Effects of NSAIDs on Differentiation and Function of Human and Murine Osteoclasts – Crucial ‘Human Osteoclastology’

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Osteoclasts Are Formed From Monocytes Stimulated by RANKL
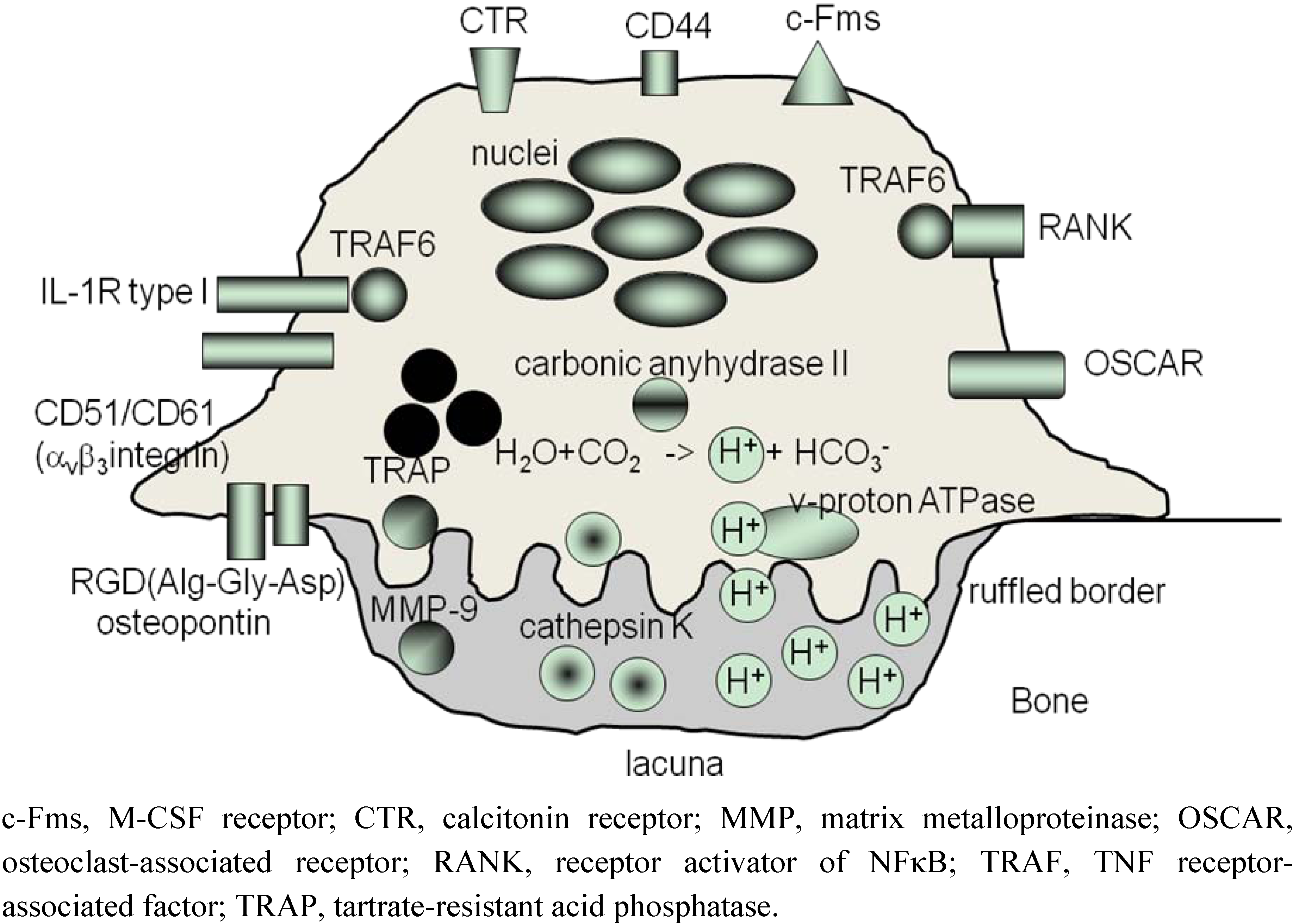
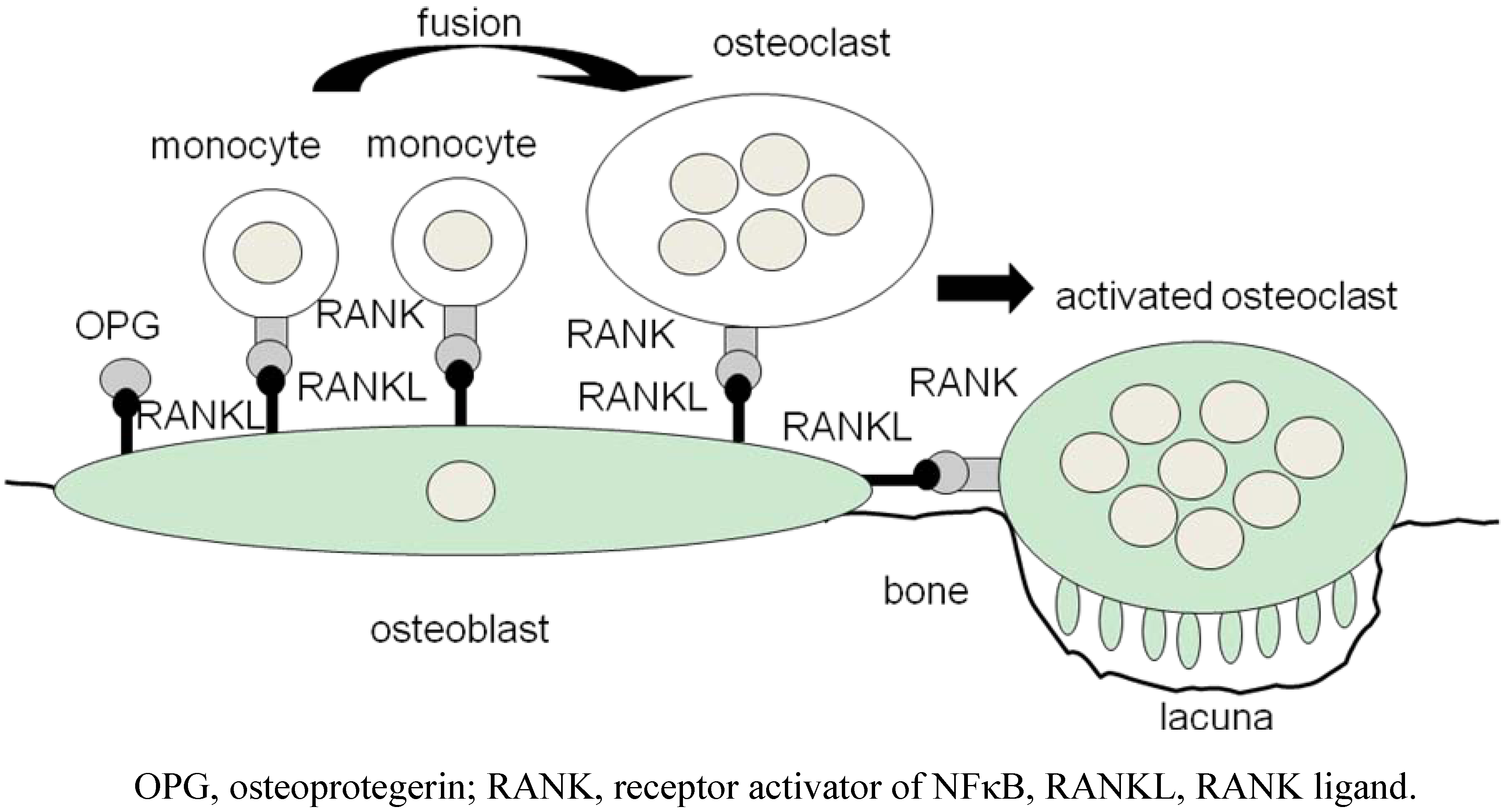
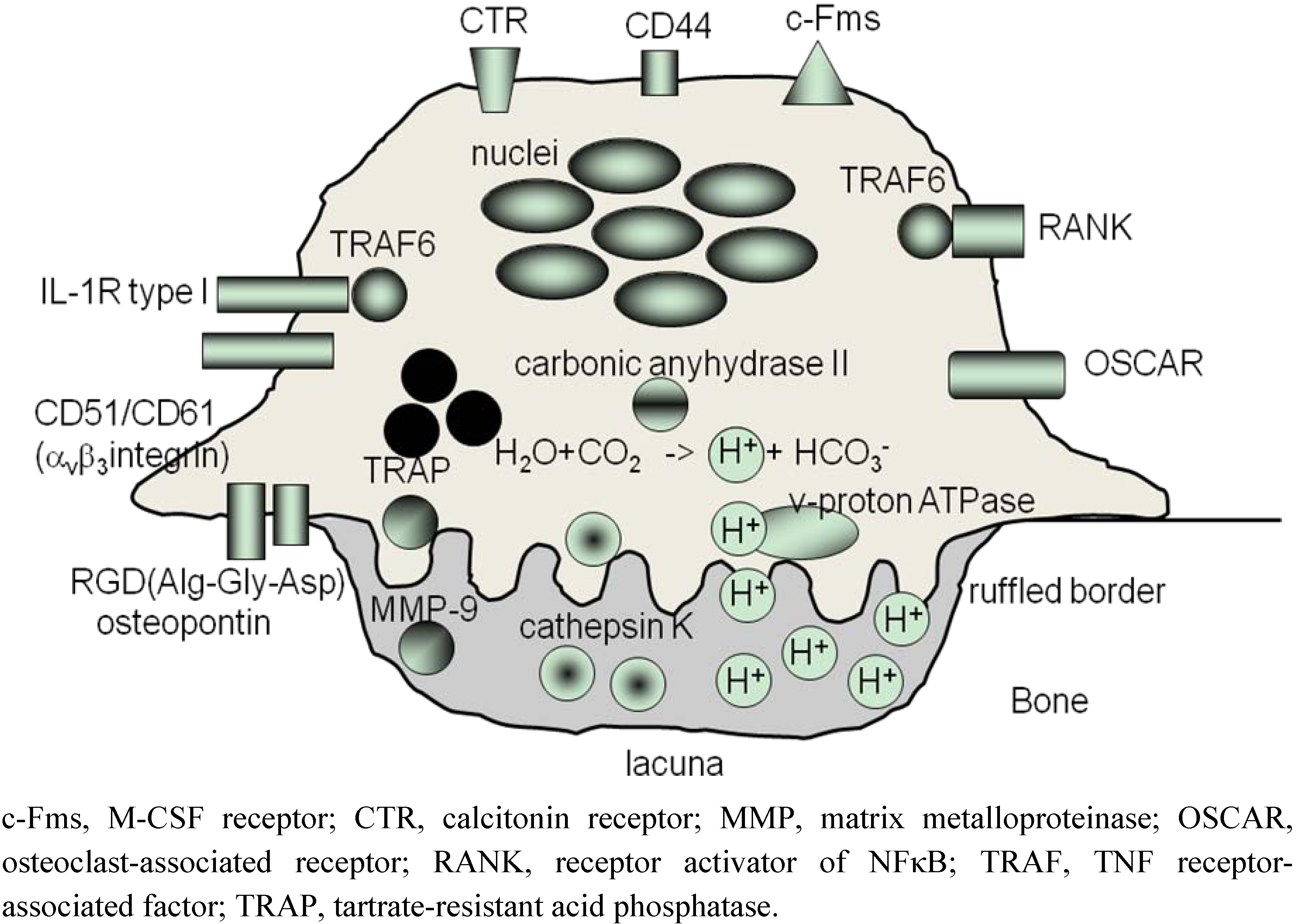
3. ‘Human Osteoclastology’
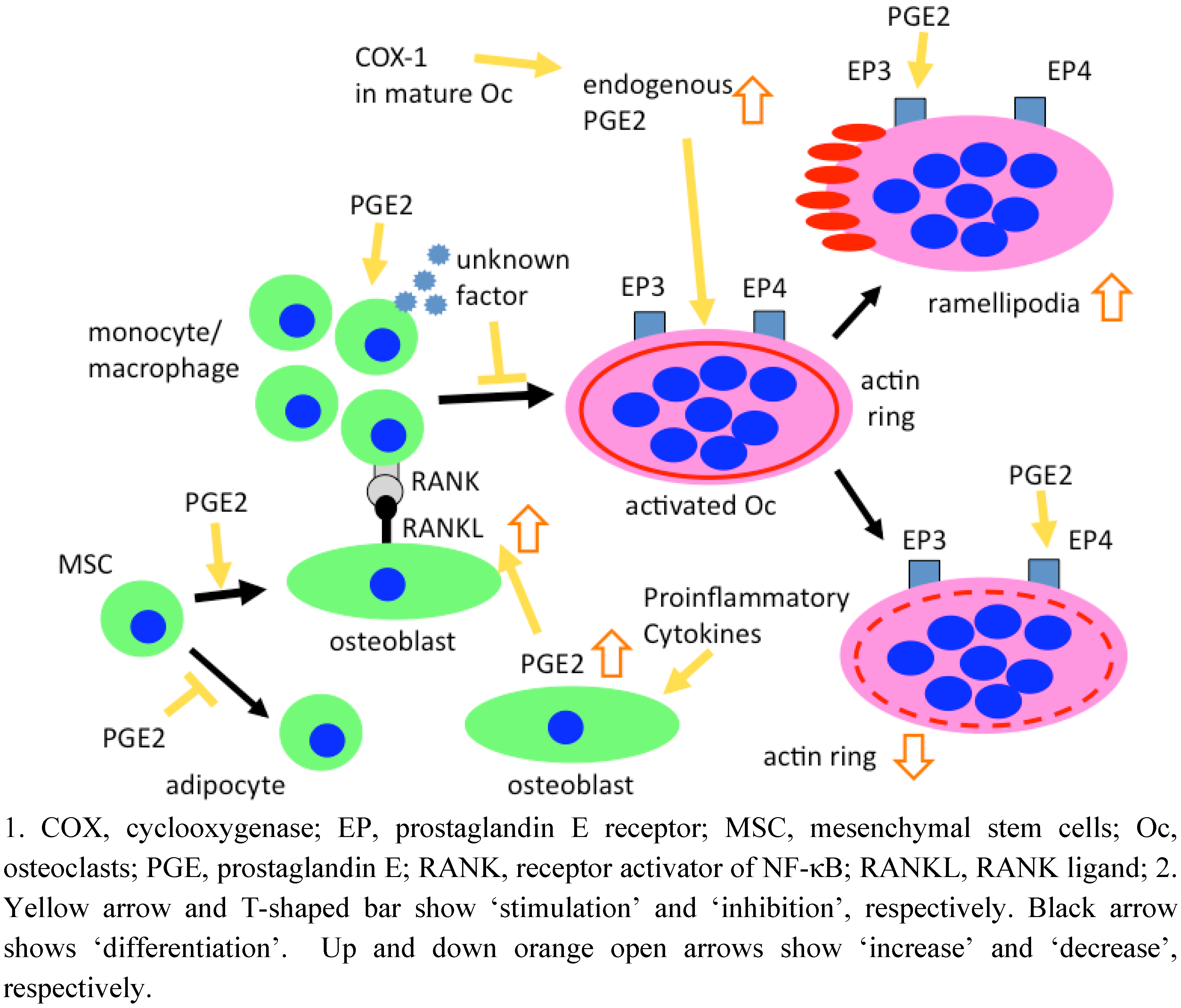
4. Roles of PGE2 in Osteoclastogenesis
4.1. Development of culture systems to form osteoclasts in vitro
4.2. Role of PGE2 in murine osteoclastogenesis
4.3. Role of PGE2 in human osteoclastogenesis

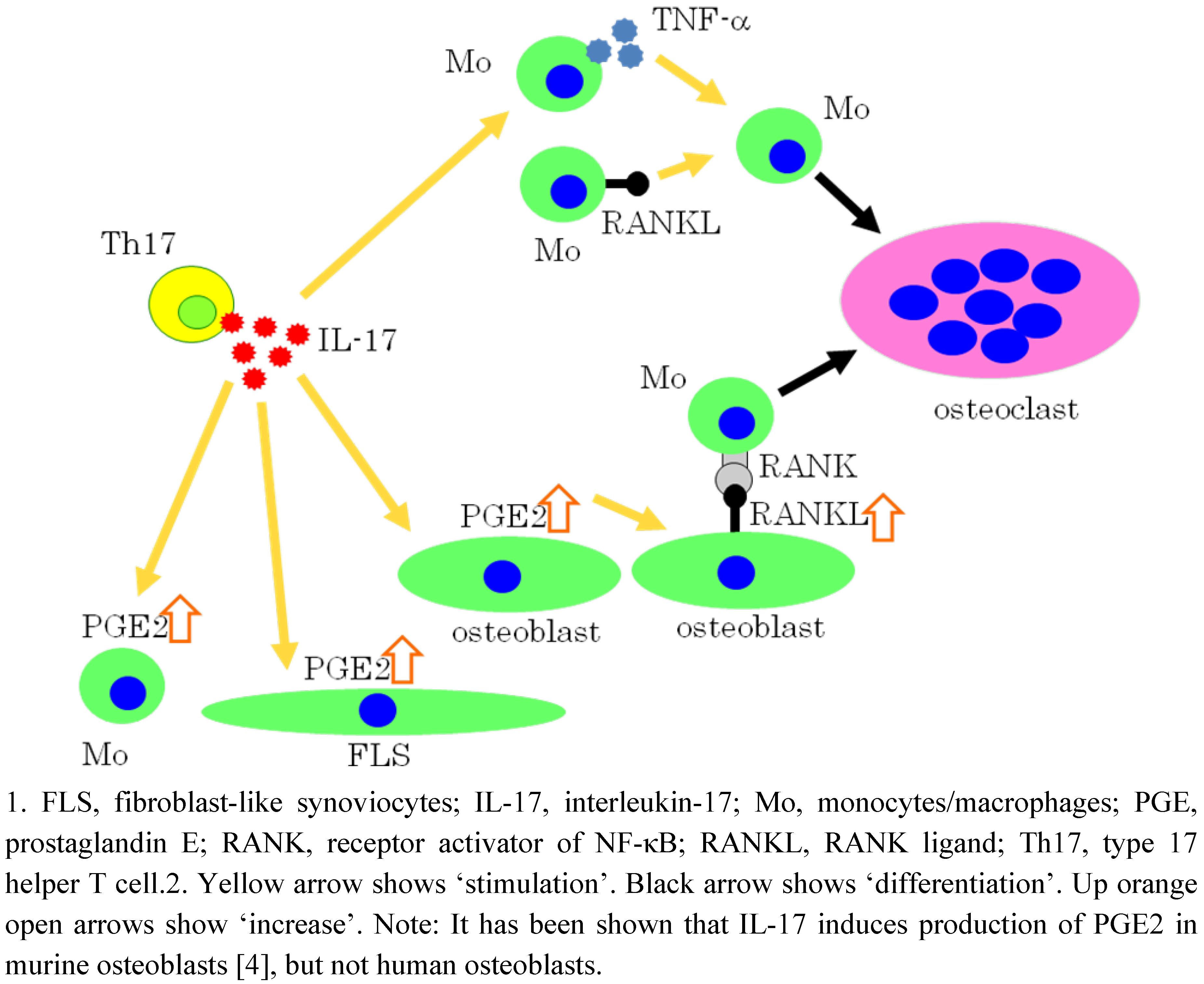
5. IL-17 Induces Osteoclastogenesis via the Expression of PGE2 in Osteoblastic cells
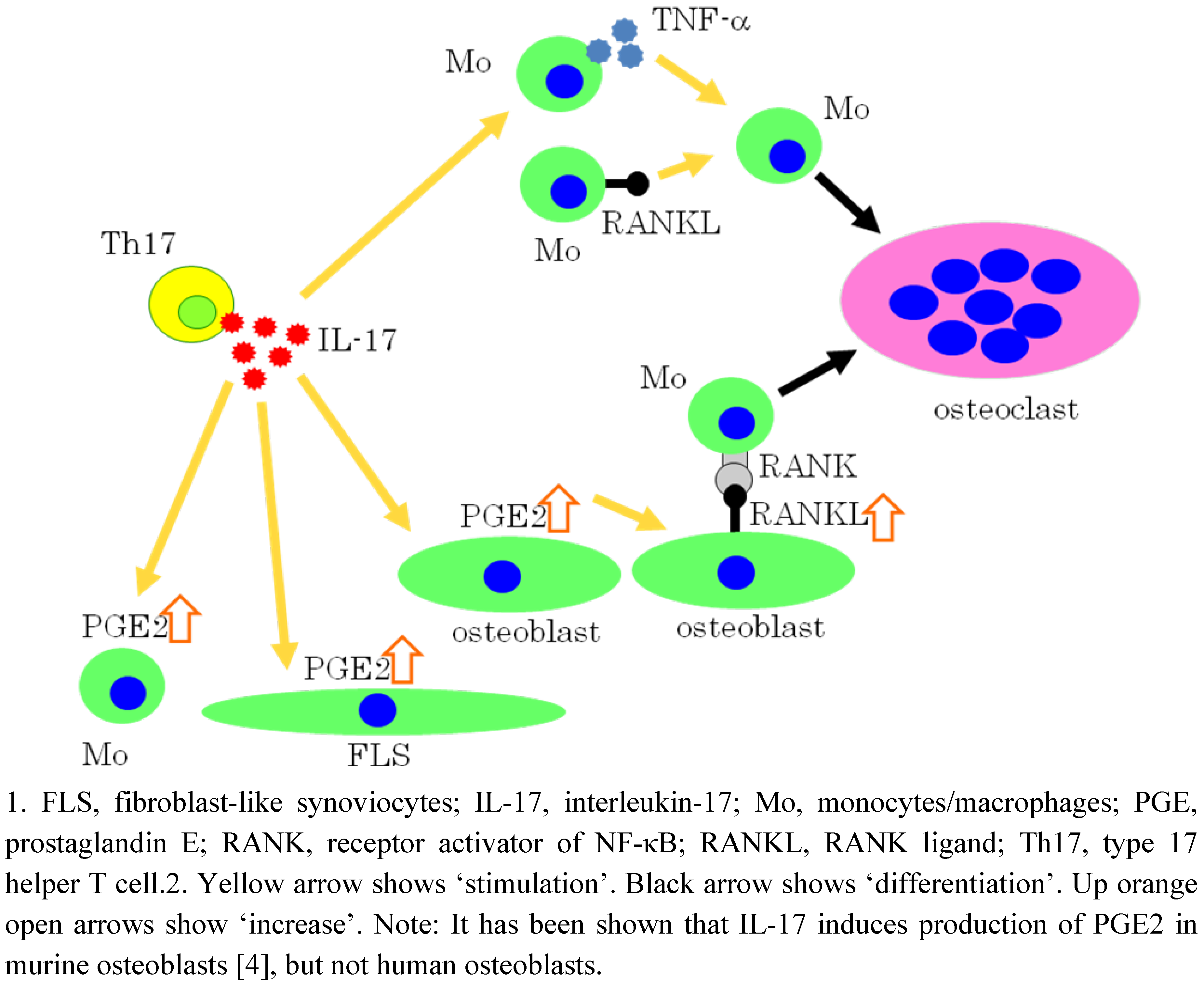
6. Effects of PGE2 on Mature Osteoclasts
7. COX-2 Inhibitor and Nonselective NSAIDs
7.1. Diclofenac sodium
7.2. Etodolac
7.3. Celecoxib
7.4. Flurbiprofen derivative HCT1026
8. Effect of NSAIDs on Osteoblasts
9. Systemic Effects of NSAIDs on Bone Metabolism
10. Geranylgeranylacetone Inhibits the Formation and Function of Human Osteoclasts
11. Conclusions
References and Notes
- Kong, Y.Y.; Feige, U.; Sarosi, I.; Bolon, B.; Tafuri, A.; Morony, S.; Capparelli, C.; Li, J.; Elliott, R.; McCabe, S.; Wong, T.; Campagnuolo, G.; Moran, E.; Bogoch, E.R.; Van, G.; Nguyen, L.T.; Ohashi, P.S.; Lacey, D.L.; Fish, E.; Boyle, W.J.; Penninger, J.M. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature 1999, 402, 304–309. [Google Scholar]
- Horwood, N.J.; Kartsogiannis, V.; Quinn, J.M.; Romas, E.; Martin, T.J.; Gillespie, M.T. Activated T lymphocytes support osteoclast formation in vitro. Biochem. Biophys. Res. Commun. 1999, 265, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Kotake, S.; Udagawa, N.; Hakoda, M.; Mogi, M.; Yano, K.; Tsuda, E.; Takahashi, K.; Furuya, T.; Ishiyama, S.; Kim, K.J.; Saito, S.; Nishikawa, T.; Takahashi, N.; Togari, A.; Tomatsu, T.; Suda, T.; Kamatani, N. Activated human T cells directly induce osteoclastogenesis from human monocytes: possible role of T cells in bone destruction in rheumatoid arthritis patients. Arthritis Rheum. 2001, 44, 1003–1012. [Google Scholar]
- Kotake, S.; Udagawa, N.; Takahashi, N.; Matsuzaki, K.; Itoh, K.; Ishiyama, S.; Saito, S.; Inoue, K.; Kamatani, N.; Gillespie, M.T.; Martin, T.J.; Suda, T. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J. Clin. Invest. 1999, 103, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Kotake, S.; Nanke, Y.; Yago, T.; Kawamoto, M.; Yamanaka, H. Human osteoclastogenic T cells and human osteoclastology (Editorial). Arthritis. Rheum. 2009, 60, 3158–3163. [Google Scholar]
