotoxicity through enhancement of oxidative stress and suppression of the antioxidant protective system. Free Radic. Biol. Med. 2010, 49, 1471–1479. [Google Scholar] [CrossRef]
- Verbon, E.H.; Post, J.A.; Boonstra, J. The influence of reactive oxygen species on cell cycle progression in mammalian cells. Gene 2012, 511, 1–6. [Google Scholar] [CrossRef]
- Eli, R.; Fasciano, J.A. An adjunctive preventive treatment for cancer: Ultraviolet light and ginkgo biloba, together with other antioxidants, are a safe and powerful, but largely ignored, treatment option for the prevention of cancer. Med. Hypotheses 2006, 66, 1152–1156. [Google Scholar] [CrossRef]
- Abbasi, A.; Mostafavi-Pour, Z.; Amiri, A.; Keshavarzi, F.; Nejabat, N.; Ramezani, F.; Sardarian, A.; Zal, F. Chemoprevention of Prostate Cancer Cells by Vitamin C plus Quercetin: Role of Nrf2 in Inducing Oxidative Stress. Nutr. Cancer 2021, 73, 2003–2013. [Google Scholar] [CrossRef]
- Gupta-Elera, G.; Garrett, A.R.; Robison, R.A.; O’neill, K.L. The role of oxidative stress in prostate cancer. Eur. J. Cancer Prev. 2012, 21, 155–162. [Google Scholar] [CrossRef]
- Sikka, S.C. Role of oxidative stress response elements and antioxidants in prostate cancer pathobiology and chemoprevention–a mechanistic approach. Curr. Med. Chem. 2003, 10, 2679–2692. [Google Scholar] [CrossRef]
- Perillo, B.; Donato, M.D.; Pezone, A.; Zazzo, E.D.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Sahoo, B.M.; Banik, B.K.; Borah, P.; Jain, A. Reactive Oxygen Species (ROS): Key Components in Cancer Therapies. Anticancer. Agents Med. Chem. 2022, 22, 215–222. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shih, L.-J.; Hsu, P.-C.; Chuu, C.-P.; Shui, H.-A.; Yeh, C.-C.; Chen, Y.-C.; Kao, Y.-H. Epigallocatechin-3-gallate Synergistically Enhanced Arecoline-Induced Cytotoxicity by Redirecting Cycle Arrest to Apoptosis. Curr. Issues Mol. Biol. 2024, 46, 1516-1529. https://doi.org/10.3390/cimb46020098
Shih L-J, Hsu P-C, Chuu C-P, Shui H-A, Yeh C-C, Chen Y-C, Kao Y-H. Epigallocatechin-3-gallate Synergistically Enhanced Arecoline-Induced Cytotoxicity by Redirecting Cycle Arrest to Apoptosis. Current Issues in Molecular Biology. 2024; 46(2):1516-1529. https://doi.org/10.3390/cimb46020098
Chicago/Turabian StyleShih, Li-Jane, Po-Chi Hsu, Chih-Pin Chuu, Hao-Ai Shui, Chien-Chih Yeh, Yueh-Chung Chen, and Yung-Hsi Kao. 2024. "Epigallocatechin-3-gallate Synergistically Enhanced Arecoline-Induced Cytotoxicity by Redirecting Cycle Arrest to Apoptosis" Current Issues in Molecular Biology 46, no. 2: 1516-1529. https://doi.org/10.3390/cimb46020098
APA StyleShih, L.-J., Hsu, P.-C., Chuu, C.-P., Shui, H.-A., Yeh, C.-C., Chen, Y.-C., & Kao, Y.-H. (2024). Epigallocatechin-3-gallate Synergistically Enhanced Arecoline-Induced Cytotoxicity by Redirecting Cycle Arrest to Apoptosis. Current Issues in Molecular Biology, 46(2), 1516-1529. https://doi.org/10.3390/cimb46020098