Mitochondrial Targeted Endonuclease III DNA Repair Enzyme Protects against Ventilator Induced Lung Injury in Mice

Abstract
:1. Introduction
2. Experimental Section
2.1. Fusion Protein Constructs
2.2. Treatment with mt-Targeted EndoIII
2.3. Experimental Protocols
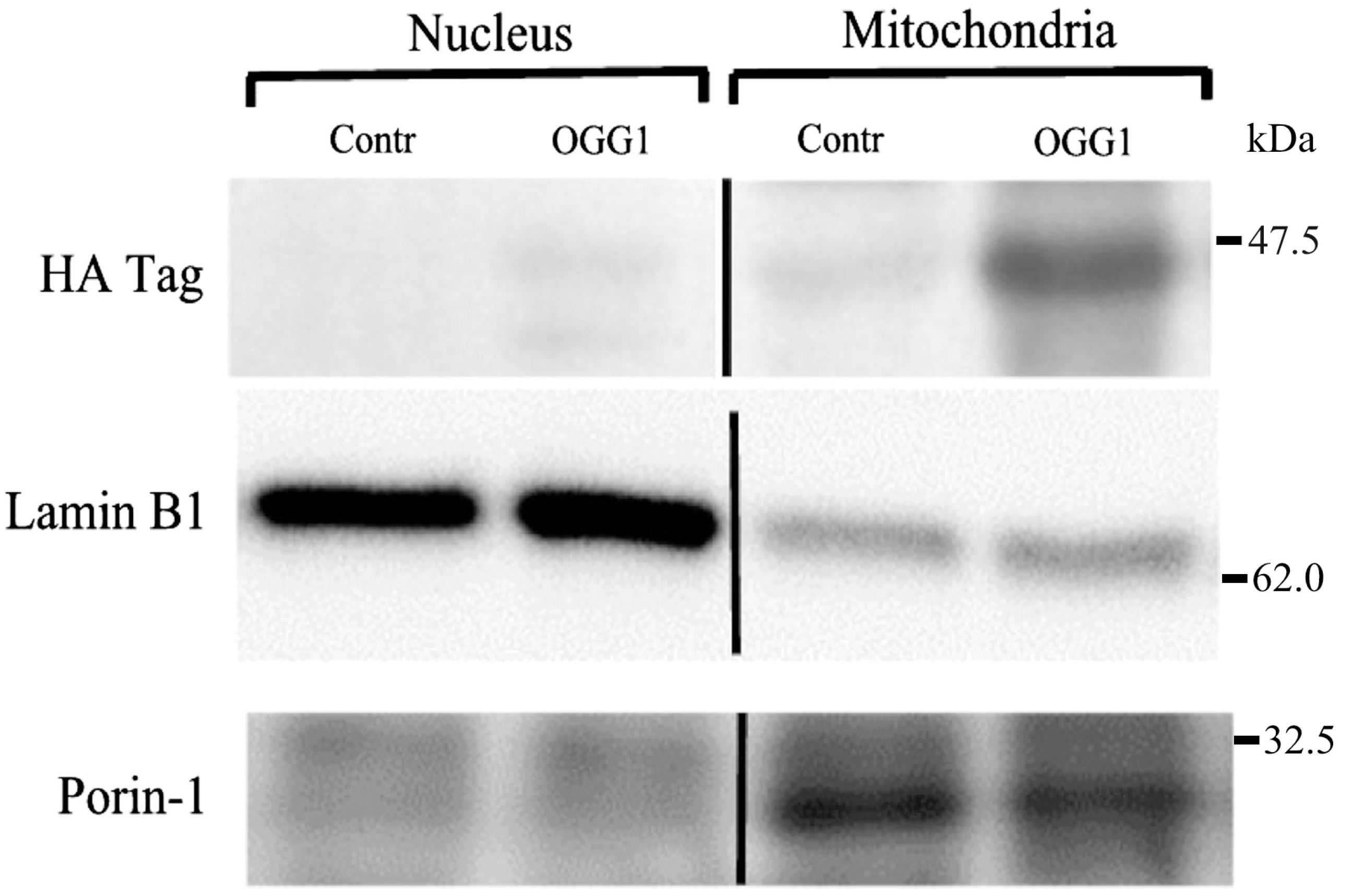
2.4. Western Immuno-Blot Analysis of Sub-Cellular Fusion Protein Localization
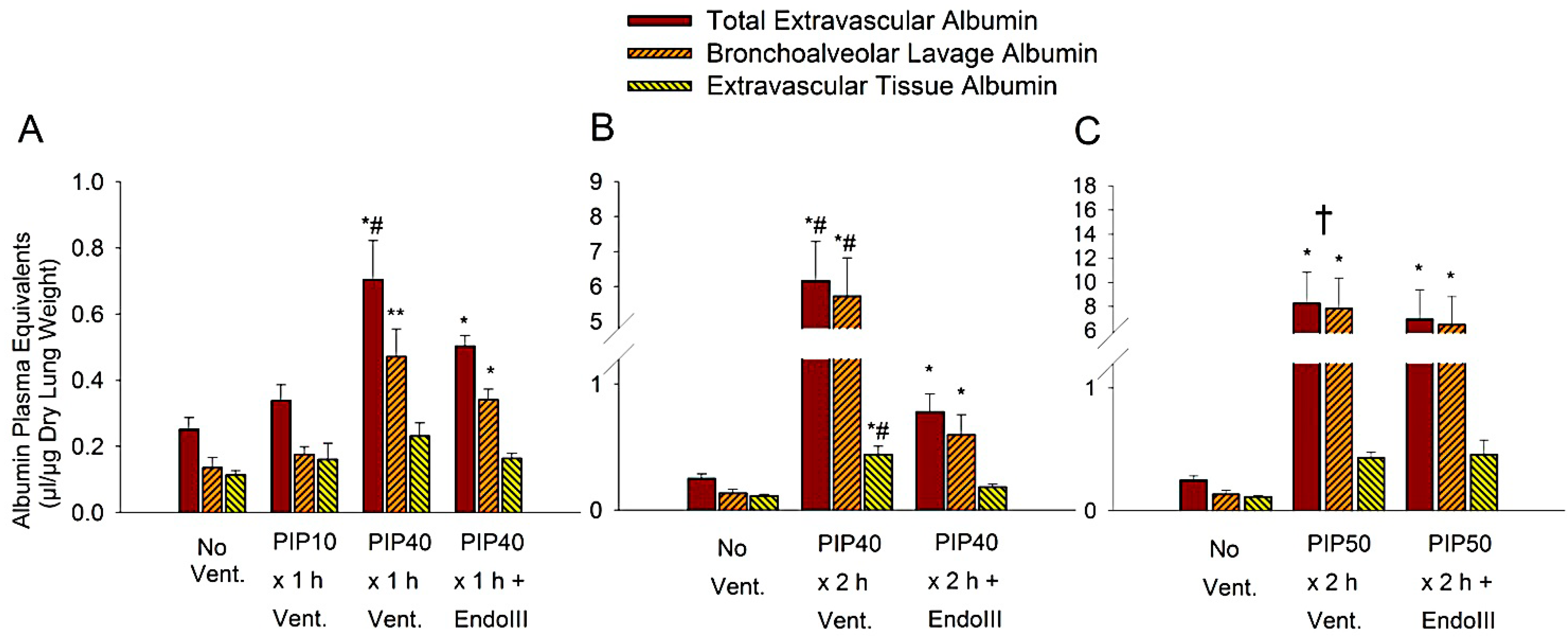
2.5. Measurement of Albumin Plasma Equivalents
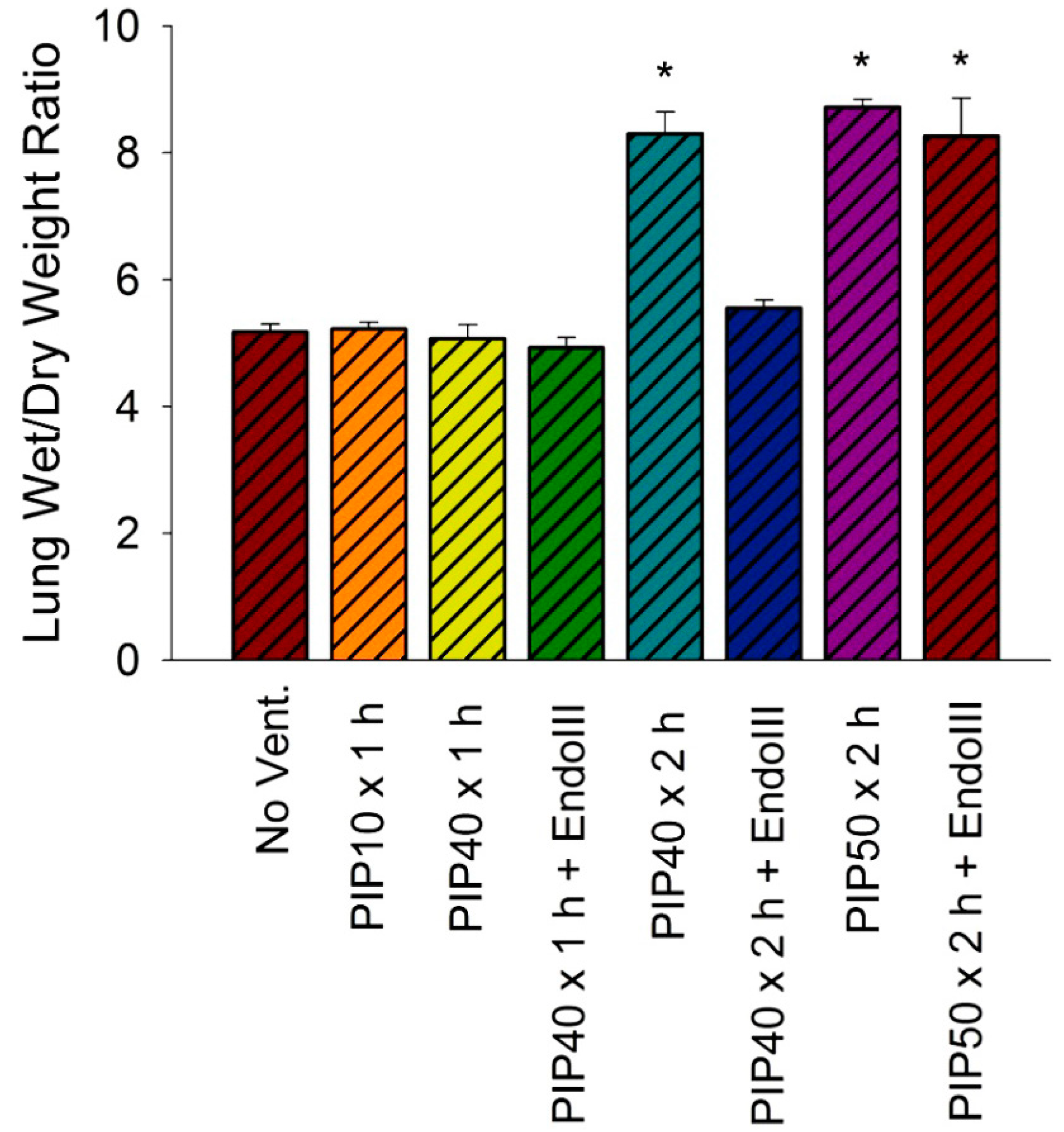
2.6. Lung Wet-To-Dry Weight Ratios
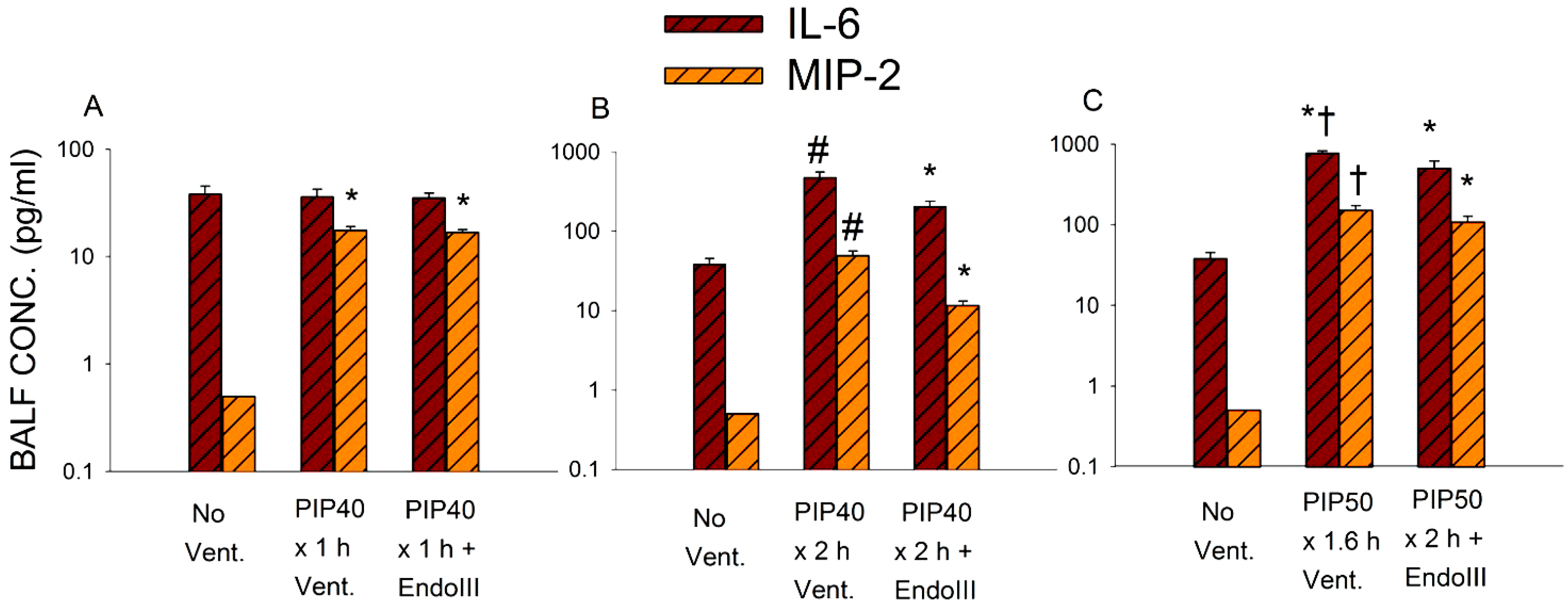
2.7. Measurement of MIP-2 and IL-6
2.8. Analysis of mtDNA Content and Oxidative Damage
2.9. Statistical Analysis
3. Results and Discussion
3.1. Ventilation Parameters and Arterial Blood Gasses

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | PIP (cmH2O) | Vent. Time (min) | PaO2 (mmHg) | PaCO2 (mmHg) | pH | Vent. Rate Breaths/min |
|---|---|---|---|---|---|---|
| NV | 10 | 11.2 ± 1.2 | 211 ± 11 | 36.0 ± 1.2 | 7.28 ± 0.01 | 110 |
| Low PIP | 10 | 60 | 293 ± 7 | 34.5 ± 2.4 | 7.33 ± 0.01 | 110 ± 2 |
| PIP40 | 40 | 60 | 466 ± 14* | 27.6 ± 1.1 | 7.35 ± 0.01 | 28.3 ± 0.5 |
