

MAP3K19 Affects TWEAK-Induced Response in Cultured Bronchial Epithelial Cells and Regulates Allergic Airway Inflammation in an Asthma Murine Model
 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Mice
2.4. Preparation of Cell Lysates and Western Blotting
2.5. RNA Interference Assay
2.6. Induction of Airway Inflammation
2.7. Characterization of Bronchoalveolar Lavage (BAL) Fluids
2.8. Airway Hyperresponsiveness (AHR) Measurements
2.9. Stimulation of Lung-Draining Lymph Node Cells In Vitro
2.10. Histological Analysis of Lung Sections
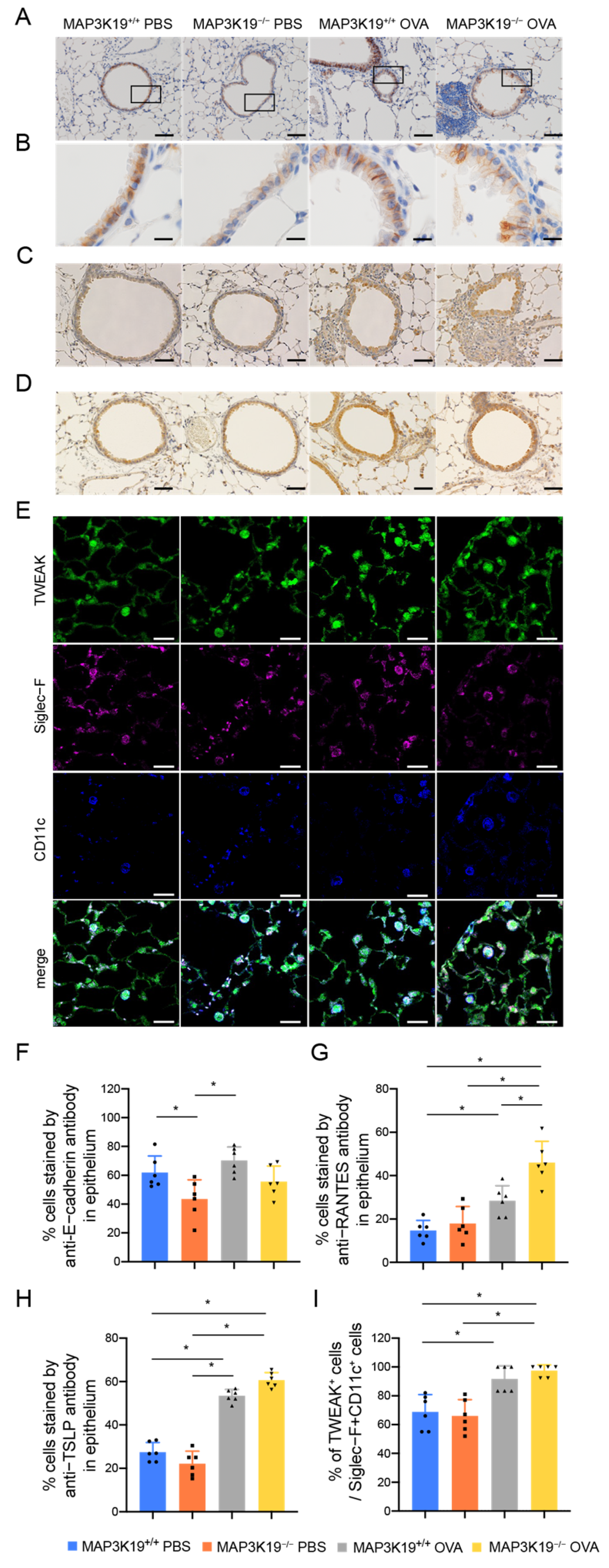
2.11. Immunofluorescence Staining of Lung Sections for TWEAK
2.12. Immunohistochemical Staining of Lung Sections
2.13. RNA Isolation and Quantitative RT-PCR
2.14. RANTES Measurements
2.15. Statistical Analysis
3. Results
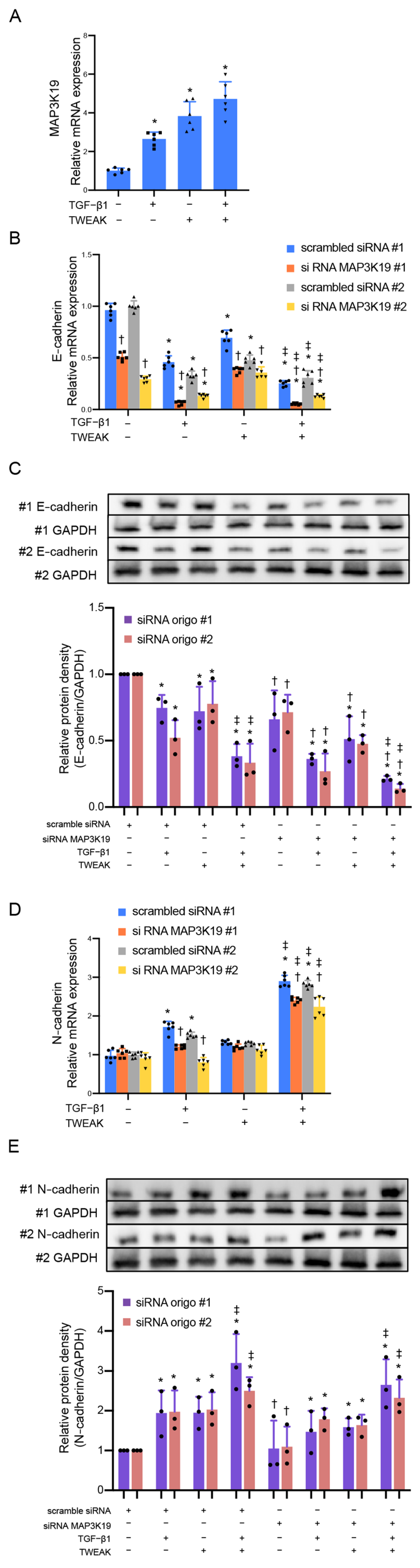
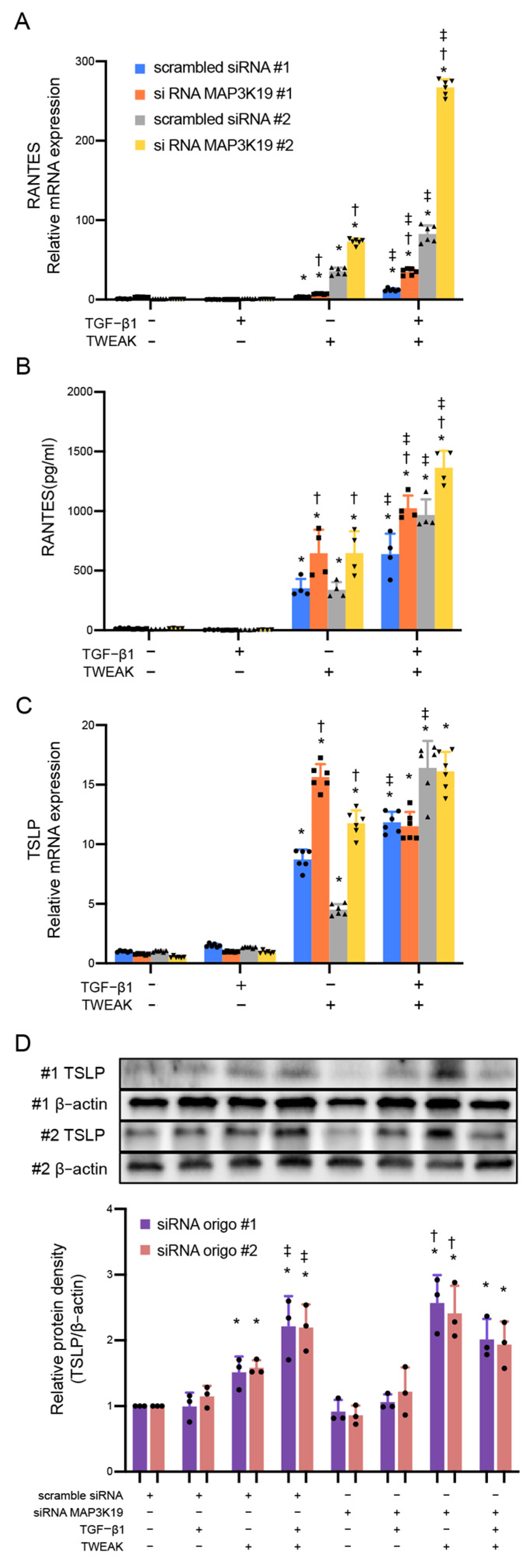
3.1. MAP3K19 Suppresses TWEAK Stimulation and RANTES Production
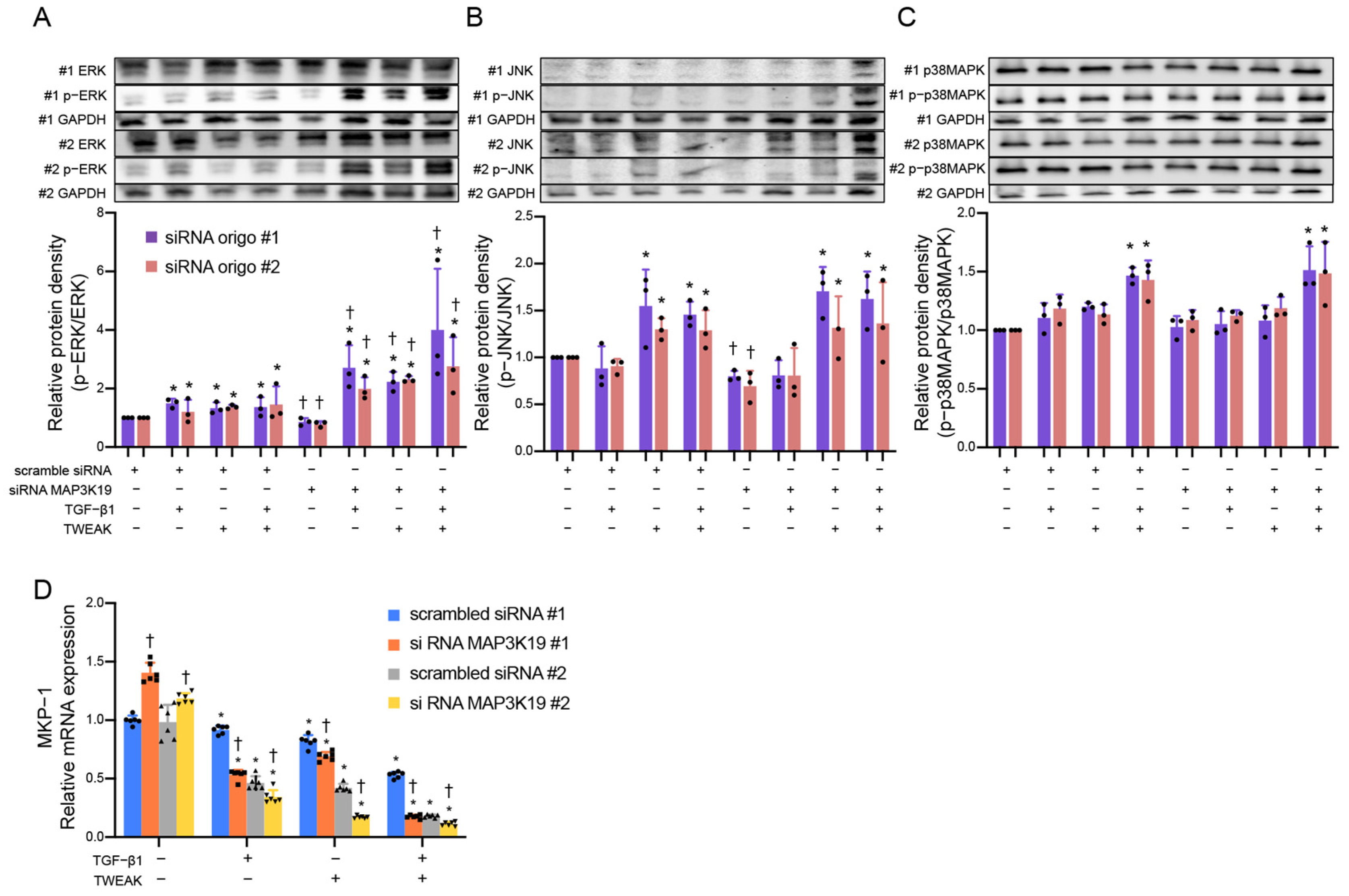
3.2. MAP3K19 Inhibits Phosphorylation of Extracellular Signal-Regulated Kinases (ERKs) by Inducing MAPK Phosphatases-1 (MKP-1)
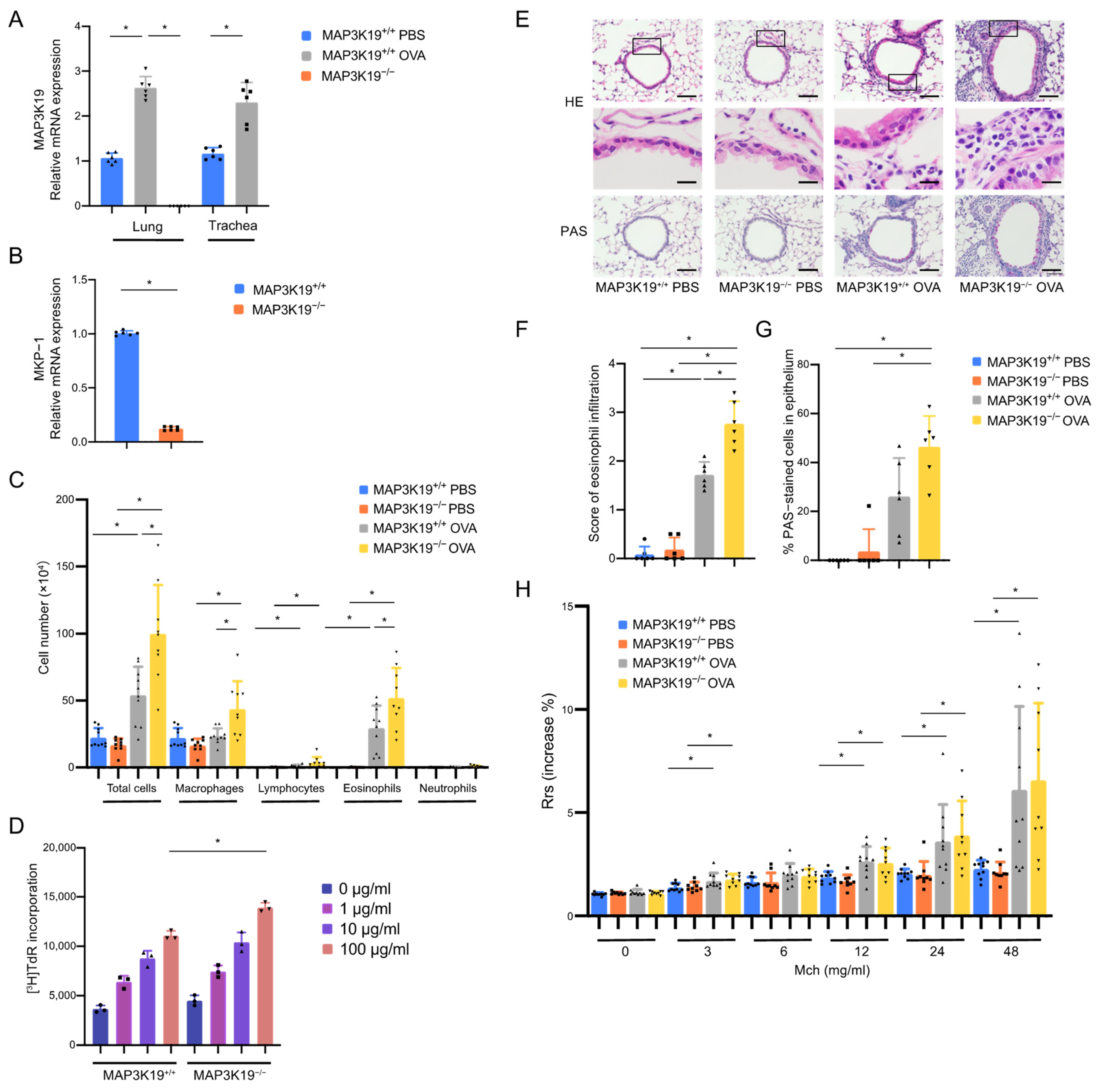
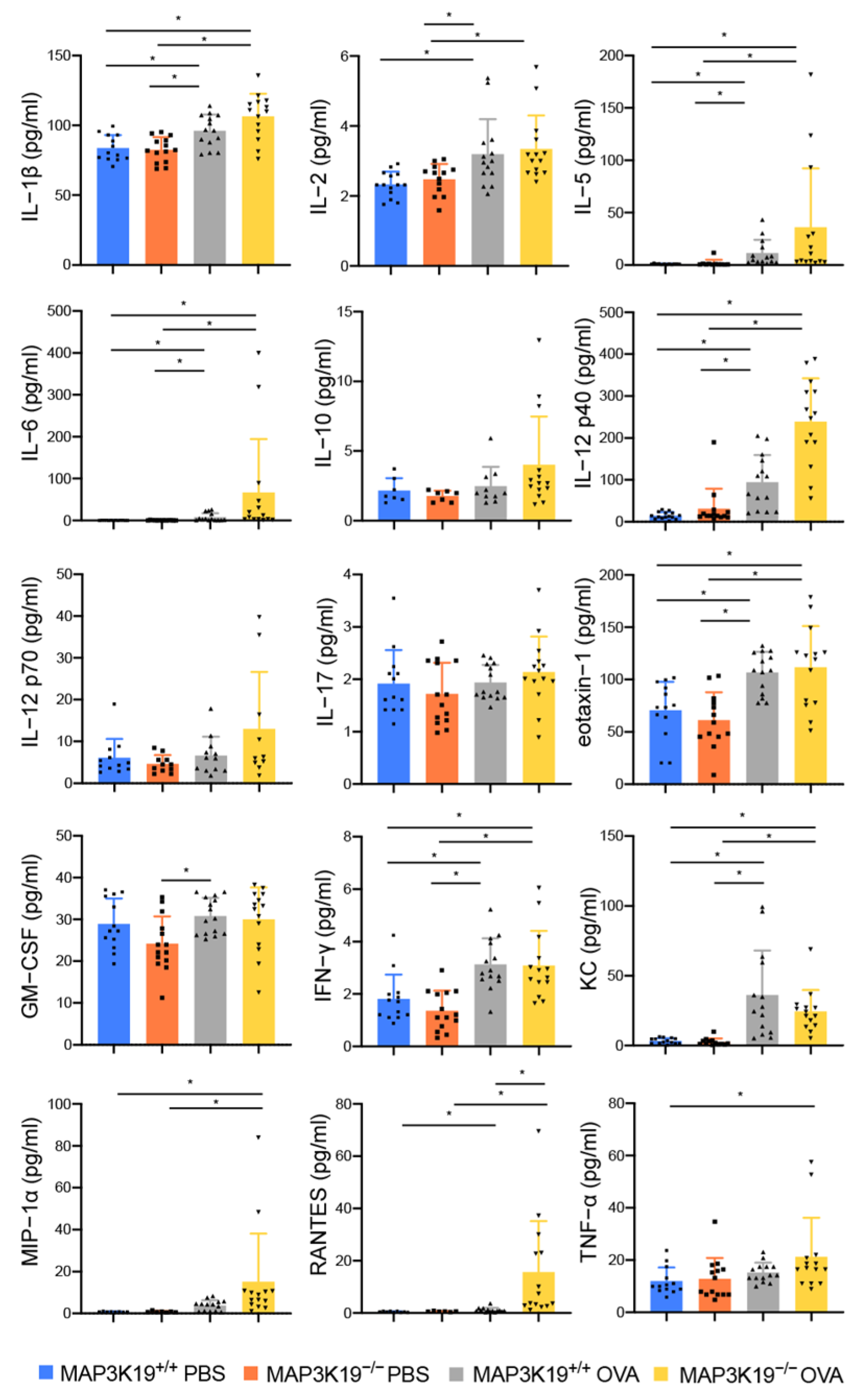
3.3. MAP3K19 Suppresses Airway Inflammation in OVA-Induced Asthma Murine Model
3.4. MAP3K19 Suppresses RANTES Production in OVA-Induced Asthma Murine Model
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Arthur, J.S.; Ley, S.C. Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 2013, 13, 679–692. [Google Scholar] [CrossRef]
- Meister, M.; Tomasovic, A.; Banning, A.; Tikkanen, R. Mitogen-Activated Protein (MAP) Kinase Scaffolding Proteins: A Recount. Int. J. Mol. Sci. 2013, 14, 4854–4884. [Google Scholar] [CrossRef] [PubMed]
