NSAIDs, Opioids, Cannabinoids and the Control of Pain by the Central Nervous System

Abstract
:
{kind=link}
{kind=link}
1. Introduction
2. NSAIDs and Descending Inhibition of Pain
3. The Descending Pain Control System and NSAID-Induced Tolerance
4. Are the Endocannabinoids Involved?
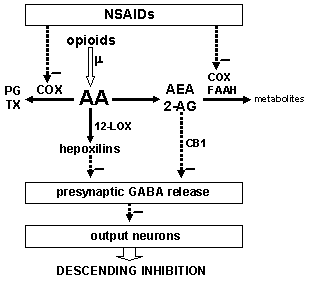
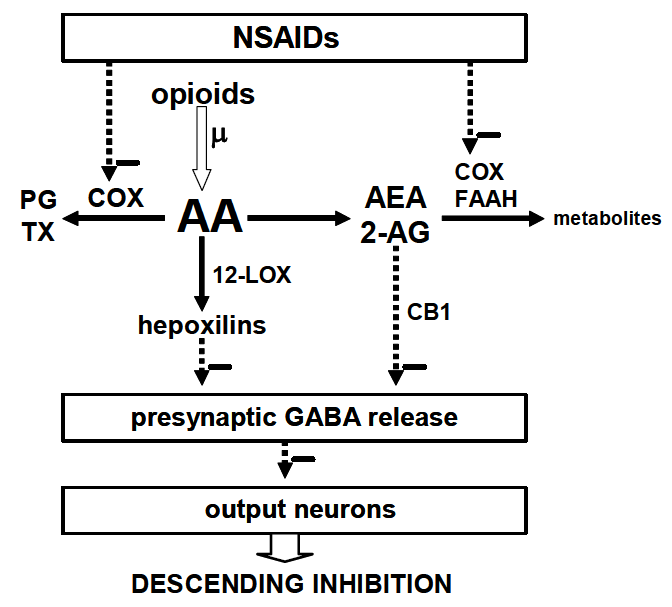
5. Possible Mechanisms of Interaction
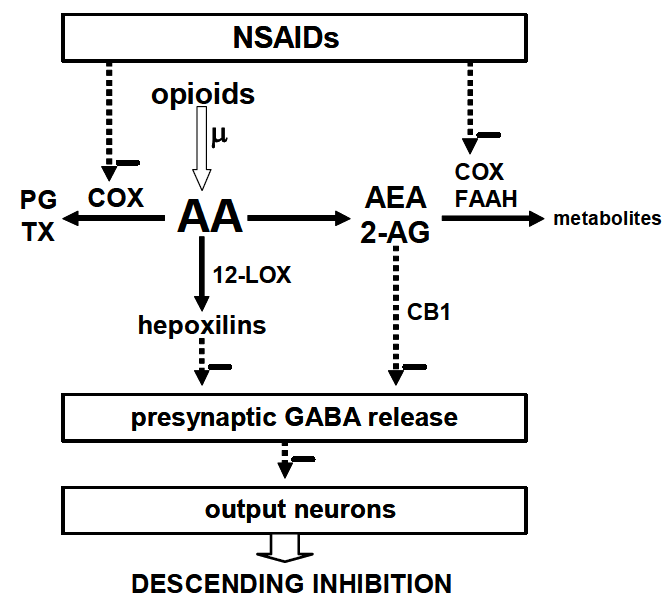
6. Conclusion
References
- Fields, H.L. Pain; McGraw-Hill: New York, NY, USA, 1987. [Google Scholar]
- Fields, H.L.; Basbaum, A.I. Brainstem control of spinal pain transmission neurons. Annu. Rev. Physiol. 1978, 40, 217–248. [Google Scholar]
- Ren, K.; Dubner, R. Enhanced descending modulation of nociception in rats with persistent hindpaw inflammation. J. Neurophysiol. 1996, 76, 3025–3037. [Google Scholar]
- Schaible, H.-G.; Neugebauer, V.; Cervero, F.; Schmidt, R.F. Changes in tonic descending inhibition of spinal neurons with articular input during the development of acute arthritis in the cat. J. Neurophysiol. 1991, 66, 1021–1032. [Google Scholar]
- Danziger, N.; Weil-Fugazza, J.; Le Bars, D.; Bouhassira, D. Stage-dependent changes in the modulation of spinal nociceptive neuronal activity during the course of inflammation. Eur. J. Neurosci. 2001, 13, 230–240. [Google Scholar]
- Morgan, M.M.; Gold, M.S.; Liebeskind, J.C.; Stein, C. Periaqueductal gray stimulation produces a spinally mediated, opioid antinociception for the inflamed hindpaw of the rat. Brain Res. 1991, 545, 17–23. [Google Scholar]
- Kovelowski, C.J.; Ossipov, M.H.; Sun, H.; Lai, J.; Malan, T.P.; Porreca, F. Supraspinal cholecystokinin may drive tonic descending facilitation mechanisms to maintain neuropathic pain in the rat. Pain 2000, 87, 265–273. [Google Scholar]
- Pertovaara, A.; Wei, H.; Hämäläinen, M.M. Lidocaine in the rostroventromedial medulla and the periaqueductal gray attenuates allodynia in neuropathic rats. Neurosci. Lett. 1996, 218, 127–130. [Google Scholar]
- Burgess, S.E.; Gardell, L.R.; Ossipov, M.H.; Malan, T.P.; Vanderah, T.W.; Lai, J.; Porreca, F. Time-dependent descending facilitation from the rostral ventromedial medulla maintains, but does not initiate, neuropathic pain. J. Neurosci. 2002, 22, 5129–5136. [Google Scholar] [PubMed]
- Vanegas, H.; Schaible, H.-G. Descending control of persistent pain: Inhibitory or facilitatory? Brain Res. Rev. 2004, 46, 295–309. [Google Scholar] [CrossRef]
- Fields, H. State-dependent opioid control of pain. Nat. Rev. Neurosci. 2004, 5, 565–575. [Google Scholar]
- Hohmann, A.G.; Suplita, R.L.; Bolton, N.M.; Neely, M.H.; Fegley, D.; Mangieri, R.; Krey, J.F.; Walker, J.M.; Holmes, P.V.; Crystal, J.D.; Duranti, A.; Tontini, A.; Mor, M.; Trazia, G.; Piomelli, D. An endocannabinoid mechanism for stress-induced analgesia. Nature 2005, 435, 1108–1112. [Google Scholar]