| PIP40 EndoIII | 40 | 60 | 457 ± 12* | 22.0 ± 1.6* | 7.40 ± 0.02 | 30.4 ± O.2 |
| PIP40 | 40 | 120 | 344 ± 4 | 26.3 ± 0.5 | 7.36 ± 0.01 | 28.3 ± 0.3 |
| PIP40 EndoIII | 40 | 120 | 478 ± 5* | 21.7 ± 1.0** | 7.35 ± 0.04 | 31.8 ± 0.2 |
| PIP50 | 50 | 96.6 ± 7 | NM | NM | NM | 25.6 ± 0.2 |
| PIP50 EndoIII | 50 | 120 | 335 ± 11 | 17.2 ± 1.3** | 7.29 ± 0.08 | 26.6 ± 1.0 |
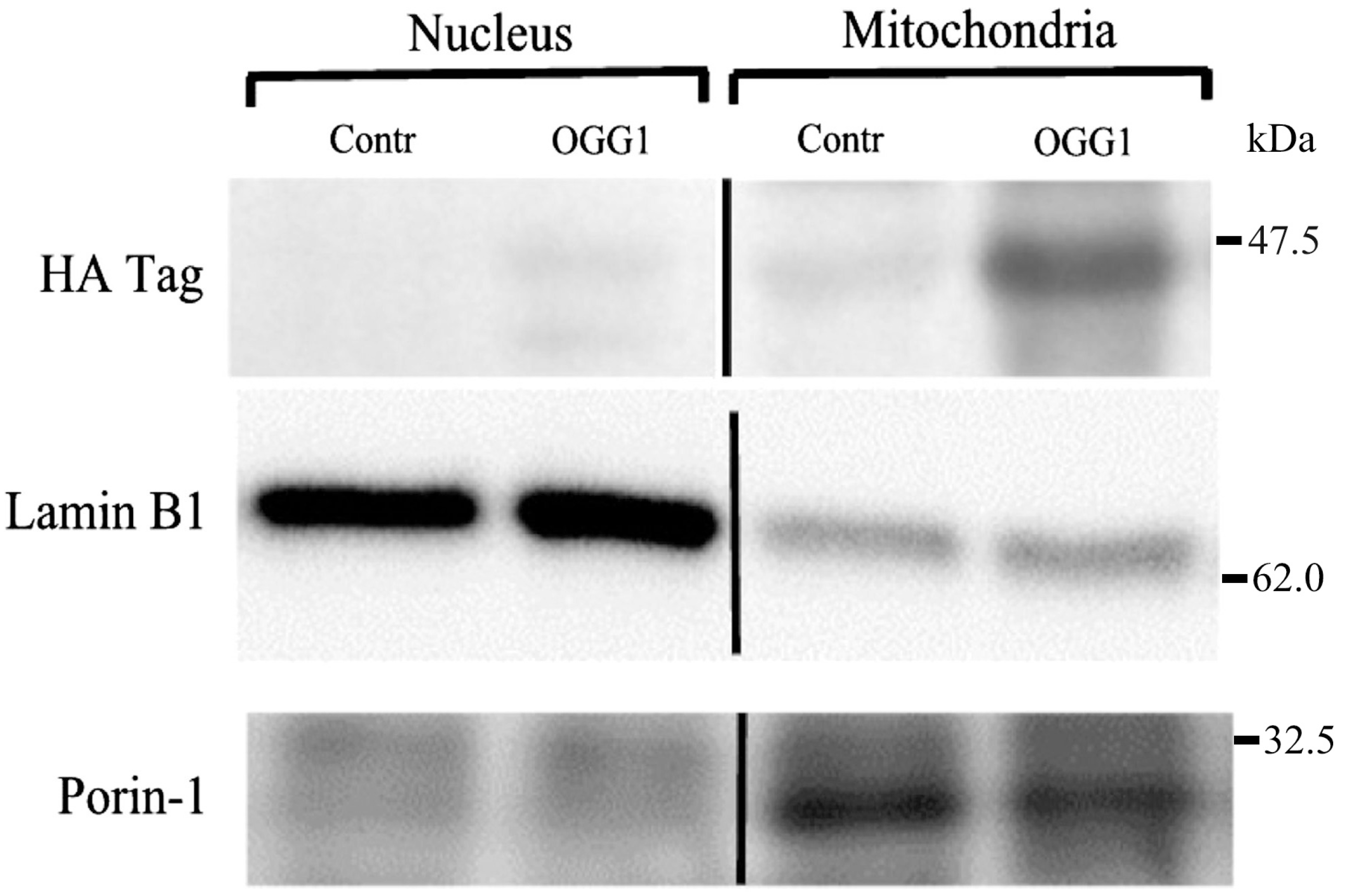
3.2. Subcellular Localization of the Fusion Protein
3.3. Lung Extravascular Albumin Spaces
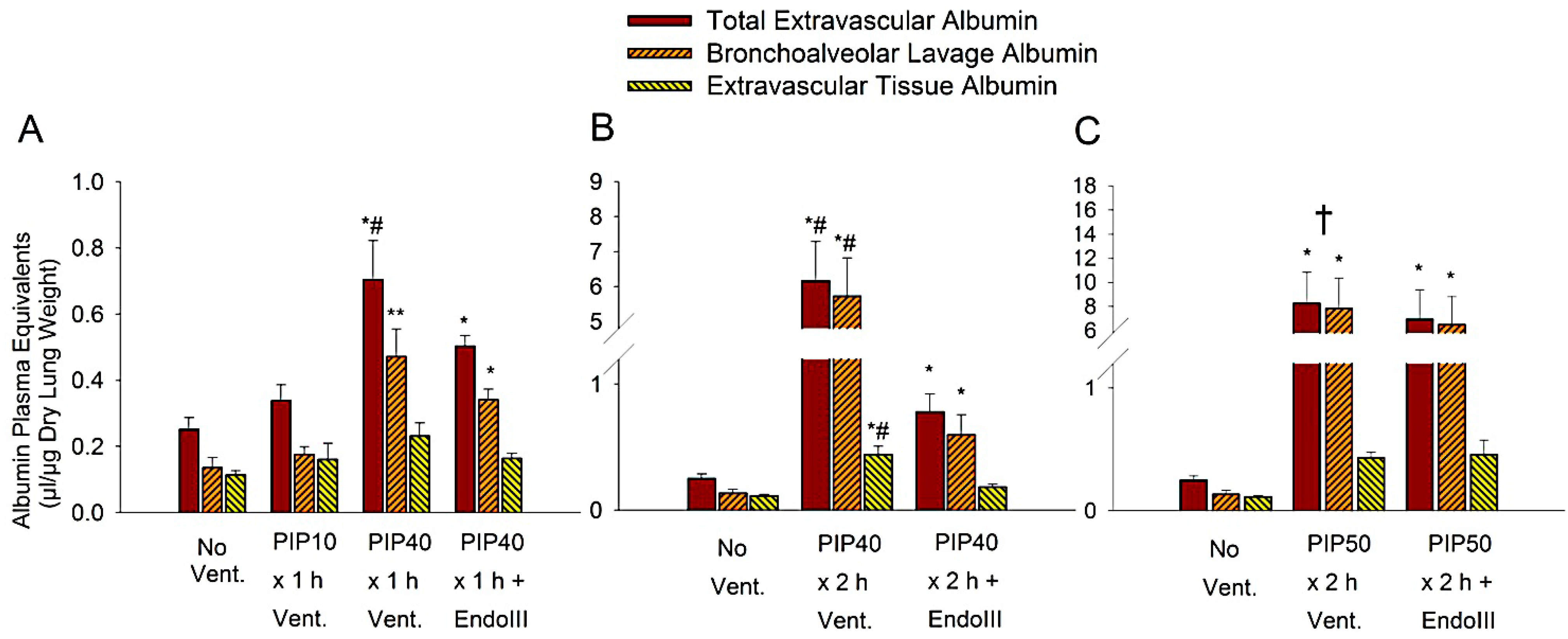
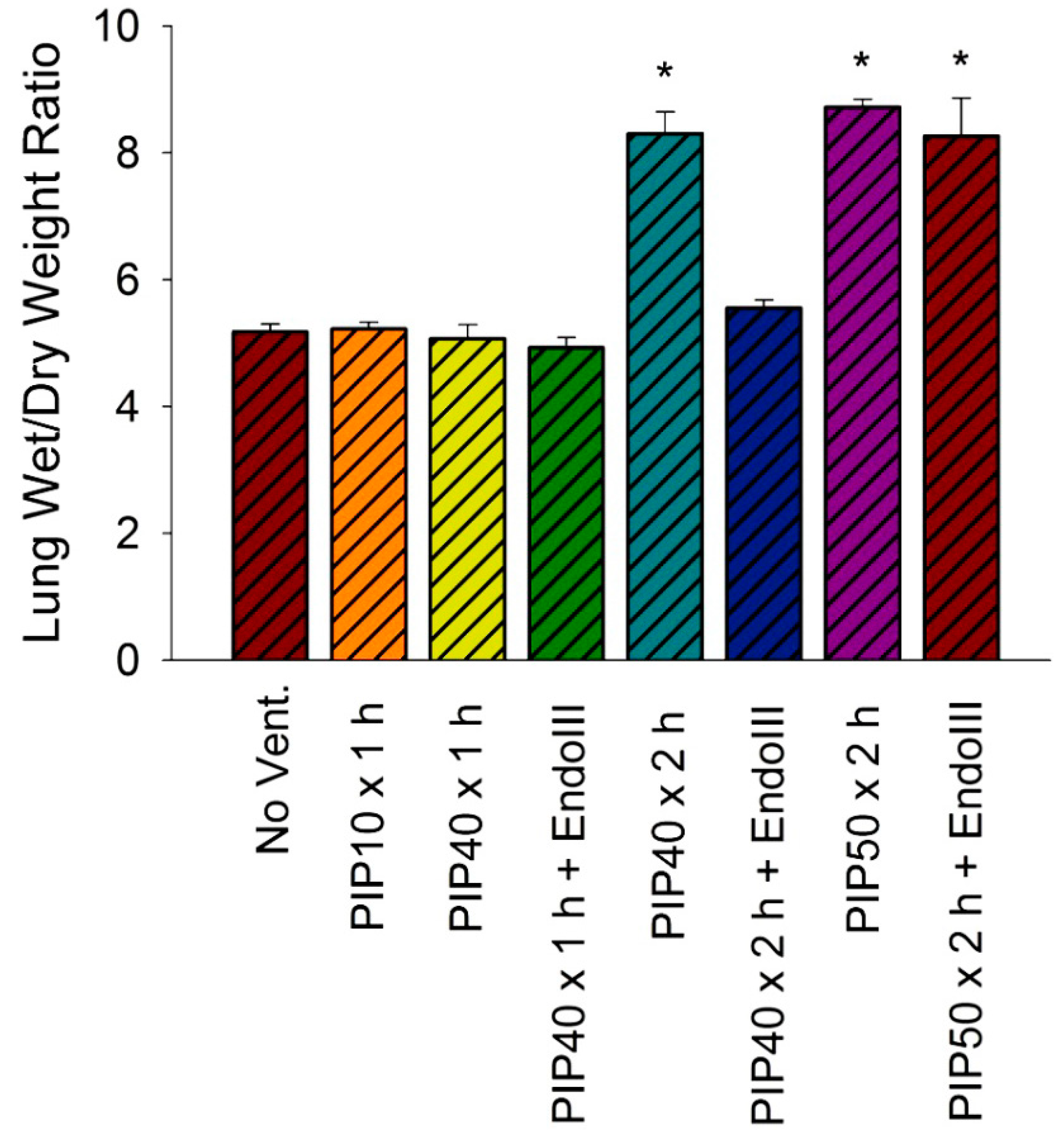
3.4. Lung Wet/Dry Weight Ratios
3.5. Bronchoalveolar Lavage Inflammatory Cytokines
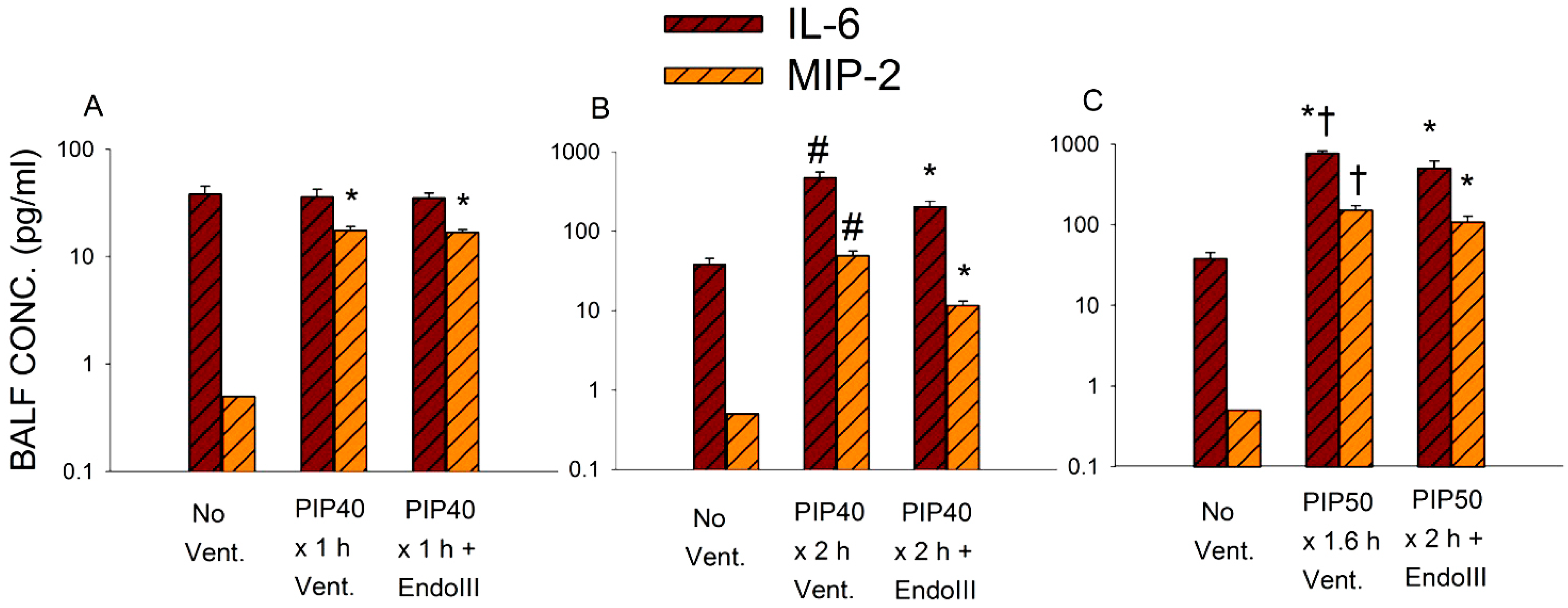
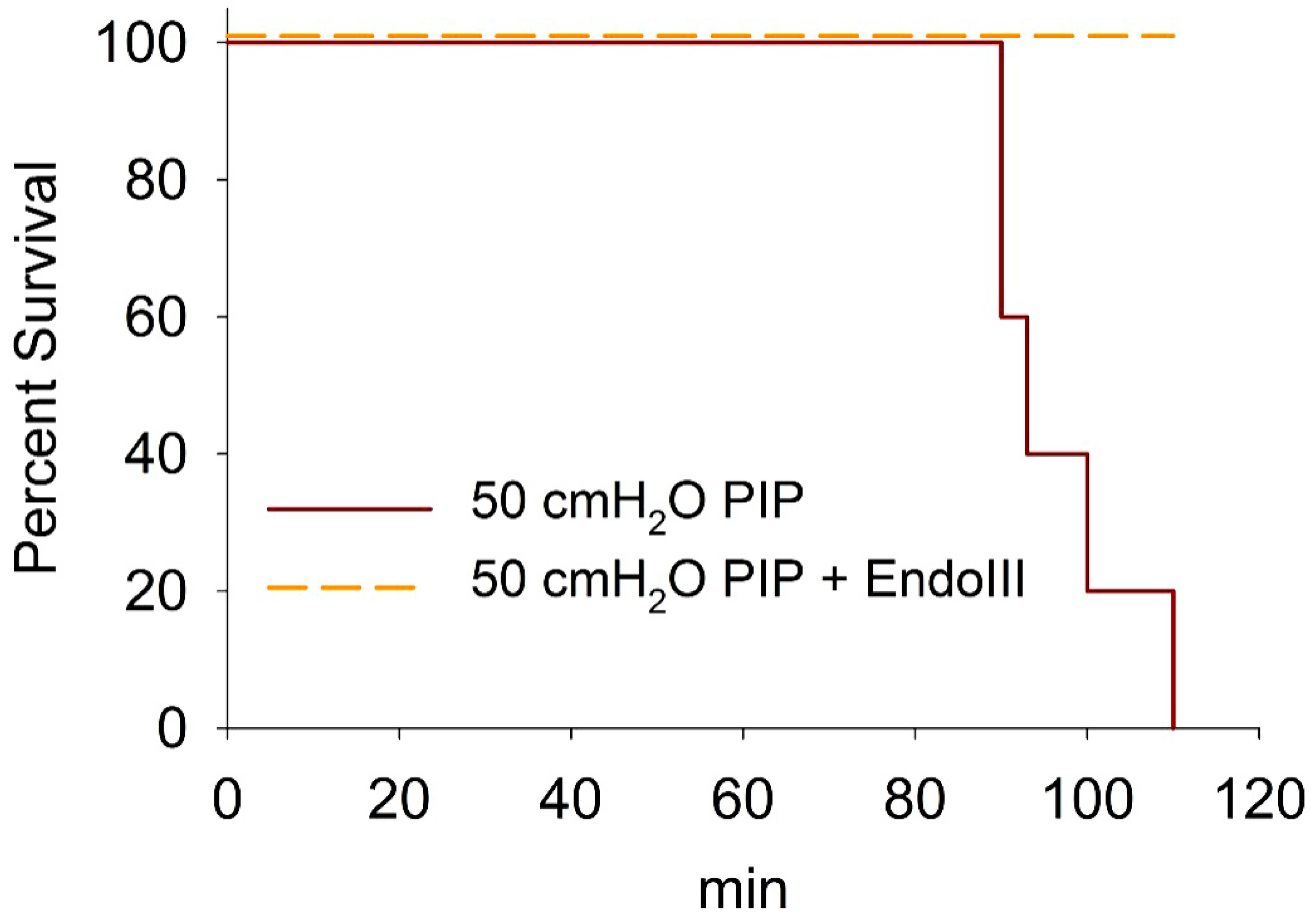
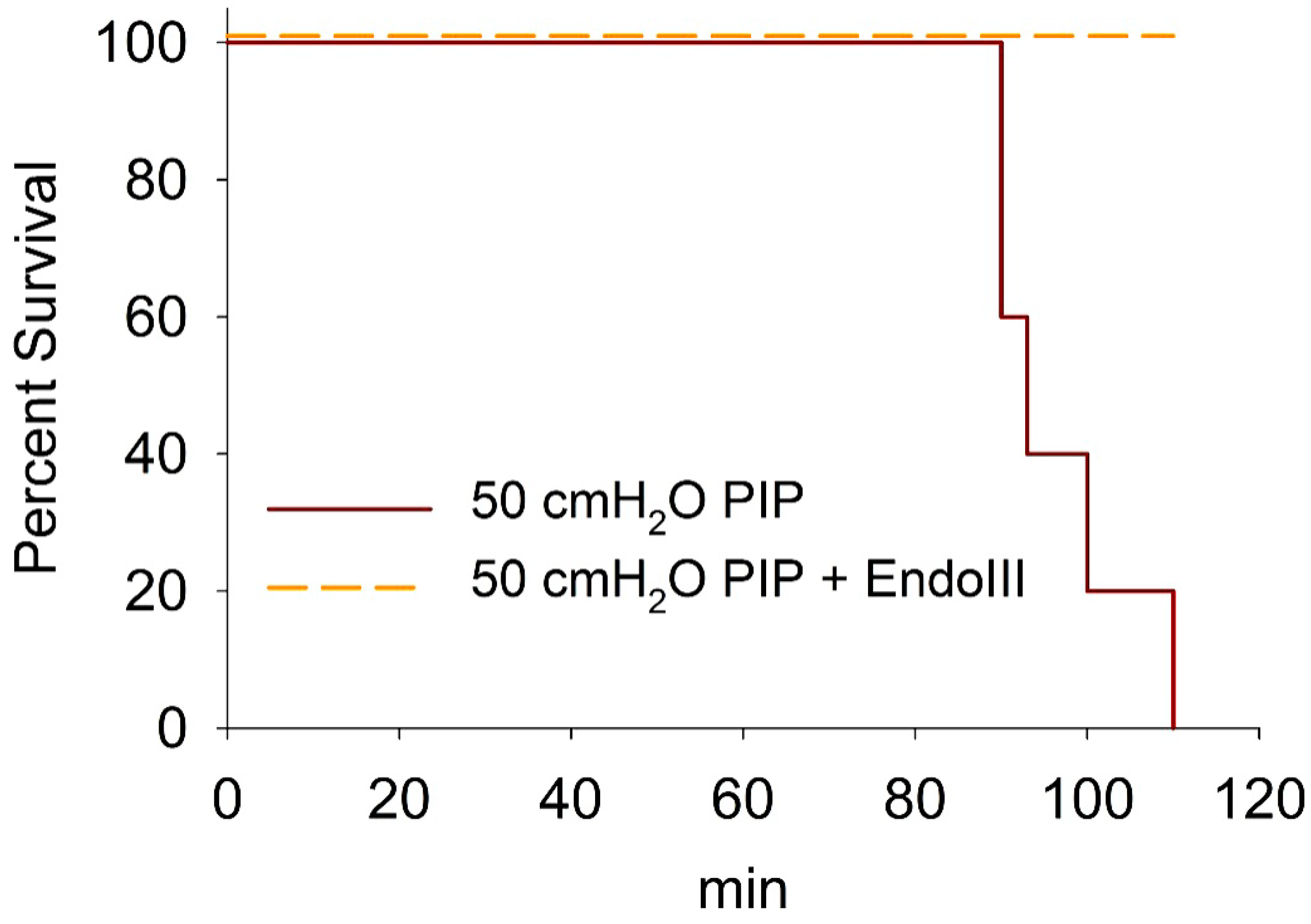
3.6. Survival During 2 h Ventilation at 50 cmH2O PIP
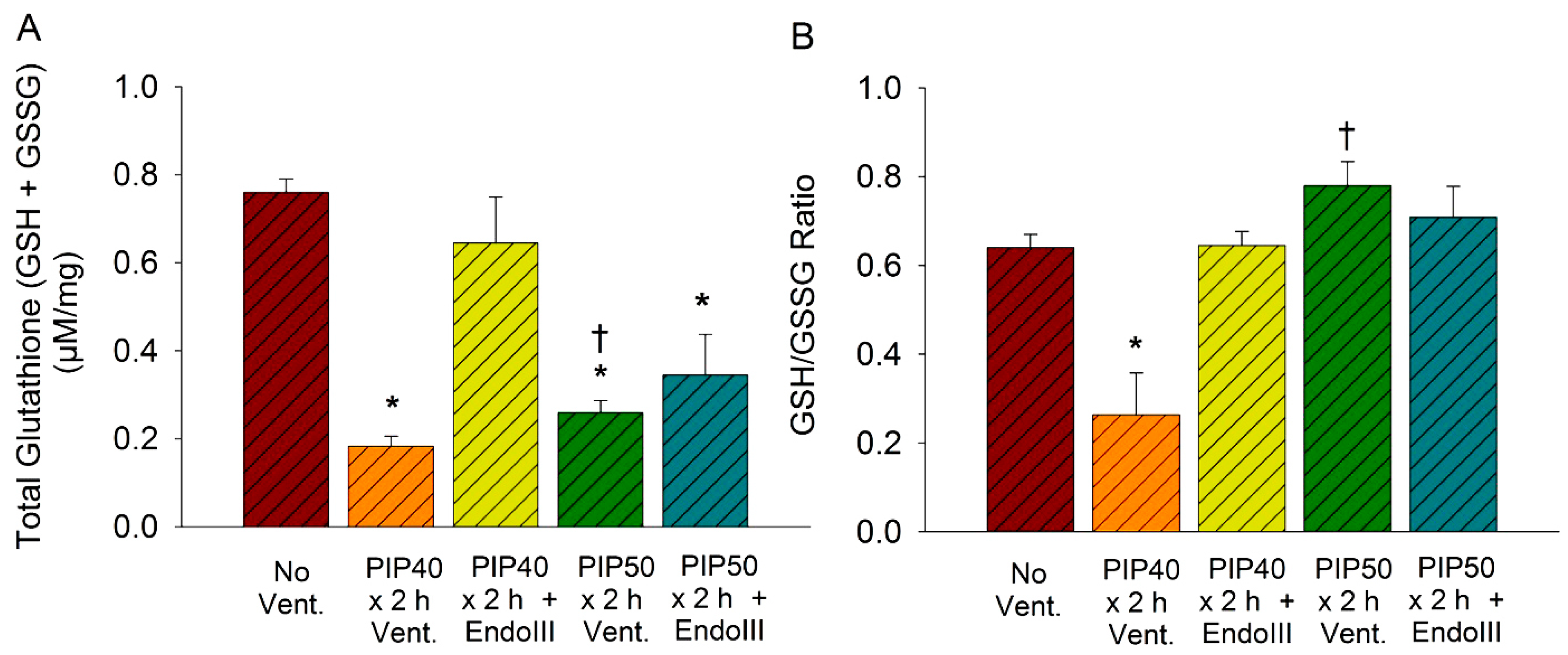
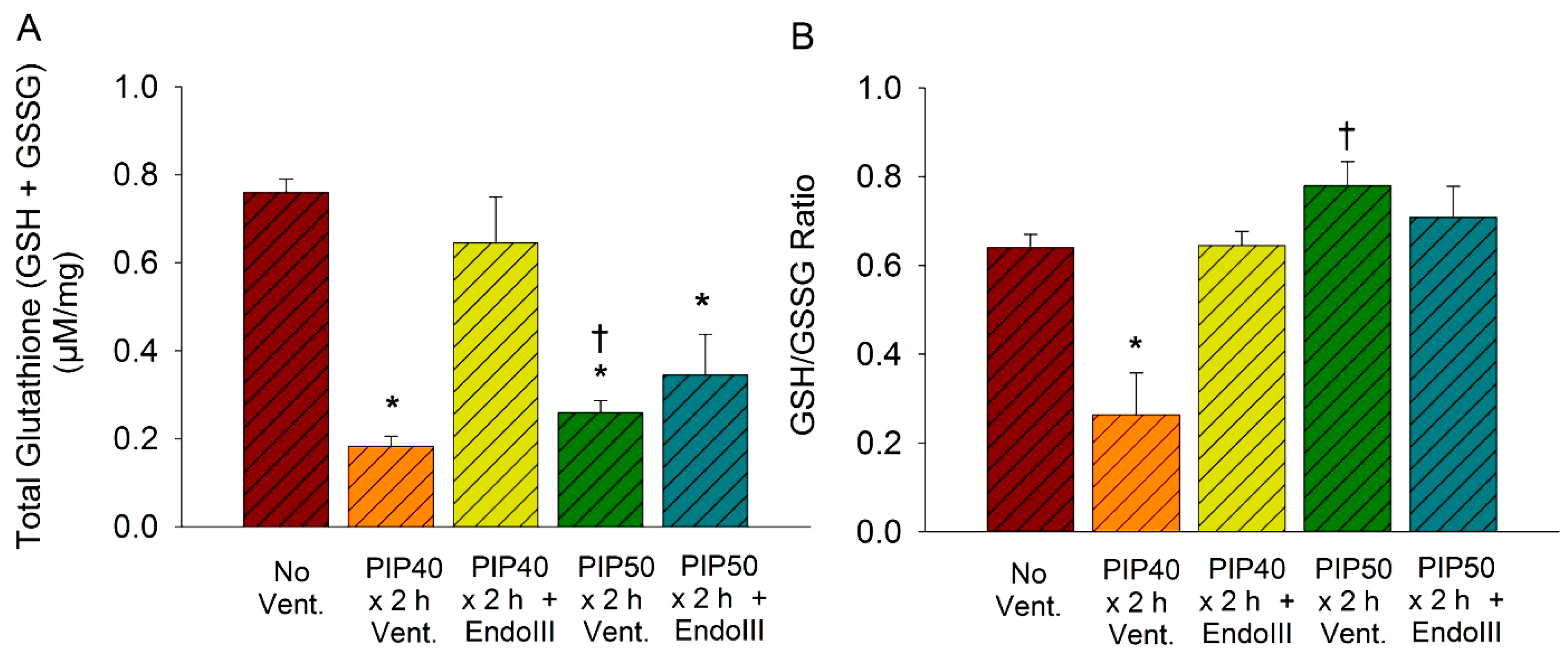
3.7. Lung Glutathione Concentrations
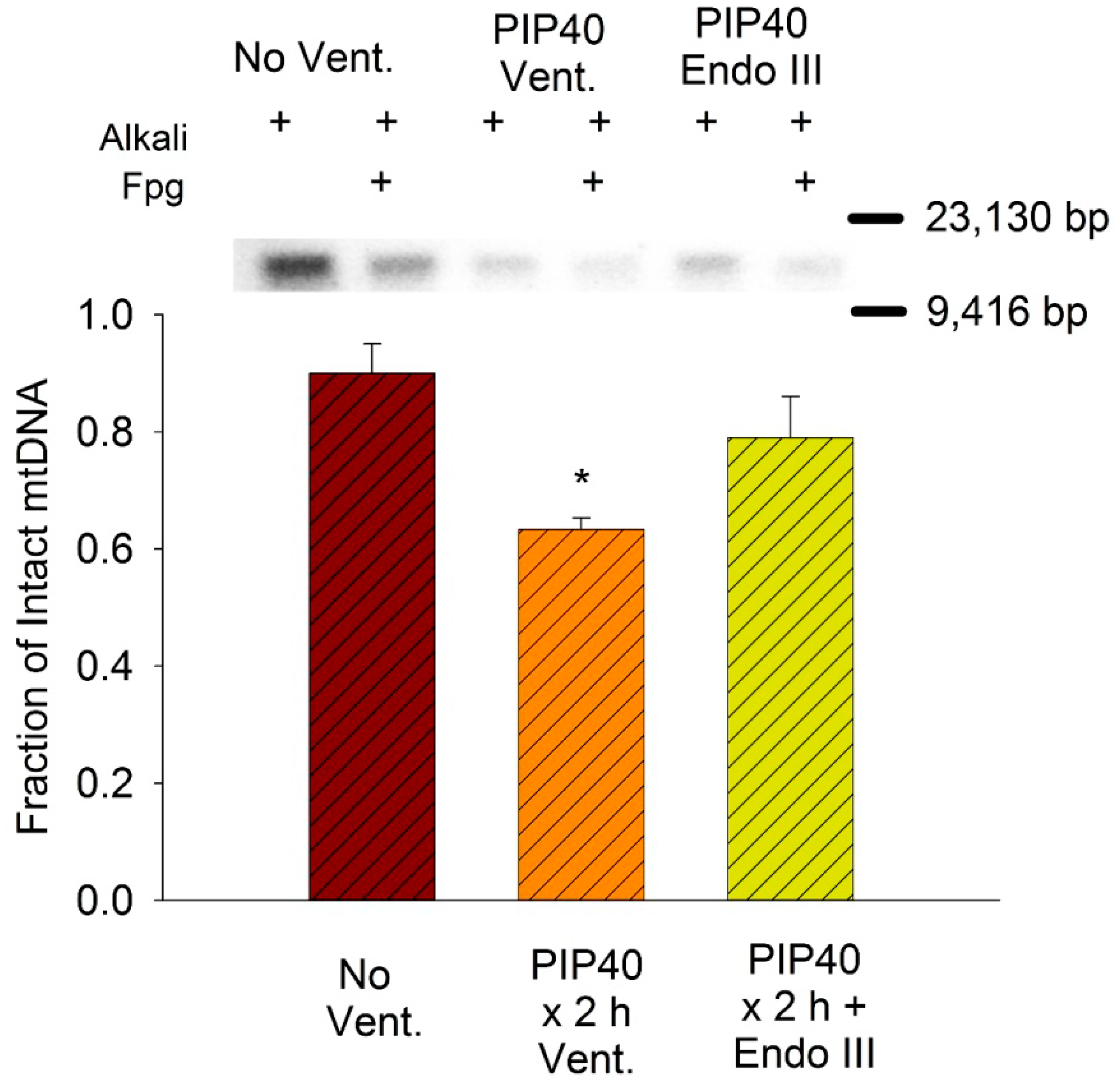
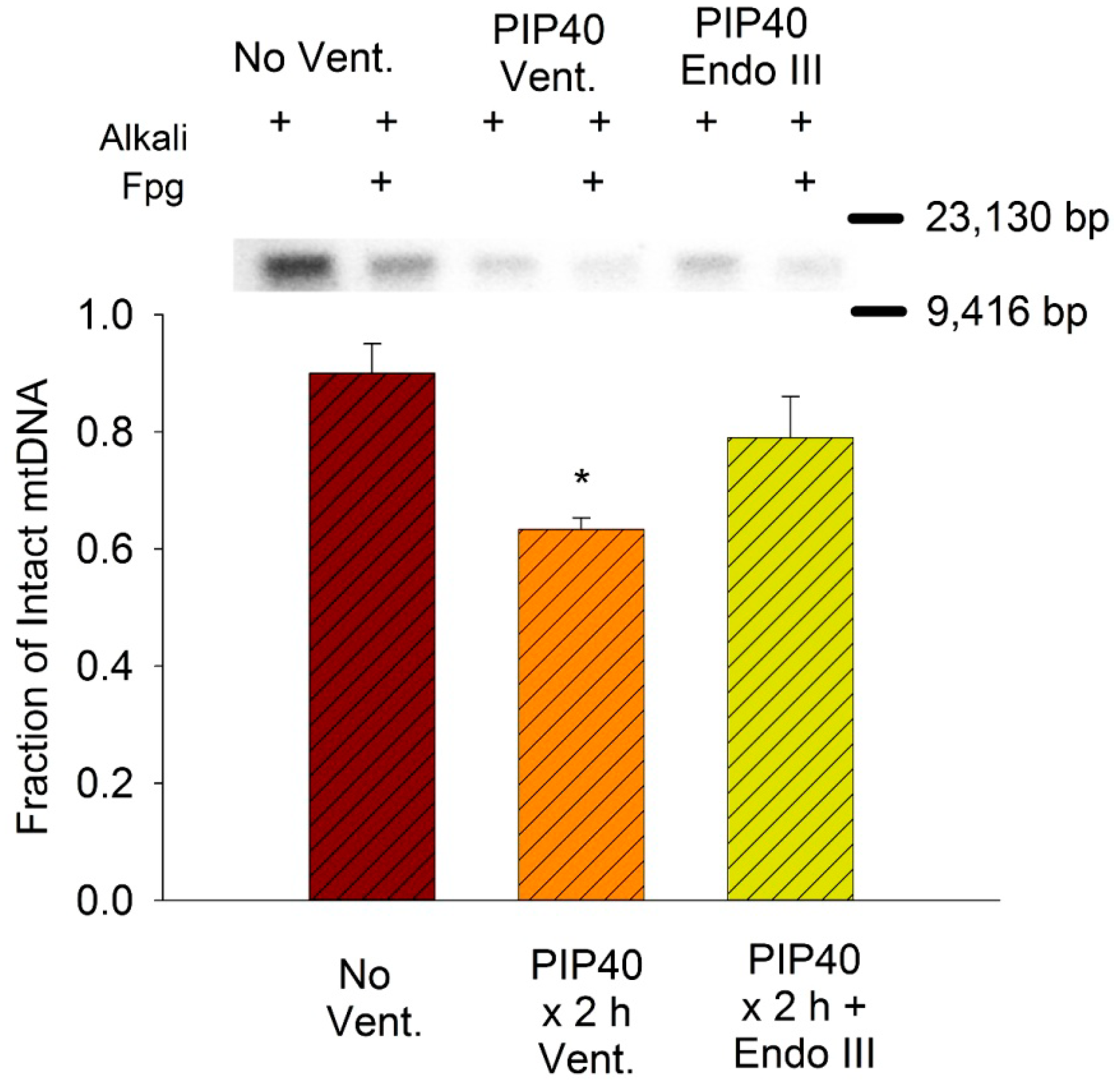
3.8. Lung Mitochondrial DNA Damage
4. Discussion
5. Conclusions
Acknowledgements
Author Contributions
Conflict of Interest