- Take, I.; Kobayashi, Y.; Yamamoto, Y.; Tsuboi, H.; Ochi, T.; Uematsu, S.; Okafuji, N.; Kurihara, S.; Udagawa, N.; Takahashi, N. Prostaglandin E2 strongly inhibits human osteoclast formation. Endocrinology 2005, 146, 5204–5214. [Google Scholar]
- Kotake., S.; Nanke, Y.; Mogi, M.; Kawamoto, M.; Furuya, T.; Yago, T.; Kobashigawa, T.; Togari, A.; Kamatani, N. IFN-gamma-producing human T cells directly induce osteoclastogenesis from human monocytes via the expression of RANKL. Eur. J. Immunol. 2005, 35, 3353–3363. [Google Scholar]
- Luxenburg, C.; Geblinger, D.; Klein, E.; Anderson, K.; Hanein, D.; Geiger, B.; Addadi, L. The architecture of the adhesive apparatus of cultured osteoclasts: From podosome formation to sealing zone assembly. PLoS ONE 2007, 2, e179. [Google Scholar]
- Fuller, K.; Kirstein, B.; Chambers, T.J. Regulation and enzymatic basis of bone resorption by human osteoclasts. Clin. Sci. (Lond). 2007, 112, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Shiozawa, S.; Shiozawa, K.; Imura, S.; Fujita, T. Quantitative histologic studies on the pathogenesis of periarticular osteoporosis in rheumatoid arthritis. Arthritis Rheum. 1985, 28, 25–31. [Google Scholar]
- Kotake, S.; Sato, K.; Kim, K.J.; Takahashi, N.; Udagawa, N.; Nakamura, I.; Yamaguchi, A.; Kishimoto, T.; Suda, T.; Kashiwazaki, S. Interleukin-6 and soluble interleukin-6 receptors in the synovial fluids from rheumatoid arthritis patients are responsible for osteoclast-like cell formation. J. Bone Miner. Res. 1996, 11, 88–95. [Google Scholar]
- Kotake, S.; Higaki, M.; Sato, K.; Himeno, S.; Morita, H.; Kim, K.J.; Nara, N.; Miyasaka, N.; Nishioka, K.; Kashiwazaki, S. Detection of myeloid precursors (granulocyte/macrophage colony forming units) in the bone marrow adjacent to rheumatoid arthritis joints. J. Rheumatol. 1992, 19, 1511–1516. [Google Scholar]
- Kotake, S.; Schumacher, H.R., Jr.; Yarboro, C.H.; Arayssi, T.K.; Pando, J.A.; Kanik, K.S.; Gourley, M.F.; Klippel, J.H.; Wilder, R.L. In vivo gene expression of type 1 and type 2 cytokines in synovial tissues from patients in early stages of rheumatoid, reactive, and undifferentiated arthritis. Proc. Assoc. Am. Physicians 1997, 109, 286–301. [Google Scholar] [PubMed]