- Wessendorf, M.W.; Vaughan, C.W.; Vanegas, H. Rethinking the PAG and RVM: Supraspinal modulation of nociception by opioids and non-opioids. In Proceedings of the 11th World Congress on Pain; Flor, H., Kalso, E., Dostrovsky, J.O., Eds.; IASP Press: Seattle, WA, USA, 2006; pp. 311–320. [Google Scholar]
- Fields, H.L.; Vanegas, H.; Hentall, I.D.; Zorman, G. Evidence that disinhibition of brain stem neurones contributes to morphine analgesia. Nature 1983, 306, 684–686. [Google Scholar]
- Heinricher, M.M.; Morgan, M.M.; Fields, H.L. Direct and indirect actions of morphine on medullary neurons that modulate nociception. Neuroscience 1992, 48, 533–543. [Google Scholar]
- Yaksh, T.L.; Yeung, J.C.; Rudy, T.A. Systematic examination in the rat of brain sites sensitive to the direct application of morphine: Observation of differential effects within the periaqueductal gray. Brain Res. 1976, 114, 83–103. [Google Scholar]
- Bodnar, R.J.; Williams, C.L.; Lee, S.J.; Pasternak, G.W. Role of μ1-opiate receptors in supraspinal opiate analgesia: A microinjection study. Brain Res. 1988, 447, 25–34. [Google Scholar]
- Smith, D.J.; Perrotti, J.M.; Crisp, T.; Cabral, M.E.; Long, J.T.; Scalzitti, J.M. The mu opiate receptor is responsible for descending pain inhibition originating in the periaqueductal gray region of the rat brain. Eur. J. Pharmacol. 1988, 156, 47–54. [Google Scholar]
- Rossi, G.C.; Pasternak, G.W.; Bodnar, R.J. μ and δ opioid synergy between the periaqueductal gray and the rostro-ventral medulla. Brain Res. 1994, 665, 85–93. [Google Scholar]
- Fang, F.G.; Haws, C.M.; Drasner, K.; Williamson, A.; Fields, H.L. Opioid peptides (DAGO-enkephalin, dynorphin A(1-13), BAM 22P) microinjected into the rat brainstem: comparison of their antinociceptive effect and their effect on neuronal firing in the rostral ventromedial medulla. Brain Res. 1989, 501, 116–128. [Google Scholar] [PubMed]
- Brunton, L.L.; Lazo, J.S.; Parker, K.L. Goodman & Gilman's The Pharmacological Basis of Therapeutics, 11st ed.; McGraw-Hill: New York, NY, USA, 2006. [Google Scholar]
- Menassé, R.; Hedwall, P.R.; Kraetz, J.; Pericin, C.; Riesterer, L.; Sallmann, A.; Ziel, R.; Jaques, R. Pharmacological properties of diclofenac sodium and its metabolites. Scand. J. Rheumatol. 1978, 7 (Suppl. 22), 5–16. [Google Scholar]
- Vane, J.R. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nature New Biol. 1971, 231, 232–235. [Google Scholar]
- Campos, C.; de Gregorio, R.; Garcia-Nieto, R.; Gago, F.; Ortiz, P.; Alemany, S. Regulation of cyclooxygenase activity by metamizol. Eur. J. Pharmacol. 1999, 378, 339–347. [Google Scholar]
- Pierre, S.C.; Schmidt, R.; Brenneis, C.; Michaelis, M.; Geisslinger, G.; Scholich, K. Inhibition of cyclooxygenases by dipyrone. Br. J. Pharmacol. 2007, 151, 494–503. [Google Scholar]
- Björkman, R.L.; Hedner, T.; Hallman, K.M.; Henning, M.; Hedner, J. Localization of the central antinociceptive effects of diclofenac in the rat. Brain Res. 1992, 590, 66–73. [Google Scholar]