References
- Brower, R.G.; Matthay, M.A.; Morris, A.; Schoenfeld, D.; Thompson, B.T. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and respiratory distress syndrome. New Eng. J. Med. 2000, 342, 1301–1308. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; Zimmerman, G.A. Acute lung injury and the acute respiratory distress syndrome: Four decades of inquiry into pathogenesis and rational management. Am. J. Respir. Cell. Mol. Biol. 2005, 33, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, L.A.; Coker, P.J.; May, S.; Thompson, A.L.; Parker, J.C. Mechanical ventilation increases microvascular permeability in oleic acid-injured lungs. J. Appl. Physiol. 1990, 69, 2057–2061. [Google Scholar] [PubMed]
- Dhanireddy, S.; Altemeier, W.A.; Matute-Bello, G.; O'Mahony, D.S.; Glenny, R.W.; Martin, T.R.; Liles, W.C. Mechanical ventilation induces inflammation, lung injury, and extra-pulmonary organ dysfunction in experimental pneumonia. Lab. Invest. 2006, 86, 790–799. [Google Scholar] [CrossRef] [PubMed]
- Levine, G.K.; Deutschman, C.S.; Helfaer, M.A.; Margulies, S.S. Sepsis-induced lung injury in rats increases alveolar epithelial vulnerability to stretch. Crit. Care Med. 2006, 34, 1746–1751. [Google Scholar]