- Kotake, S.; Schumacher, H.R., Jr.; Arayssi, T.K.; Gerard, H.C.; Branigan, P.J.; Hudson, A.P.; Yarboro, C.H.; Klippel, J.H.; Wilder, R.L. Gamma interferon and interleukin-10 gene expression in synovial tissues from patients with early stages of Chlamydia-associated arthritis and undifferentiated oligoarthritis and from healthy volunteers. Infect. Immun. 1999, 67, 2682–2686. [Google Scholar]
- Kotake, S.; Schumacher, H.R., Jr.; Wilder, R.L. A simple nested RT-PCR method for quantitation of the relative amounts of multiple cytokine mRNAs in small tissue samples. J. Immunol. Methods 1996, 199, 193–203. [Google Scholar]
- Takahashi, N.; Akatsu, T.; Udagawa, N.; Sasaki, T.; Yamaguchi, A.; Moseley, J.M.; Martin, T.J.; Suda, T. Osteoblastic cells are involved in osteoclast formation. Endocrinology 1988, 123, 2600–2602. [Google Scholar]
- Udagawa, N.; Takahashi, N.; Akatsu, T.; Tanaka, H.; Sasaki, T.; Nishihara, T.; Koga, T.; Martin, T.J.; Suda, T. Origin of osteoclasts: Mature monocytes and macrophages are capable of differentiating into osteoclasts under a suitable microenvironment prepared by bone marrow-derived stromal cells. Proc. Natl. Acad. Sci. USA 1990, 87, 7260–7264. [Google Scholar]
- Suda, T.; Udagawa, N.; Nakamura, I.; Miyaura, C.; Takahashi, N. Modulation of osteoclast differentiation by local factors. Bone 1995, 17 (2 Suppl.1), 87–91. [Google Scholar] [CrossRef] [PubMed]
- Udagawa, N.; Kotake, S.; Kamatani, N.; Takahashi, N.; Suda, T. The molecular mechanism of osteoclastogenesis in rheumatoid arthritis. Arthritis. Res. 2002, 4, 281–289. [Google Scholar]
- Lam, J.; Nelson, C.A.; Ross, F.P.; Teitelbaum, S.L.; Fremont, D.H. Crystal structure of the TRANCE/RANKL cytokine reveals determinants of receptor-ligand specificity. J. Clin. Invest. 2001, 108, 971–979. [Google Scholar]
- Kotake, S.; Nanke, Y.; Kawamoto, M.; Yago, T.; Udagawa, N.; Ichikawa, N.; Kobashigawa, T.; Saito, S.; Momohara, S.; Kamatani, N.; Yamanaka, H. T-cell leukemia translocation-associated gene (TCTA) protein is required for human osteoclastogenesis. Bone 2009, 45, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Baecher-Allan, C.; Brown, J.A.; Freeman, G.J.; Hafler, D.A. CD4+CD25high regulatory cells in human peripheral blood. J. Immunol. 2001, 167, 1245–1253. [Google Scholar]
- Nanke, Y.; Kotake, S.; Goto, M.; Ujihara, H.; Matsubara, M.; Kamatani, N. Decreased percentages of regulatory T cells in peripheral blood of patients with Behcet's disease before ocular attack: a possible predictive marker of ocular attack. Mod. Rheumatol. 2008, 18, 354–358. [Google Scholar]
- Testa, N.G.; Allen, T.D.; Lajtha, L.G.; Onions, D.; Jarret, O. Generation of osteoclasts in vitro. J. Cell. Sci. 1981, 47, 127–137. [Google Scholar] [PubMed]