- Tortorici, V.; Vanegas, H. Putative role of medullary off- and on-cells in the antinociception produced by dipyrone (metamizol) administered systemically or microinjected into PAG. Pain 1994, 57, 197–205. [Google Scholar]
- Tortorici, V.; Vanegas, H. Antinociception induced by systemic or PAG-microinjected lysine-acetylsalicylate in rats: Effects on tail-flick related activity of medullary off- and on-cells. Eur. J. Neurosci. 1995, 7, 1857–1865. [Google Scholar]
- Jones, S.L. Dipyrone into the nucleus raphe magnus inhibits the rat nociceptive tail-flick reflex. Eur. J. Pharmacol. 1996, 318, 37–40. [Google Scholar]
- Pini, L.A.; Vitale, G.; Sandrini, M. Serotonin and opiate involvement in the antinociceptive effect of acetylsalicylic acid. Pharmacology 1997, 54, 84–91. [Google Scholar]
- Tortorici, V.; Vasquez, E.; Vanegas, H. Naloxone partial reversal of the antinociception produced by dipyrone microinjected in the periaqueductal gray of rats: Possible involvement of medullary off- and on-cells. Brain Res. 1996, 725, 106–110. [Google Scholar]
- Vazquez, E.; Hernandez, N.; Escobar, W.; Vanegas, H. Antinociception induced by intravenous dipyrone (metamizol) upon dorsal horn neurons: involvement of endogenous opioids at the periaqueductal gray matter, the nucleus raphe magnus, and the spinal cord in rats. Brain Res. 2005, 1048, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Pernia-Andrade, A.J.; Tortorici, V.; Vanegas, H. Induction of opioid tolerance by lysine-acetylsalicylate in rats. Pain 2004, 111, 191–200. [Google Scholar]
- Vanegas, H.; Tortorici, V.; Eblen-Zajjur, A.; Vasquez, E. PAG-microinjected dipyrone (metamizol) inhibits responses of spinal dorsal horn neurons to natural noxious stimulation in rats. Brain Res. 1997, 759, 171–174. [Google Scholar]
- Vazquez, E.; Escobar, W.; Ramirez, K.; Vanegas, H. A nonopioid analgesic acts upon the PAG-RVM axis to reverse inflammatory hyperalgesia. Eur. J. Neurosci. 2007, 25, 471–479. [Google Scholar]
- Vasquez, E.; Vanegas, H. The antinociceptive effect of PAG-microinjected dipyrone in rats is mediated by endogenous opioids of the rostral ventromedial medulla. Brain Res. 2000, 854, 249–252. [Google Scholar]
- Siuciak, J.A.; Advokat, C. Tolerance to morphine microinjections in the periaqueductal gray (PAG) induces tolerance to systemic, but not intrathecal morphine. Brain Res. 1987, 424, 311–319. [Google Scholar]
- Jacquet, Y.F.; Lajtha, A. The periaqueductal gray: site of morphine analgesia and tolerance as shown by 2-way cross tolerance between systemic and intracerebral injections. Brain Res. 1976, 103, 501–513. [Google Scholar]
- Tortorici, V.; Robbins, C.S.; Morgan, M.M. Tolerance to the antinociceptive effect of morphine microinjections into the ventral but not lateral-dorsal periaqueductal gray of the rat. Behav. Neurosci. 1999, 113, 833–839. [Google Scholar]
