
SMYD5-BRD4 Interaction Drives Hepatocellular Carcinoma Progression: A Combined in Silico and Experimental Analysis

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
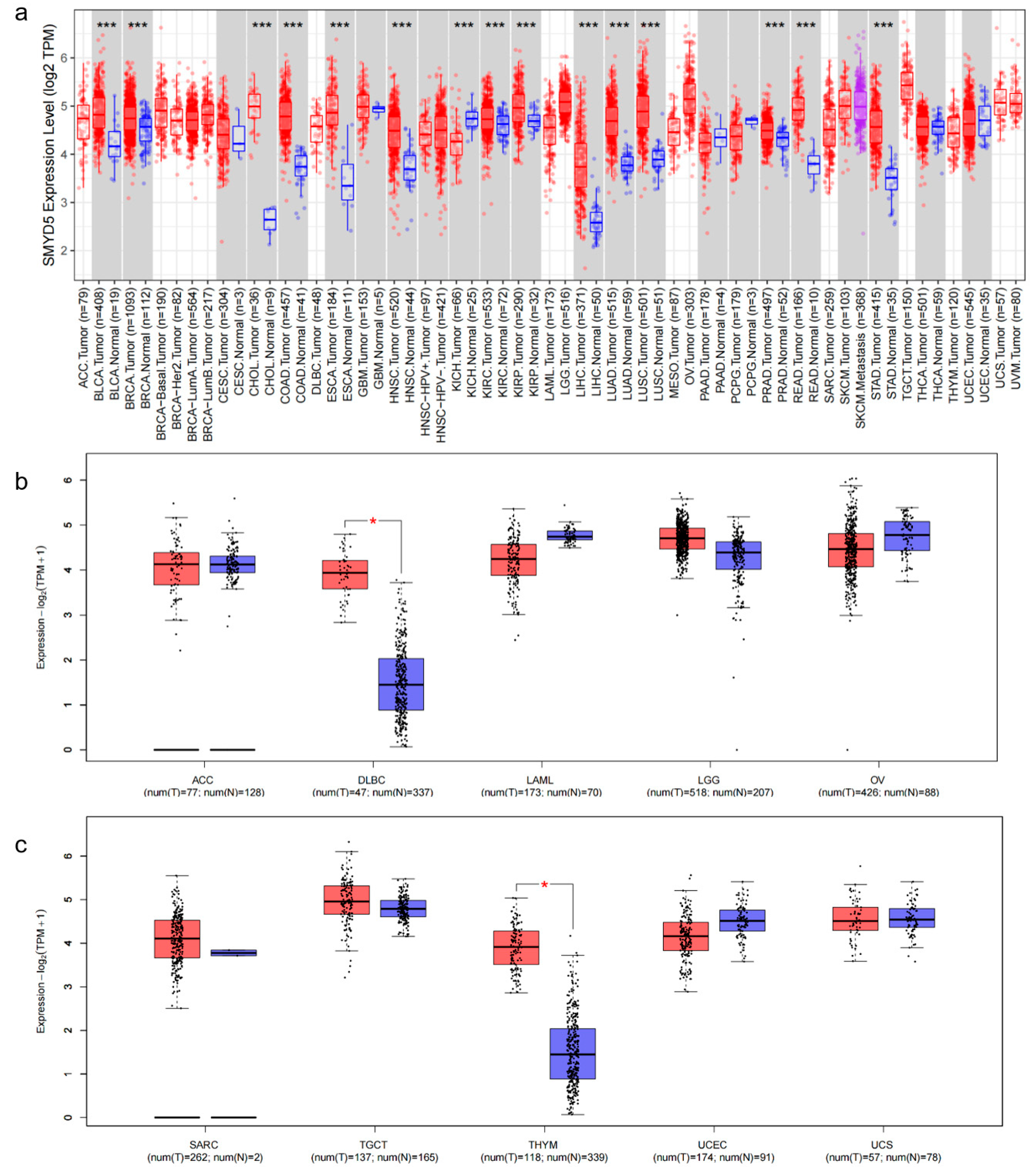
2.1. Analysis of SMYD5 Expression Across Cancer Types
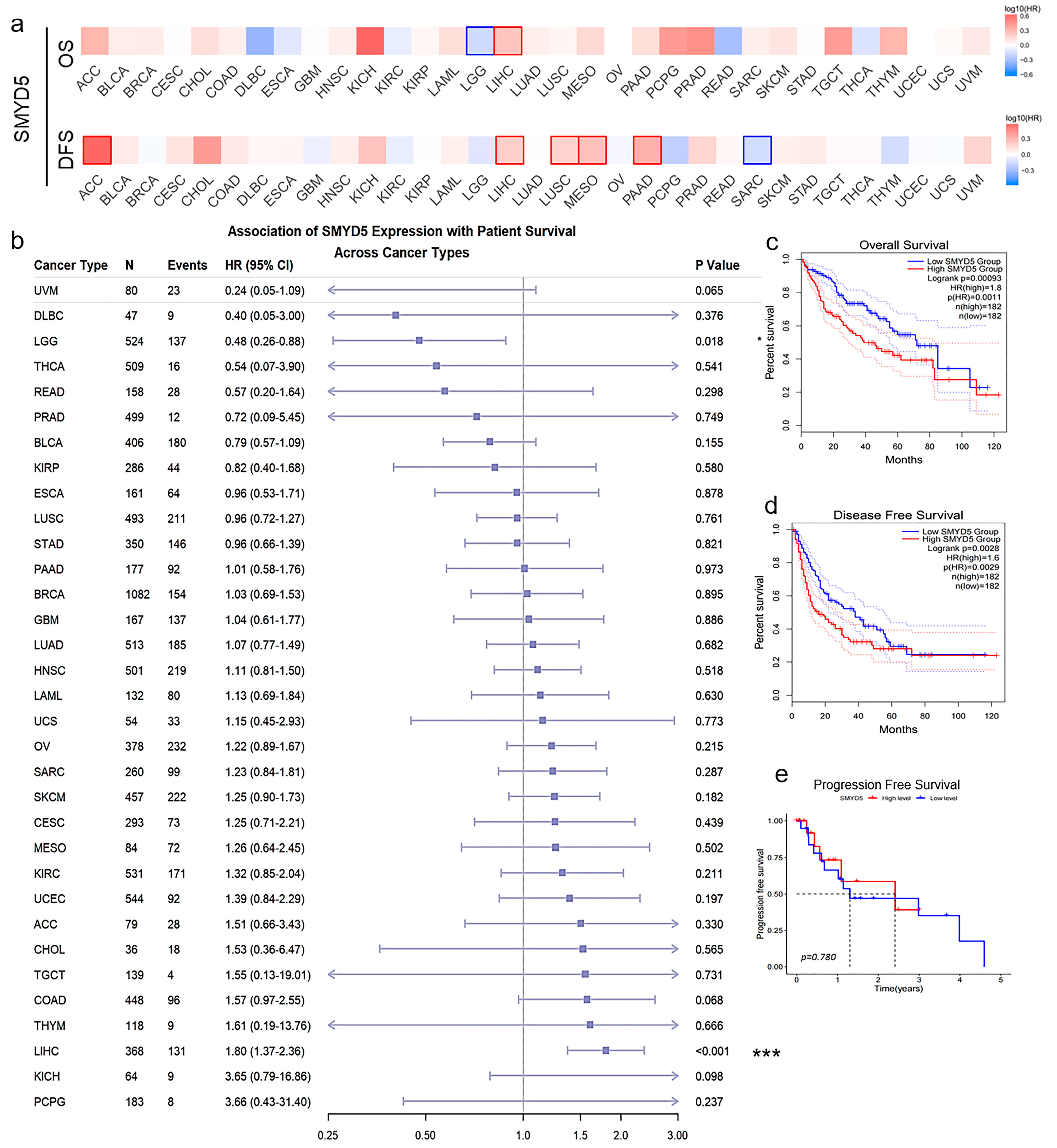
2.2. Elevated SMYD5 Levels Correlate with Poor Prognosis in LIHC Patients
2.3. Role of SMYD5 in Tumor Immune Microenvironment in LIHC
2.4. Characterization of SMYD5-Related Pathways in LIHC
2.5. SMYD5 Knockdown Inhibits Proliferation and Promotes Apoptosis in LIHC Cells
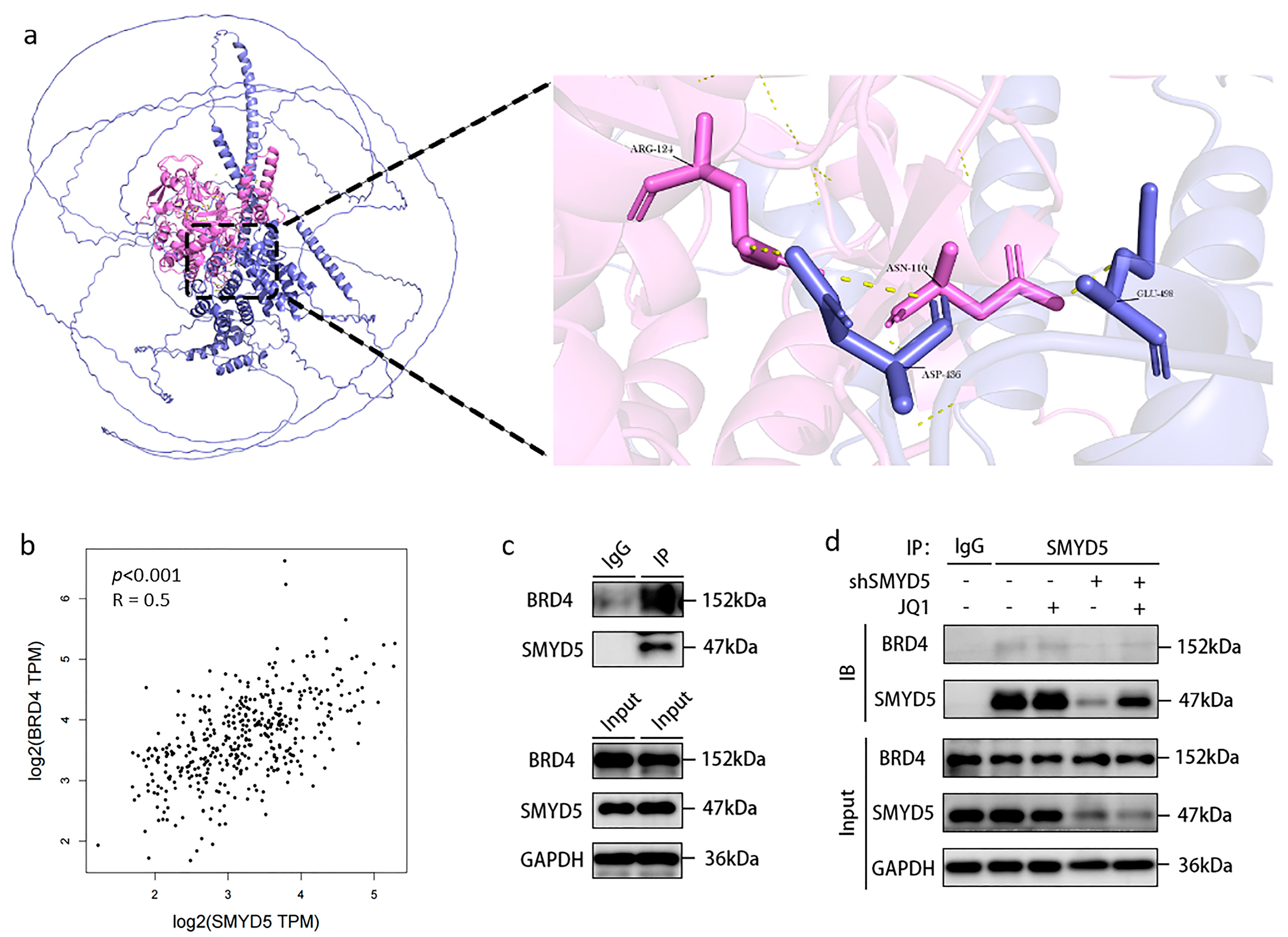
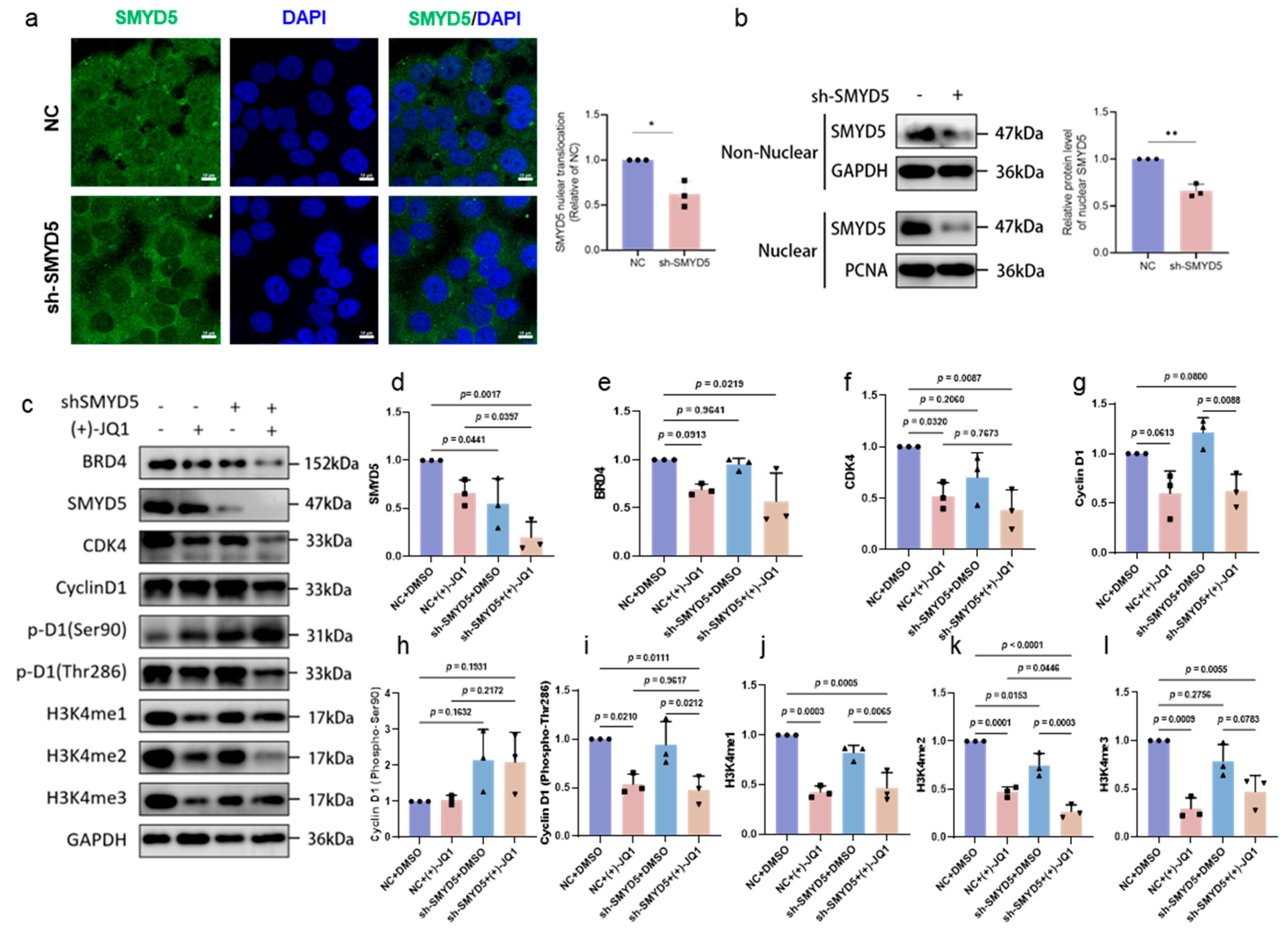
2.6. Interaction Between SMYD5 and BRD4 in LIHC
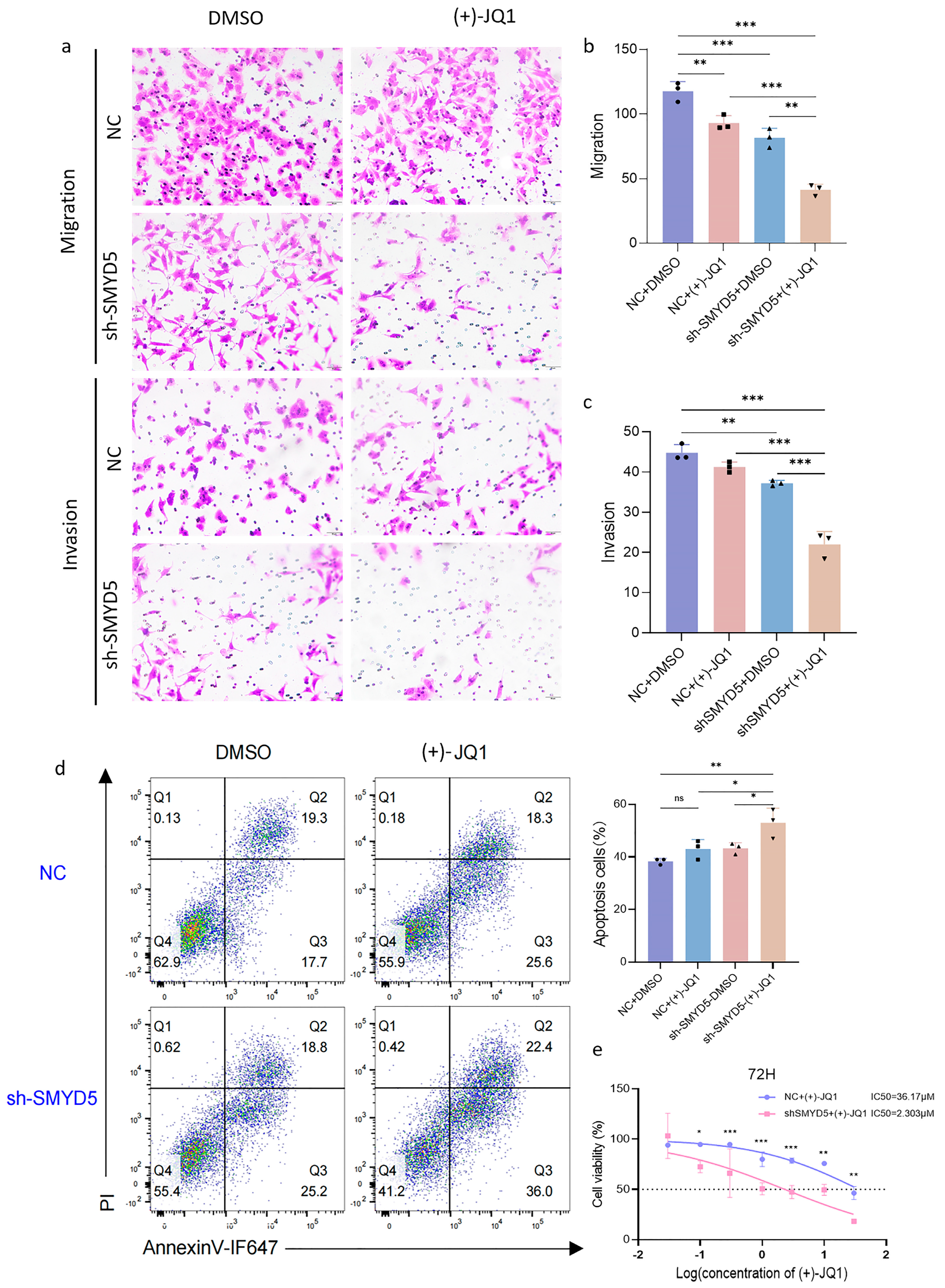
2.7. Targeting SMYD5 Enhances BRD4 Inhibition Efficacy in Suppressing LIHC Migration and Promoting Apoptosis
2.8. Impact of SMYD5 and BRD4 Targeting on Cell Cycle Regulators and Methylation Modification in Lihc Progression
3. Discussion
4. Materials and Methods
4.1. SMYD5 Pan-Cancer Analysis
4.2. SMYD5 Single-Gene Analysis in LIHC
4.3. PPI Network Development
4.4. Cell Culture
4.5. shRNAs and Plasmid Extraction