- Boehme, S.A.; Franz-Bacon, K.; DiTirro, D.N.; Ly, T.W.; Bacon, K.B. MAP3K19 Is a Novel Regulator of TGF-beta Signaling That Impacts Bleomycin-Induced Lung Injury and Pulmonary Fibrosis. PLoS ONE 2016, 11, e0154874. [Google Scholar] [CrossRef] [PubMed]
- Boehme, S.A.; Franz-Bacon, K.; Ludka, J.; DiTirro, D.N.; Ly, T.W.; Bacon, K.B. MAP3K19 Is Overexpressed in COPD and Is a Central Mediator of Cigarette Smoke-Induced Pulmonary Inflammation and Lower Airway Destruction. PLoS ONE 2016, 11, e0167169. [Google Scholar] [CrossRef]
- Jones, I.C.; Espindola, M.S.; Narayanan, R.; Coelho, A.L.; Habiel, D.M.; Boehme, S.A.; Ly, T.W.; Bacon, K.B.; Hogaboam, C.M. Targeting MAP3K19 prevents human lung myofibroblast activation both in vitro and in a humanized SCID model of idiopathic pulmonary fibrosis. Sci. Rep. 2019, 9, 19796. [Google Scholar] [CrossRef] [PubMed]
- Itoigawa, Y.; Harada, N.; Harada, S.; Katsura, Y.; Makino, F.; Ito, J.; Nurwidya, F.; Kato, M.; Takahashi, F.; Atsuta, R.; et al. TWEAK enhances TGF-beta-induced epithelial-mesenchymal transition in human bronchial epithelial cells. Respir. Res. 2015, 16, 48. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.; Tran, M.N.; Rivera, A.; Cheng, T.; Windsor, G.O.; Chabot, A.B.; Cavanaugh, J.E.; Collins-Burow, B.M.; Lee, S.B.; Drewry, D.H.; et al. MAP3K Family Review and Correlations with Patient Survival Outcomes in Various Cancer Types. Front. Biosci. 2022, 27, 167. [Google Scholar] [CrossRef] [PubMed]
- Ijaz, T.; Pazdrak, K.; Kalita, M.; Konig, R.; Choudhary, S.; Tian, B.; Boldogh, I.; Brasier, A.R. Systems biology approaches to understanding Epithelial Mesenchymal Transition (EMT) in mucosal remodeling and signaling in asthma. World Allergy Organ. J. 2014, 7, 13. [Google Scholar] [CrossRef]