- Morgan, M.M.; Clayton, C.C.; Boyer-Quick, J.S. Differential susceptibility of the PAG and RVM to tolerance to the antinociceptive effect of morphine in the rat. Pain 2005, 113, 91–98. [Google Scholar]
- Lane, D.A.; Patel, P.A.; Morgan, M.M. Evidence for an intrinsic mechanism of antinociceptive tolerance within the ventrolateral periaqueductal gray of rats. Neuroscience 2005, 135, 227–234. [Google Scholar]
- Tsiklauri, N.; Viatchenko-Karpinski, V.; Voitenko, N.; Tsagareli, M.G. Non-opioid tolerance in juvenile and adult rats. Eur. J. Pharmacol. 2010, 629, 68–72. [Google Scholar]
- Tortorici, V.; Vanegas, H. Opioid tolerance induced by metamizol (dipyrone) microinjections into the periaqueductal gray of rats. Eur. J. Neurosci. 2000, 12, 4074–4080. [Google Scholar]
- Rainsford, K.D. Ibuprofen: Pharmacology, efficacy and safety. Immunopharmacology 2009, 17, 275–342. [Google Scholar]
- Fuh, J.-L.; Wang, S.-J.; Lu, S.-R.; Juang, K.-D. Does medication overuse headache represent a behavior of dependence? Pain 2005, 119, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Pullar, T.; Myall, O.; Haigh, J.R.M.; Lowe, J.R.; Dixon, J.S.; Bird, H.A. The effect of indomethacin on the psychomotor function of patients with rheumatic disease. Br. J. Rheumatol. 1988, 27, 227–229. [Google Scholar]
- Roberts, M.S.; Owen, S.G.; Friesen, W.T.; Francis, H.; Flux, W. Community Surveillance Study—Perceived response of rheumatoid arthritis patients to NSAIDs: In Non-steroidal Anti-inflammatory Drugs. Basis for Variability in Response. Agents Actions Suppl. 1985, 17, 41–54. [Google Scholar] [PubMed]
- Limmroth, V.; Katsarava, Z. Medication overuse headache. Curr. Opin. Neurol. 2004, 17, 301–306. [Google Scholar]
- Walker, J.S.; Levy, G. Effect of multiple dosing on the analgesic action of diflunisal in rats. Life Sci. 1990, 46, 737–742. [Google Scholar]
- Walker, J.S. NSAID: An update on their analgesic effects. Clin. Exp. Pharmacol. Physiol. 1995, 22, 855–860. [Google Scholar]
- Wiesenfeld-Hallin, Z.; Alster, P.; Grass, S.; Hoffmann, O.; de Araujo Lucas, G.; Plesan, A.; Xu, X.-J. Opioid sensitivity in antinociception: role of anti-opioid systems with emphasis on cholecystokinin and NMDA receptors. Prog. Pain Res. Manage. 1999, 14, 237–252. [Google Scholar]
- Watkins, L.R.; Kinscheck, I.B.; Mayer, D.J. Potentiation of opiate analgesia and apparent reversal of morphine tolerance by proglumide. Science 1984, 224, 395–396. [Google Scholar]
- Dourish, C.T.; O'Neill, M.F.; Coughlan, J.; Kitchener, S.J.; Hawley, D.; Iversen, S.D. The selective CCK-B receptor antagonist L-365,260 enhances morphine analgesia and prevents morphine tolerance in the rat. Eur. J. Pharmacol. 1990, 176, 35–44. [Google Scholar]