- Gutierrez, J.; Ballinger, S.W.; Rley-Usmar, V.M.; Landar, A. Free radicals, mitochondria, and oxidized lipids: The emerging role in signal transduction in vascular cells. Circ. Res. 2006, 99, 924–932. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.C. Acute lung injury and pulmonary vascular permeability: Use of transgenic models. Compr. Physiol. 2011, 2011, 835–882. [Google Scholar]
- Dikalov, S. Cross talk between mitochondria and NADPH oxidases. Free Radical Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef]
- Yakes, F.G.; VanGÇëHouten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidativeGÇëstress. Proc. Nat. Acad. Sci. 1997, 94, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Ruchko, M.; Gorodnya, O.; LeDoux, S.P.; Alexeyev, M.F.; Al Mehdi, A.B.; Gillespie, M.N. Mitochondrial DNA damage triggers mitochondrial dysfunction and apoptosis in oxidant-challenged lung endothelial cells. Am. J. Physiol. 2005, 288, L530–L535. [Google Scholar] [CrossRef]
- Dobson, A.W.; Grishko, V.; LeDoux, S.P.; Kelley, M.R.; Wilson, G.L.; Gillespie, M.N. Enhanced mtDNA repair capacity protects pulmonary artery endothelial cells from oxidant-mediated death. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 283, L205–L210. [Google Scholar]
- Druzhyna, N.M.; Hollensworth, S.B.; Kelley, M.R.; Wilson, G.L.; LeDoux, S.P. Targeting human 8-oxoguanine glycosylase to mitochondria of oligodendrocytes protects against menadione-induced oxidative stress. Glia 2003, 42, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, M.; Mouner, M.; Chouteau, J.M.; Gorodnya, O.M.; Ruchko, M.V.; Potter, B.J.; Wilson, G.L.; Gillespie, M.N.; Parker, J.C. Mitochondrial targeted DNA repair enzyme 8-oxoguanine DNA glycosylase 1 protects against ventilator induced lung injury in intact mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 304, L287–L297. [Google Scholar] [PubMed]
- Koczor, C.A.; Snyder, J.W.; Shokolenko, I.N.; Dobson, A.W.; Wilson, G.L.; LeDoux, S.P. Targeting repair proteins to the mitochondria of mammalian cells through stable transfection, transient transfection, viral transduction, and TAT-mediated protein transduction. Methods Mol. Biol. 2009, 554, 233–249. [Google Scholar] [PubMed]