- Lambrecht, B.N.; Hammad, H. The airway epithelium in asthma. Nat. Med. 2012, 18, 684–692. [Google Scholar] [CrossRef]
- Johnson, J.R.; Roos, A.; Berg, T.; Nord, M.; Fuxe, J. Chronic respiratory aeroallergen exposure in mice induces epithelial-mesenchymal transition in the large airways. PLoS ONE 2011, 6, e16175. [Google Scholar] [CrossRef]
- Yasukawa, A.; Hosoki, K.; Toda, M.; Miyake, Y.; Matsushima, Y.; Matsumoto, T.; Boveda-Ruiz, D.; Gil-Bernabe, P.; Nagao, M.; Sugimoto, M.; et al. Eosinophils promote epithelial to mesenchymal transition of bronchial epithelial cells. PLoS ONE 2013, 8, e64281. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Puddicombe, S.M.; Polosa, R.; Richter, A.; Krishna, M.T.; Howarth, P.H.; Holgate, S.T.; Davies, D.E. Involvement of the epidermal growth factor receptor in epithelial repair in asthma. FASEB J. 2000, 14, 1362–1374. [Google Scholar] [CrossRef] [PubMed]
- Burkly, L.C.; Michaelson, J.S.; Zheng, T.S. TWEAK/Fn14 pathway: An immunological switch for shaping tissue responses. Immunol. Rev. 2011, 244, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Harada, N.; Nakayama, M.; Nakano, H.; Fukuchi, Y.; Yagita, H.; Okumura, K. Pro-inflammatory effect of TWEAK/Fn14 interaction on human umbilical vein endothelial cells. Biochem. Biophys. Res. Commun. 2002, 299, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, K.; Harada, N.; Harada, S.; Takeshige, T.; Ishimori, A.; Itoigawa, Y.; Katsura, Y.; Kodama, Y.; Makino, F.; Ito, H.; et al. Combination of TWEAK and TGF-beta1 induces the production of TSLP, RANTES, and TARC in BEAS-2B human bronchial epithelial cells during epithelial-mesenchymal transition. Exp. Lung Res. 2018, 44, 332–343. [Google Scholar] [CrossRef]