4.6. Cell Transfection to Establish SMYD5 Knockdown Cell Lines
4.7. Colony Formation Assay
4.8. Acquiring Protein Lysate
4.9. Transwell Assay
4.10. MTT Assay
4.11. Western Blot
4.12. Co-IP Assay
4.13. Flow Cytometry
4.14. Immunofluorescence for SMYD5 Nucleus Translocation
4.15. Nucleus Protein Extraction
4.16. Image Processing
4.17. Statistical Analysis
4.18. Antibodies in This Research
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yu, Z.; Bai, X.; Zhou, R.; Ruan, G.; Guo, M.; Han, W.; Jiang, S.; Yang, H. Differences in the incidence and mortality of digestive cancer between Global Cancer Observatory 2020 and Global Burden of Disease 2019. Int. J. Cancer 2024, 154, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Yang, X.-R.; Chung, W.-Y.; Dennison, A.R.; Zhou, J. Targeted therapy for hepatocellular carcinoma. Signal Transduct. Target. Ther. 2020, 5, 146. [Google Scholar] [CrossRef]
- Mao, X.; Cheung, K.S.; Peng, C.; Mak, L.Y.; Cheng, H.M.; Fung, J.; Peleg, N.; Leung, H.H.; Kumar, R.; Lee, J.H.; et al. Steatosis, HBV-related HCC, cirrhosis, and HBsAg seroclearance: A systematic review and meta-analysis. Hepatology 2023, 77, 1735–1745. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Pan, Y.; Kong, R.; Shu, S. Therapy of Primary Liver Cancer. Innovation 2020, 1, 100032. [Google Scholar] [CrossRef] [PubMed]
- Miao, B.; Ge, L.; He, C.; Wang, X.; Wu, J.; Li, X.; Chen, K.; Wan, J.; Xing, S.; Ren, L.; et al. SMYD5 is a ribosomal methyltransferase that catalyzes RPL40 lysine methylation to enhance translation output and promote hepatocellular carcinoma. Cell Res. 2024, 34, 648–660. [Google Scholar] [CrossRef] [PubMed]
- Robichaud, N.; Sonenberg, N.; Ruggero, D.; Schneider, R.J. Translational Control in Cancer. Cold Spring Harb. Perspect. Biol. 2019, 11, a032896. [Google Scholar] [CrossRef] [PubMed]
- Kovalski, J.R.; Kuzuoglu-Ozturk, D.; Ruggero, D. Protein synthesis control in cancer: Selectivity and therapeutic targeting. EMBO J. 2022, 41, e109823. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Wu, J.; Szkop, K.J.; Jeong, J.; Jovanovic, P.; Husmann, D.; Flores, N.M.; Francis, J.W.; Chen, Y.-J.C.; Benitez, A.M.; et al. SMYD5 methylation of rpL40 links ribosomal output to gastric cancer. Nature 2024, 632, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fang, Y.; Tang, Y.; Han, S.; Jia, J.; Wan, X.; Chen, J.; Yuan, Y.; Zhao, B.; Fang, D. SMYD5 catalyzes histone H3 lysine 36 trimethylation at promoters. Nat. Commun. 