- Ibbotson, K.J.; Roodman, G.D.; McManus, L.M.; Mundy, G.R. Identification and characterization of osteoclast-like cells and their progenitors in cultures of feline marrow mononuclear cells. J. Cell. Biol. 1984, 471–480. [Google Scholar]
- Ibbotson, K.J.; Roodman, G.D.; McManus, L.M.; Mundy, G.R. Identification and characterization of osteoclast-like cells and their progenitors in cultures of feline marrow mononuclear cells. J. Cell. Biol. 1984, 471–480. [Google Scholar]
- Takahashi, N.; Yamana, H.; Yoshiki, S.; Roodman, G.D.; Mundy, G.R.; Jones, S.J.; Boyde, A.; Suda, T. Osteoclast-like cell formation and its regulation by osteotropic hormones in mouse bone marrow cultures. Endocrinology 1988, 122, 1373–1382. [Google Scholar]
- Hattersley, G.; Chambers, T.J. Calcitonin receptors as markers for osteoclastic differentiation: correlation between generation of bone-resorptive cells and cells that express calcitonin receptors in mouse bone marrow cultures. Endocrinology 1989, 125, 1606–1612. [Google Scholar]
- Sato, K.; Fujii, Y.; Kasono, K.; Saji, M.; Tsushima, T.; Shizume, K. Stimulation of prostaglandin bone resorption by recombinant human interleukin 1 in fetal mouse bone. Biochem. Biophys. Res. Commun. 1986, 138, 618–624. [Google Scholar]
- Liu, X.H.; Kirschenbaum, A.; Yao, S.; Levine, A.C. Cross-talk between the interleukin-6 and prostaglandin E(2) signaling systems results in enhancement of osteoclastogenesis through effects on the osteoprotegerin/receptor activator of nuclear factor-κB (RANK) ligand/RANK system. Endocrinology 2005, 146, 1991–1998. [Google Scholar]
- Wani, M.R.; Fuller, K.; Kim, N.S.; Choi, Y.; Chambers, T. Prostaglandin E2 cooperates with TRANCE in osteoclast induction from hemopoietic precursors: synergistic activation of differentiation, cell spreading, and fusion. Endocrinology 1999, 140, 1927–1935. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Mizoguchi, T.; Take, I.; Kurihara, S.; Udagawa, N.; Takahashi, N. Prostaglandin E2 enhances osteoclastic differentiation of precursor cells through protein kinase A-dependent phosphorylation of TAK1. J. Biol. Chem. 2005, 280, 11395–11403. [Google Scholar]
- Han, S.Y.; Lee, N.K.; Kim, K.H.; Jang, I.W.; Yim, M.; Kim, J.H.; Lee, W.J.; Lee, S.Y. Transcriptional induction of cyclooxygenase-2 in osteoclast precursors is involved in RANKL-induced osteoclastogenesis. Blood 2005, 106, 1240–1245. [Google Scholar]