- Zhu, C.; Zhang, L.; Liu, Z.; Li, C.; Bai, Y. TWEAK/Fn14 interaction induces proliferation and migration in human airway smooth muscle cells via activating the NF-kappaB pathway. J. Cell Biochem. 2018, 119, 3528–3536. [Google Scholar] [CrossRef]
- Kamijo, S.; Nakajima, A.; Kamata, K.; Kurosawa, H.; Yagita, H.; Okumura, K. Involvement of TWEAK/Fn14 interaction in the synovial inflammation of RA. Rheumatology 2008, 47, 442–450. [Google Scholar] [CrossRef]
- Schwartz, N.; Su, L.; Burkly, L.C.; Mackay, M.; Aranow, C.; Kollaros, M.; Michaelson, J.S.; Rovin, B.; Putterman, C. Urinary TWEAK and the activity of lupus nephritis. J. Autoimmun. 2006, 27, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Serafini, B.; Magliozzi, R.; Rosicarelli, B.; Reynolds, R.; Zheng, T.S.; Aloisi, F. Expression of TWEAK and its receptor Fn14 in the multiple sclerosis brain: Implications for inflammatory tissue injury. J. Neuropathol. Exp. Neurol. 2008, 67, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Winkles, J.A. The TWEAK-Fn14 cytokine-receptor axis: Discovery, biology and therapeutic targeting. Nat. Rev. Drug Discov. 2008, 7, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Soumelis, V.; Reche, P.A.; Kanzler, H.; Yuan, W.; Edward, G.; Homey, B.; Gilliet, M.; Ho, S.; Antonenko, S.; Lauerma, A.; et al. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat. Immunol. 2002, 3, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, S.F.; Artis, D. Sensing the outside world: TSLP regulates barrier immunity. Nat. Immunol. 2010, 11, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Carr, T.F.; Berdnikovs, S.; Simon, H.U.; Bochner, B.S.; Rosenwasser, L.J. Eosinophilic bioactivities in severe asthma. World Allergy Organ. J. 2016, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Chvatchko, Y.; Proudfoot, A.E.I.; Buser, R.; Juillard, P.; Alouani, S.; Kosco-Vilbois, M.; Coyle, A.J.; Nibbs, R.J.; Graham, G.; Offord, R.E.; et al. Inhibition of airway inflammation by amino-terminally modified RANTES/CC chemokine ligand 5 analogues is not mediated through CCR3. J. Immunol. 