2022, 13, 3190. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Postel-Vinay, M.C.; Finidori, J. Growth hormone receptor: Structure and signal transduction. Eur. J. Endocrinol. 1995, 133, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, P.A.; Kopchick, J.J.; Thorgeirsson, S.; LeRoith, D.; Yakar, S. Role of Growth Hormone (GH) in Liver Regeneration. Endocrinology 2004, 145, 4748–4755. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.J.; Lu, L.; Leung, K.H.; Wong, C.C.L.; Peng, C.; Yan, S.C.; Ma, D.L.; Cai, Z.; David Wang, H.M.; Leung, C.H. An iridium(iii)-based irreversible protein-protein interaction inhibitor of BRD4 as a potent anticancer agent. Chem. Sci. 2015, 6, 5400–5408. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Prakash, C.; Sum, C.; Gong, Y.; Li, Y.; Kwok, J.J.; Thiessen, N.; Pettersson, S.; Jones, S.J.; Knapp, S.; et al. Bromodomain-containing protein 4 (BRD4) regulates RNA polymerase II serine 2 phosphorylation in human CD4+ T cells. J. Biol. Chem. 2012, 287, 43137–43155. [Google Scholar] [CrossRef] [PubMed]
- Konuma, T.; Yu, D.; Zhao, C.; Ju, Y.; Sharma, R.; Ren, C.; Zhang, Q.; Zhou, M.M.; Zeng, L. Structural Mechanism of the Oxygenase JMJD6 Recognition by the Extraterminal (ET) Domain of BRD4. Sci. Rep. 2017, 7, 16272. [Google Scholar] [CrossRef] [PubMed]
- Olley, G.; Ansari, M.; Bengani, H.; Grimes, G.R.; Rhodes, J.; von Kriegsheim, A.; Blatnik, A.; Stewart, F.J.; Wakeling, E.; Carroll, N.; et al. BRD4 interacts with NIPBL and BRD4 is mutated in a Cornelia de Lange-like syndrome. Nat. Genet. 2018, 50, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Lee, A.Y.; Lai, H.T.; Zhang, H.; Chiang, C.M. Phospho switch triggers Brd4 chromatin binding and activator recruitment for gene-specific targeting. Mol. Cell 2013, 49, 843–857. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Deng, B. Hepatocellular carcinoma: Molecular mechanism, targeted therapy, and biomarkers. Cancer Metastasis Rev. 2023, 42, 629–652. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Chi, G.; Pei, J.; Li, X.; Li, X.; Pang, H.; Cui, J.; Wu, D.; Qu, G.; He, Y. SMYD5 acts as a potential biomarker for hepatocellular carcinoma. Exp. Cell Res. 2022, 414, 113076. [Google Scholar] [CrossRef] [PubMed]