- Xu, X.-J.; Wiesenfeld-Hallin, Z.; Hughes, J.; Horwell, D.C.; Hökfelt, T. CI988, a selective antagonist of cholecystokininB receptors, prevents morphine tolerance in the rat. Br. J. Pharmacol. 1992, 105, 591–596. [Google Scholar] [PubMed]
- Hoffmann, O.; Wiesenfeld-Hallin, Z. The CCK-B receptor antagonist CI 988 reverses tolerance to morphine in rats. Neuroreport 1994, 5, 2565–2568. [Google Scholar]
- Tortorici, V.; Nogueira, L.; Salas, R.; Vanegas, H. Involvement of local cholecystokinin in the tolerance induced by morphine microinjections into the periaqueductal gray of rats. Pain 2003, 102, 9–16. [Google Scholar]
- Tortorici, V.; Nogueira, L.; Aponte, Y.; Vanegas, H. Involvement of cholecystokinin in the opioid tolerance induced by dipyrone (metamizole) microinjections into the periaqueductal gray matter of rats. Pain 2004, 112, 113–120. [Google Scholar]
- Tortorici, V.; Morgan, M.M.; Vanegas, H. Tolerance to repeated microinjection of morphine into the periaqueductal gray is associated with changes in the behavior of off- and on-cells in the rostral ventromedial medulla. Pain 2001, 89, 237–244. [Google Scholar]
- Tortorici, V.; Aponte, Y.; Acevedo, H.; Nogueira, L.; Vanegas, H. Tolerance to non-opioid analgesics in PAG involves unresponsiveness of medullary pain-modulating neurons in male rats. Eur. J. Neurosci. 2009, 29, 1188–1196. [Google Scholar]
- Fowler, C.J.; Holt, S.C.; Nilsson, O.; Jonsson, K.O.; Tiger, G.; Jacobsson, S.O.P. The endocannabinoid signaling system: Pharmacological and therapeutic aspects. Pharmacol. Biochem. Behav. 2005, 81, 248–262. [Google Scholar]
- Piomelli, D. The molecular logic of endocannabinoid signalling. Nat. Rev. Neurosci. 2003, 4, 873–884. [Google Scholar]
- Ryberg, E.; Larsson, N.; Sjögren, S.; Hjort, S.; Hermansson, N.-O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2007, 152, 1092–1101. [Google Scholar]
- Staton, P.C.; Hatcher, J.P.; Walker, D.J.; Morrison, A.D.; Shapland, E.M.; Hughes, J.P.; Chong, E.; Mander, P.K.; Green, P.J.; Billinton, A.; Fulleylove, M.; Lancaster, H.C.; Smith, J.C.; Bailey, L.T.; Wise, A.; Brown, A.J.; Richardson, J.C.; Chessell, I.P. The putative cannabinoid receptor GPR55 plays a role in mechanical hyperalgesia associated with inflammatory and neuropathic pain. Pain 2008, 139, 225–236. [Google Scholar]
- Lichtman, A.H.; Martin, B.R. Spinal and supraspinal components of cannabinoid-induced antinociception. J. Pharmacol. Exp. Ther. 1991, 258, 517–523. [Google Scholar]
- Lichtman, A.H.; Cook, S.A.; Martin, B.R. Investigation of brain sites mediating cannabinoid-induced antinociception in rats: Evidence supporting periaqueductal gray involvement. J. Pharmacol. Exp. Ther. 1996, 276, 585–593. [Google Scholar]
- Martin, W.J.; Tsou, K.; Walker, J.M. Cannabinoid receptor-mediated inhibition of the rat tail-flick reflex after microinjection into the rostral ventromedial medulla. Neurosci. Lett. 1998, 242, 33–36. [Google Scholar]