2003, 171, 5498–5506. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kim, J.D.; Sol, I.S.; Kim, M.J.; Na Kim, M.; Hong, J.Y.; Kim, H.R.; Kim, Y.H.; Lee, Y.J.; Kim, K.W.; et al. Sputum TWEAK expression correlates with severity and degree of control in non-eosinophilic childhood asthma. Pediatr. Allergy Immunol. 2018, 29, 42–49. [Google Scholar] [CrossRef]
- Cuarental, L.; Sucunza-Saenz, D.; Valino-Rivas, L.; Fernandez-Fernandez, B.; Sanz, A.B.; Ortiz, A.; José Vaquero, J.; Sanchez-Niño, M.D. MAP3K kinases and kidney injury. Nefrologia 2019, 39, 568–580. [Google Scholar] [CrossRef]
- Hoang, V.T.; Nyswaner, K.; Torres-Ayuso, P.; Brognard, J. The protein kinase MAP3K19 phosphorylates MAP2Ks and thereby activates ERK and JNK kinases and increases viability of KRAS-mutant lung cancer cells. J. Biol. Chem. 2020, 295, 8470–8479. [Google Scholar] [CrossRef]
- Hoppstadter, J.; Ammit, A.J. Role of Dual-Specificity Phosphatase 1 in Glucocorticoid-Driven Anti-inflammatory Responses. Front. Immunol. 2019, 10, 1446. [Google Scholar] [CrossRef] [PubMed]
- Hough, C.; Radu, M.; Dore, J.J. Tgf-beta induced Erk phosphorylation of smad linker region regulates smad signaling. PLoS ONE 2012, 7, e42513. [Google Scholar] [CrossRef] [PubMed]
- Hayashida, T.; Decaestecker, M.; Schnaper, H.W. Cross-talk between ERK MAP kinase and Smad signaling pathways enhances TGF-beta-dependent responses in human mesangial cells. FASEB J. 2003, 17, 1576–1578. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Guo, Z.P.; Li, M.M.; Li, J.Y.; Jiao, X.Y.; Zhang, Y.H.; Liu, H.J. Tumour necrosis factor-like weak inducer of apoptosis (TWEAK), an important mediator of endothelial inflammation, is associated with the pathogenesis of Henoch-Schonlein purpura. Clin. Exp. Immunol. 2011, 166, 64–71. [Google Scholar] [CrossRef]