- Chouteau, J.M.; Obiako, B.; Gorodnya, O.M.; Pastukh, V.M.; Ruchko, M.V.; Wright, A.J.; Wilson, G.L.; Gillespie, M.N. Mitochondrial DNA integrity may be a determinant of endothelial barrier properties in oxidant-challenged rat lungs. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 301, L892–L898. [Google Scholar] [PubMed]
- Tankersley, C.G.; Rabold, R.; Mitzner, W. Differential lung mechanics are genetically determined in inbred murine strains. J. Appl. Physiol. 1999, 86, 1764–1769. [Google Scholar]
- Wilson, M.R.; Choudhury, S.; Goddard, M.E.; O'Dea, K.P.; Nicholson, A.G.; Takata, M. High tidal volume upregulates intrapulmonary cytokines in an in vivo mouse model of ventilator-induced lung injury. J. Appl. Physiol. 2003, 95, 1385–1393. [Google Scholar] [PubMed]
- Ruchko, M.V.; Gorodnya, O.M.; Zuleta, A.; Pastukh, V.M.; Gillespie, M.N. The DNA glycosylase Ogg1 defends against oxidant-induced mtDNA damage and apoptosis in pulmonary artery endothelial cells. Free Radical Biol. Med. 2011, 50, 1107–1113. [Google Scholar] [CrossRef]
- Pastukh, V.; Ruchko, M.; Gorodnya, O.; Wilson, G.L.; Gillespie, M.N. Sequence-specific oxidative base modifications in hypoxia-inducible genes. Free Radical Biol. Med. 2007, 43, 1616–1626. [Google Scholar] [CrossRef]