- Suplita, R.L.; Farthing, J.N.; Gutierrez, T.; Hohmann, A.G. Inhibition of fatty-acid amide hydrolase enhances cannabinoid stress-induced analgesia: sites of action in the dorsolateral periaqueductal gray and rostral ventromedial medulla. Neuropharmacology 2005, 49, 1201–1209. [Google Scholar]
- Meng, I.A.; Manning, B.H.; Martin, W.J.; Fields, H.L. An analgesia circuit activated by cannabinoids. Nature 1998, 395, 381–383. [Google Scholar]
- Ottani, A.; Leone, S.; Sandrini, M.; Ferrari, A.; Bertolini, A. The analgesic activity of paracetamol is prevented by the blockade of cannabinoid CB1 receptors. Eur. J. Pharmacol. 2006, 531, 280–281. [Google Scholar]
- Naidu, P.S.; Booker, L.; Cravatt, B.F.; Lichtman, A.H. Synergy between enzyme inhibitors of fatty acid amide hydrolase and cyclooxygenase in visceral nociception. J. Pharmacol. Exp. Ther. 2009, 329, 48–56. [Google Scholar]
- Mallet, C.; Daulhac, L.; Bonnefont, J.; Ledent, C.; Etienne, M.; Chapuy, E.; Libert, F.; Eschalier, A. Endocannabinoid and serotonergic systems are needed for acetaminophen-induced analgesia. Pain 2008, 139, 190–200. [Google Scholar]
- Guindon, J.; De Lean, A.; Beaulieu, P. Local interaction between anandamide, an endocannabinoid, and ibuprofen, a nonsteroidal anti-inflammatory drug, in acute inflammatory pain. Pain 2006, 121, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Guindon, J.; LoVerme, J.; De Lean, A.; Piomelli, D.; Beaulieu, P. Synergistic antinociceptive effects of anandamide, an endocannabinoid, and nonsteroidal anti-inflammatory drugs in peripheral tissue: A role for endogenous fatty-acid ethanolamides? Eu. J. Pharmacol. 2006, 550, 68–77. [Google Scholar] [CrossRef]
- Gühring, H.; Hamza, M.; Sergejeva, M.; Ates, M.; Kotalla, C.E.; Ledent, K.; Brune, K. A role for endocannabinoids in indomethacin-induced spinal antinociception. Eur. J. Pharmacol. 2002, 454, 153–163. [Google Scholar]
- Telleria-Diaz, A.; Schmidt, M.; Kreusch, S.; Neubert, A.K.; Schache, F.; Vazquez, E.; Vanegas, H.; Schaible, H.G.; Ebersberger, A. Spinal antinociceptive effects of cyclooxygenase inhibition during inflammation: Involvement of prostaglandins and endocannabinoids. Pain 2009, 148, 26–35. [Google Scholar]
- Vazquez-Rodriguez, E.; Escobar, W.; Ramirez, K.; Avila, C.; Vanegas, H. The antihyperalgesic effect of PAG-microinjected metamizol is mediated by endocannabinoids in the PAG-RVM axis. In 12th World Congress on Pain; IASP Press: Seattle, WA, USA, 2008. [Google Scholar]
- Vanegas, H.; Schaible, H.-G. Prostaglandins and cyclooxygenases in the spinal cord. Prog. Neurobiol. 2001, 64, 327–363. [Google Scholar]
- Hamza, M.; Dionne, R.A. Mechanisms of non-opioid analgesics beyond cyclooxygenase enzyme inhibition. Curr. Mol. Pharmacol. 2009, 9, 1–14. [Google Scholar]
- Vaughan, C.W.; Ingram, S.L.; Connor, M.A.; Christie, M.J. How opioids inhibit GABA-mediated neurotransmission. Nature 1997, 390, 611–614. [Google Scholar]