- Ono, K.; Kaneko, H.; Choudhary, S.; Pilbeam, C.C.; Lorenzo, J.A.; Akatsu, T.; Kugai, N.; Raisz, L.G. Biphasic effect of prostaglandin E2 on osteoclast formation in spleen cell cultures: role of the EP2 receptor. J. Bone. Miner. Res. 2005, 20, 23–29. [Google Scholar]
- Leonhardt, A.; Timmermanns, G.; Roth, B.; Seyberth, H.W. Calcium homeostasis and hypercalciuria in hyperprostaglandin E syndrome. J. Pediatr. 1992, 120, 546–554. [Google Scholar]
- Neale, S.D.; Fujikawa, Y.; Sabokbar, A.; Gundle, R.; Murray, D.W.; Graves, S.E.; Howie, D.W.; Athanasou, N.A. Human bone-derived cells support formation of human osteoclasts from arthroplasty-derived cells in vitro. J. Bone. Joint. Surg. Br. 2000, 82, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Tsukii, K.; Shima, N.; Mochizuki, S.; Yamaguchi, K.; Kinosaki, M.; Yano, K.; Shibata, O.; Udagawa, N.; Yasuda, H.; Suda, T.; Higashio, K. Osteoclast differentiation factor mediates an essential signal for bone resorption induced by 1 alpha, 25-dihydroxyvitamin D3, prostaglandin E2, or parathyroid hormone in the microenvironment of bone. Biochem. Biophys. Res. Commun. 1998, 246, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Nagai, M.; Sato, N. Reciprocal gene expression of osteoclastogenesis inhibitory factor and osteoclast differentiation factor regulates osteoclast formation. Biochem. Biophys. Res. Commun. 1999, 257, 719–723. [Google Scholar]
- Lader, C.S.; Flanagan, A.M. Prostaglandin E2, interleukin 1alpha, and tumor necrosis factor-alpha increase human osteoclast formation and bone resorption in vitro. Endocrinology 1998, 139, 3157–3164. [Google Scholar] [CrossRef] [PubMed]
- Itonaga, I.; Sabokbar, A.; Neale, S.D.; Athanasou, N.A. 1,25-Dihydroxyvitamin D(3) and prostaglandin E(2) act directly on circulating human osteoclast precursors. Biochem. Biophys. Res. Commun. 1999, 264, 590–595. [Google Scholar]
- Kwan Tat, S.; Pelletier, J.P.; Lajeunesse, D.; Fahmi, H.; Duval, N.; Martel-Pelletier, J. Differential modulation of RANKL isoforms by human osteoarthriticsubchondral bone osteoblasts: Influence of osteotropic factors. Bone 2008, 43, 284–291. [Google Scholar]
- Stamp, L.K.; James, M.J.; Cleland, L.G. Paracrineupregulation of monocyte cyclooxygenase-2 by mediators produced by T lymphocytes: role of interleukin 17 and interferon-gamma. J. Rheumatol. 2004, 31, 1255–1264. [Google Scholar]
- Stamp, L.K.; Cleland, L.G.; James, M.J. Upregulation of synoviocyte COX-2 through interactions with T lymphocytes: role of interleukin 17 and tumor necrosis factor-alpha. J. Rheumatol. 2004, 31, 1246–1254. [Google Scholar]