- Tran, N.L.; McDonough, W.S.; Savitch, B.A.; Sawyer, T.F.; Winkles, J.A.; Berens, M.E. The tumor necrosis factor-like weak inducer of apoptosis (TWEAK)-fibroblast growth factor-inducible 14 (Fn14) signaling system regulates glioma cell survival via NFkappaB pathway activation and BCL-XL/BCL-W expression. J. Biol. Chem. 2005, 280, 3483–3492. [Google Scholar] [CrossRef] [PubMed]
- Vendrell, J.; Maymo-Masip, E.; Tinahones, F.; Garcia-Espana, A.; Megia, A.; Caubet, E.; García-Fuentes, E.; Chacón, M.R. Tumor necrosis-like weak inducer of apoptosis as a proinflammatory cytokine in human adipocyte cells: Up-regulation in severe obesity is mediated by inflammation but not hypoxia. J. Clin. Endocrinol. Metab. 2010, 95, 2983–2992. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Okamoto, A.; Ichikawa, J.; Ando, T.; Tasaka, K.; Masuyama, K.; Ogawa, H.; Yagita, H.; Okumura, K.; Nakao, A. TWEAK/Fn14 interaction stimulates human bronchial epithelial cells to produce IL-8 and GM-CSF. Biochem. Biophys. Res. Commun. 2004, 318, 422–427. [Google Scholar] [CrossRef]
- Azman, S.; Sekar, M.; Bonam, S.R.; Gan, S.H.; Wahidin, S.; Lum, P.T.; Dhadde, S.B. Traditional Medicinal Plants Conferring Protection Against Ovalbumin-Induced Asthma in Experimental Animals: A Review. J. Asthma Allergy 2021, 14, 641–662. [Google Scholar] [CrossRef]
- Kianmeher, M.; Ghorani, V.; Boskabady, M.H. Animal Model of Asthma, Various Methods and Measured Parameters: A Methodological Review. Iran. J. Allergy Asthma Immunol. 2016, 15, 445–465. [Google Scholar]