- Vaughan, C.W.; Christie, M.J. Presynaptic inhibitory action of opioids on synaptic transmission in the rat periaqueductal grey in vitro. J. Physiol. 1997, 498, 463–472. [Google Scholar] [PubMed]
- Vaughan, C.W. Enhancement of opioid inhibition of GABAergic synaptic transmission by cyclo-oxygenase inhibitors in rat periaqueductal grey neurones. Br. J. Pharmacol. 1998, 123, 1479–1481. [Google Scholar]
- Moreau, J.-L.; Fields, H.L. Evidence for GABA involvement in midbrain control of medullary neurons that modulate nociceptive transmission. Brain Res. 1986, 397, 37–46. [Google Scholar]
- Heinricher, M.M.; Tortorici, V. Interference with GABA transmission in the rostral ventromedial medulla: disinhibition of off-cells as a central mechanism in nociceptive modulation. Neuroscience 1994, 63, 533–546. [Google Scholar]
- Kim, J.; Alger, B.E. Inhibition of cyclooxygenase-2 potentiates retrograde endocannabinoid effects in hippocampus. Nat. Neurosci. 2004, 7, 697–698. [Google Scholar]
- Kozak, K.R.; Rowlinson, S.W.; Marnett, L.J. Oxygenation of the endocannabinoid, 2-arachidonylglycerol, to glyceryl prostaglandins by cyclooxygenase-2. J. Biol. Chem. 2000, 275, 33744–33749. [Google Scholar] [PubMed]
- Kozak, K.R.; Crews, B.C.; Morrow, J.D.; Wang, L.-H.; Ma, Y.H.; Weinander, R.; Jakobsson, P.-J.; Marnett, L.J. Metabolism of the endocannabinoids, 2-arachidonylglycerol and anandamide, into prostaglandin, thromboxane, and prostacyclin glycerol esters and ethanolamides. J. Biol. Chem. 2002, 277, 44877–44885. [Google Scholar] [PubMed]
- Vaughan, C.W.; McGregor, I.S.; Christie, M.J. Cannabinoid receptor activation inhibits GABAergic neurotransmission in rostral ventromedial medulla neurons in vitro. Br. J. Pharmacol. 1999, 127, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, C.W.; Connor, M.; Bagley, E.E.; Christie, M.J. Actions of cannabinoids on membrane properties and synaptic transmission in rat periaqueductal gray neurons in vitro. Mol. Pharmacol. 2000, 57, 288–295. [Google Scholar] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Vanegas, H.; Vazquez, E.; Tortorici, V. NSAIDs, Opioids, Cannabinoids and the Control of Pain by the Central Nervous System. Pharmaceuticals 2010, 3, 1335-1347. https://doi.org/10.3390/ph3051335
Vanegas H, Vazquez E, Tortorici V. NSAIDs, Opioids, Cannabinoids and the Control of Pain by the Central Nervous System. Pharmaceuticals. 2010; 3(5):1335-1347. https://doi.org/10.3390/ph3051335
Chicago/Turabian StyleVanegas, Horacio, Enrique Vazquez, and Victor Tortorici. 2010. "NSAIDs, Opioids, Cannabinoids and the Control of Pain by the Central Nervous System" Pharmaceuticals 3, no. 5: 1335-1347. https://doi.org/10.3390/ph3051335
APA StyleVanegas, H., Vazquez, E., & Tortorici, V. (2010). NSAIDs, Opioids, Cannabinoids and the Control of Pain by the Central Nervous System. Pharmaceuticals, 3(5), 1335-1347. https://doi.org/10.3390/ph3051335