- Rueda-Robles, A.; Audano, M.; Álvarez-Mercado, A.I.; Rubio-Tomás, T. Functions of SMYD proteins in biological processes: What do we know? An updated review. Arch. Biochem. Biophys. 2021, 712, 109040. [Google Scholar] [CrossRef] [PubMed]
- Oliveira-Santos, W.; Rabello, D.A.; Lucena-Araujo, A.R.; de Oliveira, F.M.; Rego, E.M.; Pittella Silva, F.; Saldanha-Araujo, F. Residual expression of SMYD2 and SMYD3 is associated with the acquisition of complex karyotype in chronic lymphocytic leukemia. Tumor Biol. 2016, 37, 9473–9481. [Google Scholar] [CrossRef] [PubMed]
- Fiskus, W.; Mill, C.P.; Birdwell, C.; Lara, B.H.; Bose, P.; Masarova, L.; Kadia, T.M.; Khoury, J.D.; Manshouri, T.; Verstovsek, S.; et al. Pre-Clinical Efficacy of Co-Targeting GFI1/KDM1A and BRD4 or JAK1/2 Against AML and Post-MPN Secondary AML Blast Progenitor Cells. Blood 2020, 136, 27. [Google Scholar] [CrossRef]
- Qian, H.; Zhu, M.; Tan, X.; Zhang, Y.; Liu, X.; Yang, L. Super-enhancers and the super-enhancer reader BRD4: Tumorigenic factors and therapeutic targets. Cell Death Discov. 2023, 9, 470. [Google Scholar] [CrossRef] [PubMed]