- Behrendt, A.K.; Hansen, G. CD27 costimulation is not critical for the development of asthma and respiratory tolerance in a murine model. Immunol. Lett. 2010, 133, 19–27. [Google Scholar] [CrossRef]
- Makino, F.; Ito, J.; Abe, Y.; Harada, N.; Kamachi, F.; Yagita, H.; Takahashi, K.; Okumura, K.; Akiba, H. Blockade of CD70-CD27 interaction inhibits induction of allergic lung inflammation in mice. Am. J. Respir. Cell Mol. Biol. 2012, 47, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Behrendt, A.K.; Meyer-Bahlburg, A.; Hansen, G. CD137 deficiency does not affect development of airway inflammation or respiratory tolerance induction in murine models. Clin. Exp. Immunol. 2012, 168, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Busse, M.; Krech, M.; Meyer-Bahlburg, A.; Hennig, C.; Hansen, G. ICOS mediates the generation and function of CD4+CD25+Foxp3+ regulatory T cells conveying respiratory tolerance. J. Immunol. 2012, 189, 1975–1982. [Google Scholar] [CrossRef] [PubMed]
- Jirmo, A.C.; Daluege, K.; Happle, C.; Albrecht, M.; Dittrich, A.M.; Busse, M.; Habener, A.; Skuljec, J.; Hansen, G. IL-27 Is Essential for Suppression of Experimental Allergic Asthma by the TLR7/8 Agonist R848 (Resiquimod). J. Immunol. 2016, 197, 4219–4227. [Google Scholar] [CrossRef] [PubMed]
- Happle, C.; Jirmo, A.C.; Meyer-Bahlburg, A.; Habener, A.; Hoymann, H.G.; Hennig, C.; Skuljec, J.; Hansen, G. B cells control maternofetal priming of allergy and tolerance in a murine model of allergic airway inflammation. J. Allergy Clin. Immunol. 2018, 141, 685–696.e6. [Google Scholar] [CrossRef] [PubMed]