- Yago, T.; Nanke, Y.; Ichikawa, N.; Kobashigawa, T.; Mogi, M.; Kamatani, N.; Kotake, S. IL-17 induces osteoclastogenesis from human monocytes alone in the absence of osteoblasts, which is potently inhibited by anti-TNF-alpha antibody: A novel mechanism of osteoclastogenesis by IL-17. J. Cell. Biochem. 2009, 108, 947–955. [Google Scholar]
- Lam, J.; Takeshita, S.; Barker, J.E.; Kanagawa, O.; Ross, F.P.; Teitelbaum, S.L. TNF-alpha induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J. Clin. Invest. 2000, 106, 1481–1488. [Google Scholar]
- Zou, W.; Hakim, I.; Tschoep, K.; Endres, S.; Bar-Shavit, Z. Tumor necrosis factor-alpha mediates RANK ligand stimulation of osteoclast differentiation by an autocrine mechanism. J. Cell. Biochem. 2001, 83, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Carus, M.E.; Benito-Miguel, M.; Balsa, A.; Cobo-Ibanez, T.; Perez de Ayala, C.; Pascual-Salcedo, D.; Martín-Mola, E. Peripheral blood T lymphocytes from patients with early rheumatoid arthritis express RANKL and interleukin-15 on the cell surface and promote osteoclastogenesis in autologous monocytes. Arthritis. Rheum. 2006, 54, 1151–1164. [Google Scholar]
- Raza, K.; Falciani, F.; Curnow, S.J.; Ross, E.J.; Lee, C.Y.; Akbar, A.N.; Lord, J.M.; Gordon, C.; Buckley, C.D.; Salmon, M. Early rheumatoid arthritis is characterized by a distinct and transient synovial fluid cytokine profile of T cell and stromal cell origin. Arthritis. Res. Ther. 2005, 7, R784–R795. [Google Scholar]
- Kokkonen, H.; Söderström, I.; Rocklöv, J.; Hallmans, G.; Lejon, K.; RantapääDahlqvist, S. Up-regulation of cytokines and chemokines predates the onset of rheumatoid arthritis. Arthritis. Rheum. 2010, 62, 383–391. [Google Scholar]
- Genovese, M.; Van den Bosch, F.; Roberson, S.; Bojin, S.; Biagini, I.; Ryan, P.; Sloan-Lancaster, J. LY2439821, a Humanized anti-IL-17 monoclonal antibody, in the treatment of patients with rheumatoid arthritis. Arthritis. Rheum. 2010. [Epub ahead of print]. [Google Scholar]
- Yago, T.; Nanke, Y.; Kawamoto, M.; Furuya, T.; Kobashigawa, T.; Kamatani, N.; Kotake, S. IL-23 induces human osteoclastogenesis via IL-17 in vitro, and anti-IL-23 antibody attenuates collagen-induced arthritis in rats. Arthritis. Res. Ther. 2007, 9, R96. [Google Scholar] [CrossRef] [PubMed]
- Chambers, T.J.; Fuller, K.; Athanasou, N.A. The effect of prostaglandins I2, E1, E2 and dibutyryl cyclic AMP on the cytoplasmic spreading of rat osteoclasts. Br. J. Exp. Pathol. 1984, 65, 557–566. [Google Scholar] [PubMed]