- Olarerin-George, A.O.; Jaffrey, S.R. MetaPlotR: A Perl/R pipeline for plotting metagenes of nucleotide modifications and other transcriptomic sites. Bioinformatics 2017, 33, 1563–1564. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Yang, Q.; Li, L.; Wei, Y.; Zhang, C.; Du, H.; Ren, G.; Li, H. Novel prognostic biomarker TBC1D1 is associated with immunotherapy resistance in gliomas. Front. Immunol. 2024, 15, 1372113. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Lin, D.; Zhang, W.; Chen, S.; Zhen, Y.; Akkouche, S.; Liang, X.; Chong, C.M.; Zhong, H.J. AMBRA1 drives gastric cancer progression through regulation of tumor plasticity. Front. Immunol. 2024, 15, 1494364. [Google Scholar] [CrossRef] [PubMed]
- Colwill, K.; Gräslund, S. A roadmap to generate renewable protein binders to the human proteome. Nat. Methods 2011, 8, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.Y.; Baek, S.; Cha, J.; Yang, S.; Kim, E.; Marcotte, E.M.; Hart, T.; Lee, I. HumanNet v3: An improved database of human gene networks for disease research. Nucleic Acids Res. 2022, 50, D632–D639. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, M.; Chen, S.; Zhen, Y.; Wang, X.; Zhong, Y.; Liang, X.; Chong, C.-M.; Zhong, H.-J. SMYD5-BRD4 Interaction Drives Hepatocellular Carcinoma Progression: A Combined in Silico and Experimental Analysis. Pharmaceuticals 2025, 18, 1105. https://doi.org/10.3390/ph18081105
Hu M, Chen S, Zhen Y, Wang X, Zhong Y, Liang X, Chong C-M, Zhong H-J. SMYD5-BRD4 Interaction Drives Hepatocellular Carcinoma Progression: A Combined in Silico and Experimental Analysis. Pharmaceuticals. 2025; 18(8):1105. https://doi.org/10.3390/ph18081105
Chicago/Turabian StyleHu, Mingye, Shiji Chen, Yumiao Zhen, Xin Wang, Yiwen Zhong, Xiaoxu Liang, Cheong-Meng Chong, and Hai-Jing Zhong. 2025. "SMYD5-BRD4 Interaction Drives Hepatocellular Carcinoma Progression: A Combined in Silico and Experimental Analysis" Pharmaceuticals 18, no. 8: 1105. https://doi.org/10.3390/ph18081105
APA StyleHu, M., Chen, S., Zhen, Y., Wang, X., Zhong, Y., Liang, X., Chong, C.-M., & Zhong, H.-J. (2025). SMYD5-BRD4 Interaction Drives Hepatocellular Carcinoma Progression: A Combined in Silico and Experimental Analysis. Pharmaceuticals, 18(8), 1105. https://doi.org/10.3390/ph18081105