- Belperio, J.A.; Keane, M.P.; Lynch, J.P., III; Strieter, R.M. The role of cytokines during the pathogenesis of ventilator-associated and ventilator-induced lung injury. Semin. Respir. Crit. Care Med. 2006, 27, 350–364. [Google Scholar]
- Hu, J.; Dong, L.; Outten, C.E. The Redox Environment in the Mitochondrial Intermembrane Space Is Maintained Separately from the Cytosol and Matrix. J. Biol. Chem. 2008, 283, 29126–29134. [Google Scholar] [CrossRef] [PubMed]
- Kazak, L.; Reyes, A.; Holt, I.J. Minimizing the damage: repair pathways keep mitochondrial DNA intact. Nat. Rev. Mol. Cell. Biol. 2012, 13, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Rachek, L.I.; Grishko, V.I.; Alexeyev, M.F.; Pastukh, V.V.; LeDoux, S.P.; Wilson, G.L. Endonuclease III and endonuclease VIII conditionally targeted into mitochondria enhance mitochondrial DNA repair and cell survival following oxidative stress. Nucleic Acids Res. 2004, 32, 3240–3247. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.K.; Boniface, O.; Olena, G.; Sujata, M.; Mark, N.G. C97. Mol. Mechan. Endothelial Barrier Function. Philadelphia, PA, USA, 17–22 May 2013.