- Jirmo, A.C.; Busse, M.; Happle, C.; Skuljec, J.; Dalüge, K.; Habener, A.; Grychtol, R.; DeLuca, D.S.; Breiholz, O.D.; Prinz, I.; et al. IL-17 regulates DC migration to the peribronchial LNs and allergen presentation in experimental allergic asthma. Eur. J. Immunol. 2020, 50, 1019–1033. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sandhu, Y.; Harada, N.; Harada, S.; Nishimaki, T.; Sasano, H.; Tanabe, Y.; Takeshige, T.; Matsuno, K.; Ishimori, A.; Katsura, Y.; et al. MAP3K19 Affects TWEAK-Induced Response in Cultured Bronchial Epithelial Cells and Regulates Allergic Airway Inflammation in an Asthma Murine Model. Curr. Issues Mol. Biol. 2023, 45, 8907-8924. https://doi.org/10.3390/cimb45110559
Sandhu Y, Harada N, Harada S, Nishimaki T, Sasano H, Tanabe Y, Takeshige T, Matsuno K, Ishimori A, Katsura Y, et al. MAP3K19 Affects TWEAK-Induced Response in Cultured Bronchial Epithelial Cells and Regulates Allergic Airway Inflammation in an Asthma Murine Model. Current Issues in Molecular Biology. 2023; 45(11):8907-8924. https://doi.org/10.3390/cimb45110559
Chicago/Turabian StyleSandhu, Yuuki, Norihiro Harada, Sonoko Harada, Takayasu Nishimaki, Hitoshi Sasano, Yuki Tanabe, Tomohito Takeshige, Kei Matsuno, Ayako Ishimori, Yoko Katsura, and et al. 2023. "MAP3K19 Affects TWEAK-Induced Response in Cultured Bronchial Epithelial Cells and Regulates Allergic Airway Inflammation in an Asthma Murine Model" Current Issues in Molecular Biology 45, no. 11: 8907-8924. https://doi.org/10.3390/cimb45110559
APA StyleSandhu, Y., Harada, N., Harada, S., Nishimaki, T., Sasano, H., Tanabe, Y., Takeshige, T., Matsuno, K., Ishimori, A., Katsura, Y., Ito, J., Akiba, H., & Takahashi, K. (2023). MAP3K19 Affects TWEAK-Induced Response in Cultured Bronchial Epithelial Cells and Regulates Allergic Airway Inflammation in an Asthma Murine Model. Current Issues in Molecular Biology, 45(11), 8907-8924. https://doi.org/10.3390/cimb45110559