- Fuller, K.; Chambers, T.J. Effect of arachidonic acid metabolites on bone resorption by isolated rat osteoclasts. J. Bone. Mines. Res. 1989, 4, 209–215. [Google Scholar]
- Sarrazin, P.; Hackett, J.A.; Fortier, I.; Gallant, M.A.; de Brum-Fernandes, A. Role of EP3 and EP4 prostaglandin receptors in reorganization of the cytoskeleton in mature human osteoclasts. J. Rheumatol. 2004, 31, 1598–1606. [Google Scholar]
- Hackett, J.A.; Allard-Chamard, H.; Sarrazin, P.; de Fatima Lucena, M.; Gallant, M.A.; Fortier, I.; Nader, M.; Parent, J.L.; Bkaily, G.; de Brum-Fernandes, A.J. Prostaglandin production by human osteoclasts in culture. J. Rheumatol. 2006, 33, 1320–1328. [Google Scholar]
- Yin, M.J.; Yamamoto, Y.; Gaynor, R.B. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(κ)B kinase-beta. Nature 1998, 396, 77–80. [Google Scholar]
- Jimi, E.; Aoki, K.; Saito, H.; D'Acquisto, F.; May, M.J.; Nakamura, I.; Sudo, T.; Kojima, T.; Okamoto, F.; Fukushima, H.; Okabe, K.; Ohya, K.; Ghosh, S. Selective inhibition of NF-kappa B blocks osteoclastogenesis and prevents inflammatory bone destruction in vivo. Nat. Med. 2004, 10, 617–624. [Google Scholar] [PubMed]
- Karakawa, A.; Fukawa, Y.; Okazaki, M.; Takahashi, K.; Sano, T.; Amano, H.; Yamamoto, M.; Yamada, S. Diclofenac sodium inhibits NFkappaB transcription in osteoclasts. J. Dent. Res. 2009, 88, 1042–1047. [Google Scholar]
- Feng, R.; Anderson, G.; Xiao, G.; Elliott, G.; Leoni, L.; Mapara, M.Y.; Roodman, G.D.; Lentzsch, S. SDX-308, a nonsteroidal anti-inflammatory agent, inhibits NF-kappaB activity, resulting in strong inhibition of osteoclast formation/activity and multiple myeloma cell growth. Blood 2007, 109, 2130–2138. [Google Scholar] [PubMed]
- Kawashima, M.; Fujikawa, Y.; Itonaga, I.; Takita, C.; Tsumura, H. The effect of selective cyclooxygenase-2 inhibitor on human osteoclast precursors to influence osteoclastogenesis in vitro. Mod. Rheumatol. 2009, 19, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Niki, Y.; Takaishi, H.; Takito, J.; Miyamoto, T.; Kosaki, N.; Matsumoto, H.; Toyama, Y.; Tada, N. Administration of cyclooxygenase-2 inhibitor reduces joint inflammation but exacerbates osteopenia in IL-1 alpha transgenic mice due to GM-CSF overproduction. J. Immunol. 2007, 179, 639–646. [Google Scholar]
- Idris, A.I.; Del Soldato, P.; Ralston, S.H.; van't Hof, R.J. The flurbiprofen derivatives HCT1026 and HCT1027 inhibit bone resorption by a mechanism independent of COX inhibition and nitric oxide production. Bone 2004, 35, 636–643. [Google Scholar]