- Brower, R.G.; Ware, L.B.; Berthiaume, Y.; Matthay, M.A. Treatment of ARDS. Chest 2001, 120, 1347–1367. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.C.; Vincent, J.L.; Guyatt, G.; Angus, D.C.; Abraham, E.; Bernard, G.; Bombardier, C.; Calandra, T.; Jorgensen, H.S.; Sylvester, R.; et al. Outcome measures for clinical research in sepsis: A report of the 2nd Cambridge colloquium of the international sepsis forum. Crit. Care Med. 2005, 33, 1708–1716. [Google Scholar]
- Lowes, D.A.; Webster, N.R.; Murphy, M.P.; Galley, H.F. Antioxidants that protect mitochondria reduce interleukin-6 and oxidative stress, improve mitochondrial function, and reduce biochemical markers of organ dysfunction in a rat model of acute sepsis. Br. J. Anaesth. 2013, 110, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.P.; Hassoun, P.M.; Brower, R. Redox imbalance and ventilator-induced lung injury. Antioxid. Redox Signal. 2007, 9, 2003–2012. [Google Scholar]
- Chapman, K.E.; Sinclair, S.E.; Zhuang, D.; Hassid, A.; Desai, L.P.; Waters, C.M. Cyclic mechanical strain increases reactive oxygen species production in pulmonary epithelial cells. Am. J. Physiol. 2005, 289, L834–L841. [Google Scholar] [CrossRef]
- Ali, M.H.; Pearlstein, D.P.; Mathieu, C.E.; Schumacker, P.T. Mitochondrial requirement for endothelial responses to cyclic strain: Implications for mechanotransduction. Am. J. Physiol. 2004, 287, L486–L496. [Google Scholar] [CrossRef]
- Reyes, A.; He, J.; Mao, C.C.; Bailey, L.J.; Di, R.M.; Sembongi, H.; Kazak, L.; Dzionek, K.; Holmes, J.B.; Cluett, T.J.; et al. Actin and myosin contribute to mammalian mitochondrial DNA maintenance. Nucleic Acids Res. 2011, 39, 5098–5108. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.C. Inhibitors of myosin light chain kinase, phosphodiesterase and calmodulin attenuate ventilator induced lung injury. J. Appl. Physiol. 2000, 89, 2241–2248. [Google Scholar] [PubMed]
- Mirzapoiazova, T.; Moitra, J.; Moreno-Vinasco, L.; Sammani, S.; Turner, J.R.; Chiang, E.T.; Evenoski, C.; Wang, T.; Singleton, P.A.; Huang, Y.; et al. Non-muscle myosin light chain kinase isoform is a viable molecular target in acute inflammatory lung injury. Am. J. Respir. Cell Mol. Biol. 2011, 44, 40–52. [Google Scholar] [CrossRef]
- Rossi, J.L.; Velentza, A.V.; Steinhorn, D.M.; Watterson, D.M.; Wainwright, M.S. MLCK210 gene knockout or kinase inhibition preserves lung function following endotoxin-induced lung injury in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L1327–L1334. [Google Scholar] [PubMed]
- Boldogh, I.; Hajas, G.; guilera-Aguirre, L.; Hegde, M.L.; Radak, Z.; Bacsi, A.; Sur, S.; Hazra, T.K.; Mitra, S. Activation of Ras signaling by 8-oxoguanine DNA glycosylase bound to its excision product 8-oxoguanine. J. Biol. Chem. 2012, 287, 20769–20773. [Google Scholar] [CrossRef] [PubMed]
- Miyahara, T.; Hamanaka, K.; Weber, D.S.; Drake, D.A.; Anghelescu, M.; Parker, J.C. Phosphoinositide 3-kinase, Src, and Akt modulate acute ventilation-induced vascular permeability increases in mouse lungs. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L11–L21. [Google Scholar] [PubMed]
- Lionetti, V.; Lisi, A.; Patrucco, E.; De Giuli, P.; Milazzo, M.G.; Ceci, S.; Wymann, M.; Lena, A.; Gremigni, V.; Fanelli, V.; et al. Lack of phosphoinositide 3-kinase-gamma attenuates ventilator-induced lung injury. Crit. Care Med. 2006, 34, 134–141. [Google Scholar] [PubMed]
- Fanelli, V.; Puntorieri, V.; Assenzio, B.; Martin, E.; Elia, V.; Bosco, M.; Delsedime, L.; Del Sorbo, L.; Ferrari, A.; Italiano, S.; et al. Pulmonary-derived phosphoinositide 3-kinase gamma (PI3K+¦) contributes to ventilator-induced lung injury and edema. Intens. Care Med. 2010, 36, 1935–1945. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hashizume, M.; Mouner, M.; Chouteau, J.M.; Gorodnya, O.M.; Ruchko, M.V.; Wilson, G.L.; Gillespie, M.N.; Parker, J.C. Mitochondrial Targeted Endonuclease III DNA Repair Enzyme Protects against Ventilator Induced Lung Injury in Mice. Pharmaceuticals 2014, 7, 894-912. https://doi.org/10.3390/ph7080894
Hashizume M, Mouner M, Chouteau JM, Gorodnya OM, Ruchko MV, Wilson GL, Gillespie MN, Parker JC. Mitochondrial Targeted Endonuclease III DNA Repair Enzyme Protects against Ventilator Induced Lung Injury in Mice. Pharmaceuticals. 2014; 7(8):894-912. https://doi.org/10.3390/ph7080894
Chicago/Turabian StyleHashizume, Masahiro, Marc Mouner, Joshua M. Chouteau, Olena M. Gorodnya, Mykhaylo V. Ruchko, Glenn L. Wilson, Mark N. Gillespie, and James C. Parker. 2014. "Mitochondrial Targeted Endonuclease III DNA Repair Enzyme Protects against Ventilator Induced Lung Injury in Mice" Pharmaceuticals 7, no. 8: 894-912. https://doi.org/10.3390/ph7080894
APA StyleHashizume, M., Mouner, M., Chouteau, J. M., Gorodnya, O. M., Ruchko, M. V., Wilson, G. L., Gillespie, M. N., & Parker, J. C. (2014). Mitochondrial Targeted Endonuclease III DNA Repair Enzyme Protects against Ventilator Induced Lung Injury in Mice. Pharmaceuticals, 7(8), 894-912. https://doi.org/10.3390/ph7080894