- Idris, A.I.; Ralston, S.H.; van't Hof, R.J. The nitrosylatedflurbiprofen derivative HCT1026 inhibits cytokine-induced signalling through a novel mechanism of action. Eur. J. Pharmacol. 2009, 602, 215–222. [Google Scholar]
- Kellinsalmi, M.; Parikka, V.; Risteli, J.; Hentunen, T.; Leskelä, H.V.; Lehtonen, S.; Selander, K.; Väänänen, K.; Lehenkari, P. Inhibition of cyclooxygenase-2 down-regulates osteoclast and osteoblast differentiation and favoursadipocyte formation in vitro. Eur. J. Pharmacol. 2007, 572, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Goodman, S.B.; Ma, T.; Mitsunaga, L.; Miyanishi, K.; Genovese, M.C.; Smith, R.L. Temporal effects of a COX-2-selective NSAID on bone ingrowth. J. Biomed. Mater. Res. A. 2005, 72, 279–287. [Google Scholar]
- Vuolteenaho, K.; Moilanen, T.; Moilanen, E. Non-steroidal anti-inflammatory drugs, cyclooxygenase-2 and the bone healing process. Basic. Clin. Pharmacol. Toxicol. 2008, 102, 10–14. [Google Scholar]
- Nanke, Y.; Kotake, S.; Ninomiya, T.; Furuya, T.; Ozawa, H.; Kamatani, N. Geranylgeranylacetone inhibits formation and function of human osteoclasts and prevents bone loss in tail-suspended rats and ovariectomized rats. Calcif. Tissue. Int. 2005, 77, 376–385. [Google Scholar]
- Nanke, Y.; Kawamoto, M.; Yago, T.; Chiba, J.; Yamanaka, H.; Kotake, S. Geranylgeranylacetone, a non-toxic inducer of heat shock protein, induces cell death in fibroblast-like synoviocytes from patients with rheumatoid arthritis. Mod. Rheumatol. 2009, 19, 379–383. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kotake, S.; Yago, T.; Kawamoto, M.; Nanke, Y. Effects of NSAIDs on Differentiation and Function of Human and Murine Osteoclasts – Crucial ‘Human Osteoclastology’. Pharmaceuticals 2010, 3, 1394-1410. https://doi.org/10.3390/ph3051394
Kotake S, Yago T, Kawamoto M, Nanke Y. Effects of NSAIDs on Differentiation and Function of Human and Murine Osteoclasts – Crucial ‘Human Osteoclastology’. Pharmaceuticals. 2010; 3(5):1394-1410. https://doi.org/10.3390/ph3051394
Chicago/Turabian StyleKotake, Shigeru, Toru Yago, Manabu Kawamoto, and Yuki Nanke. 2010. "Effects of NSAIDs on Differentiation and Function of Human and Murine Osteoclasts – Crucial ‘Human Osteoclastology’" Pharmaceuticals 3, no. 5: 1394-1410. https://doi.org/10.3390/ph3051394
APA StyleKotake, S., Yago, T., Kawamoto, M., & Nanke, Y. (2010). Effects of NSAIDs on Differentiation and Function of Human and Murine Osteoclasts – Crucial ‘Human Osteoclastology’. Pharmaceuticals, 3(5), 1394-1410. https://doi.org/10.3390/ph3051394