
Computationally Guided Design, Synthesis, and Evaluation of Novel Non-Hydroxamic Histone Deacetylase Inhibitors, Based on N-Trifluoroacetamide as a Zinc-Binding Group, Against Breast Cancer

 , and
, and 
Abstract

1. Introduction
2. Results and Discussion
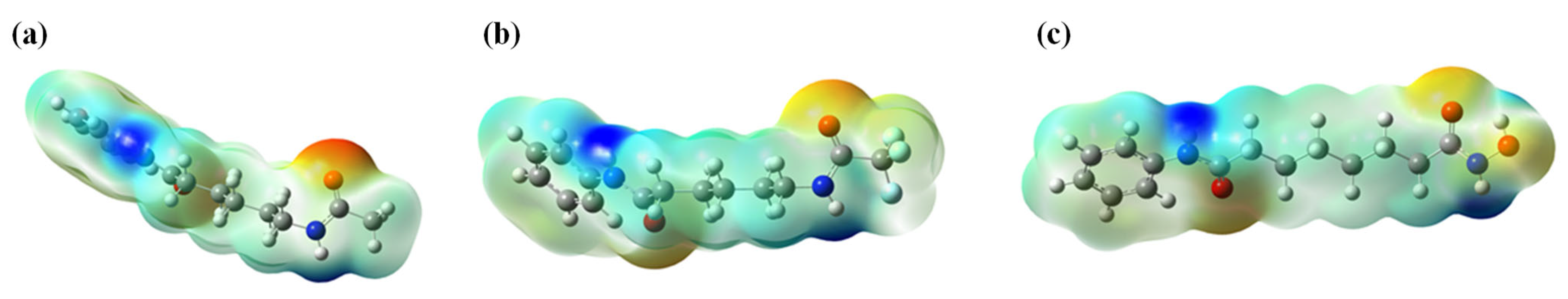
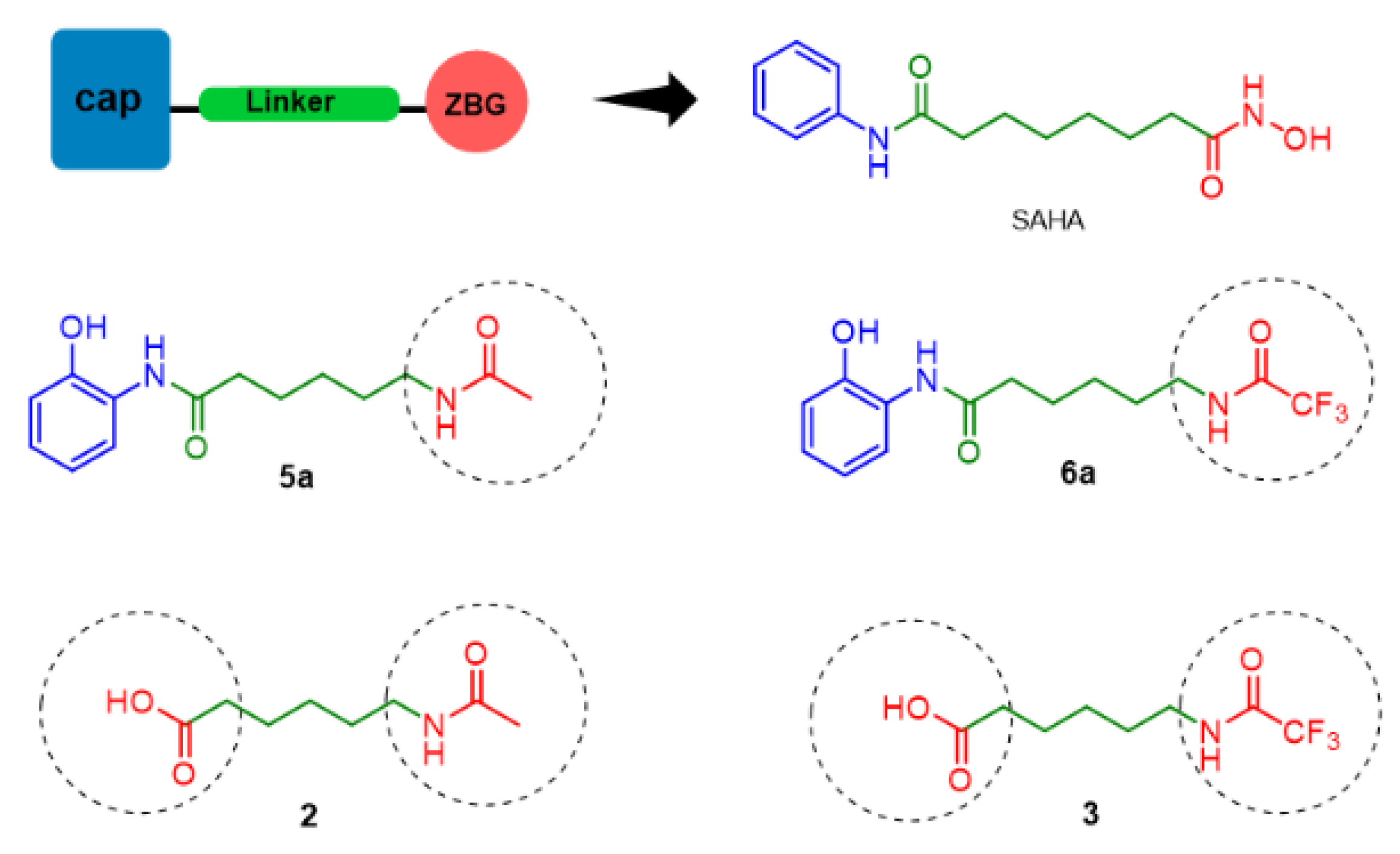
2.1. Rational Molecular Design
2.2. Computational Design
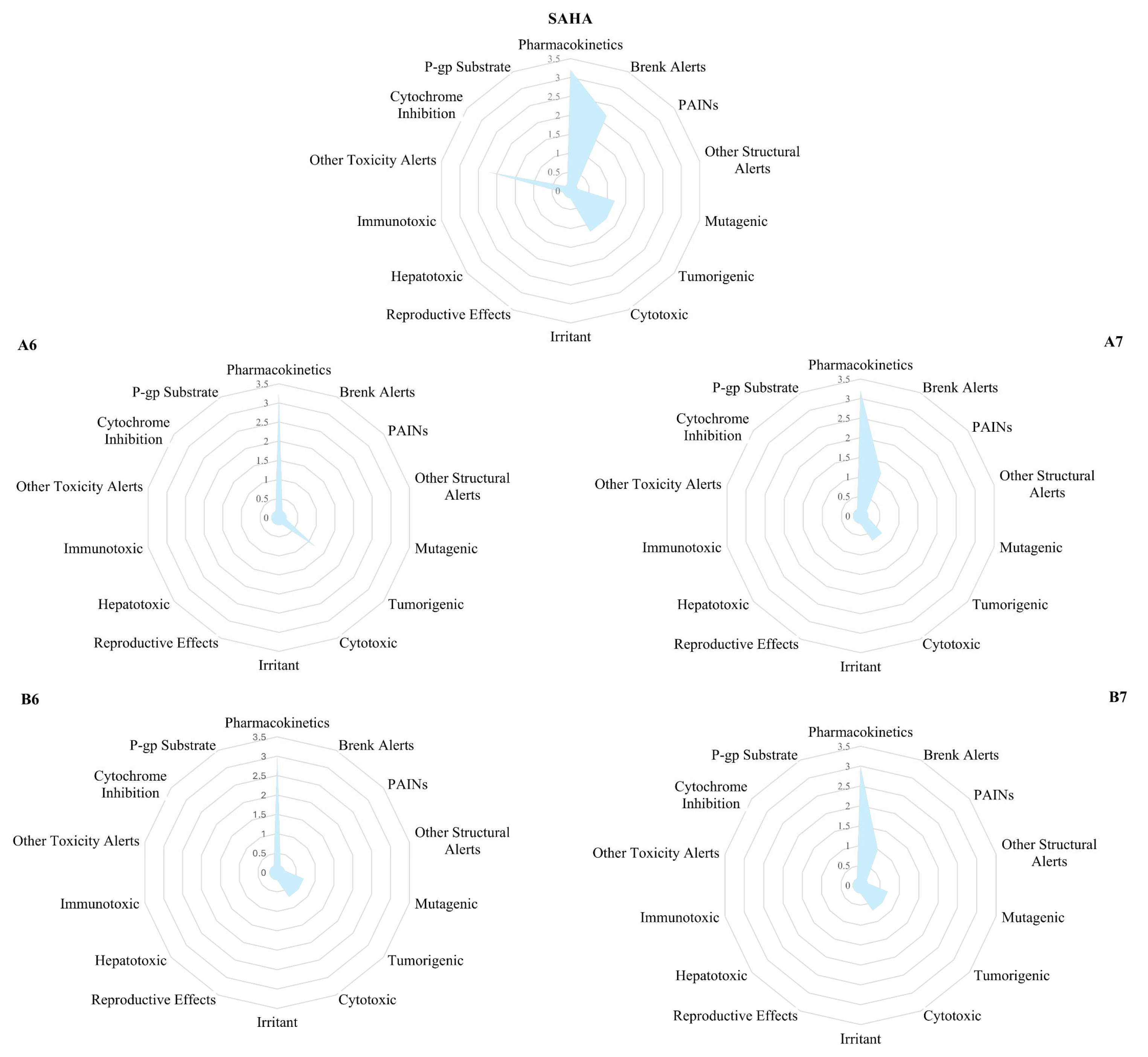
2.2.1. ADMET Prediction
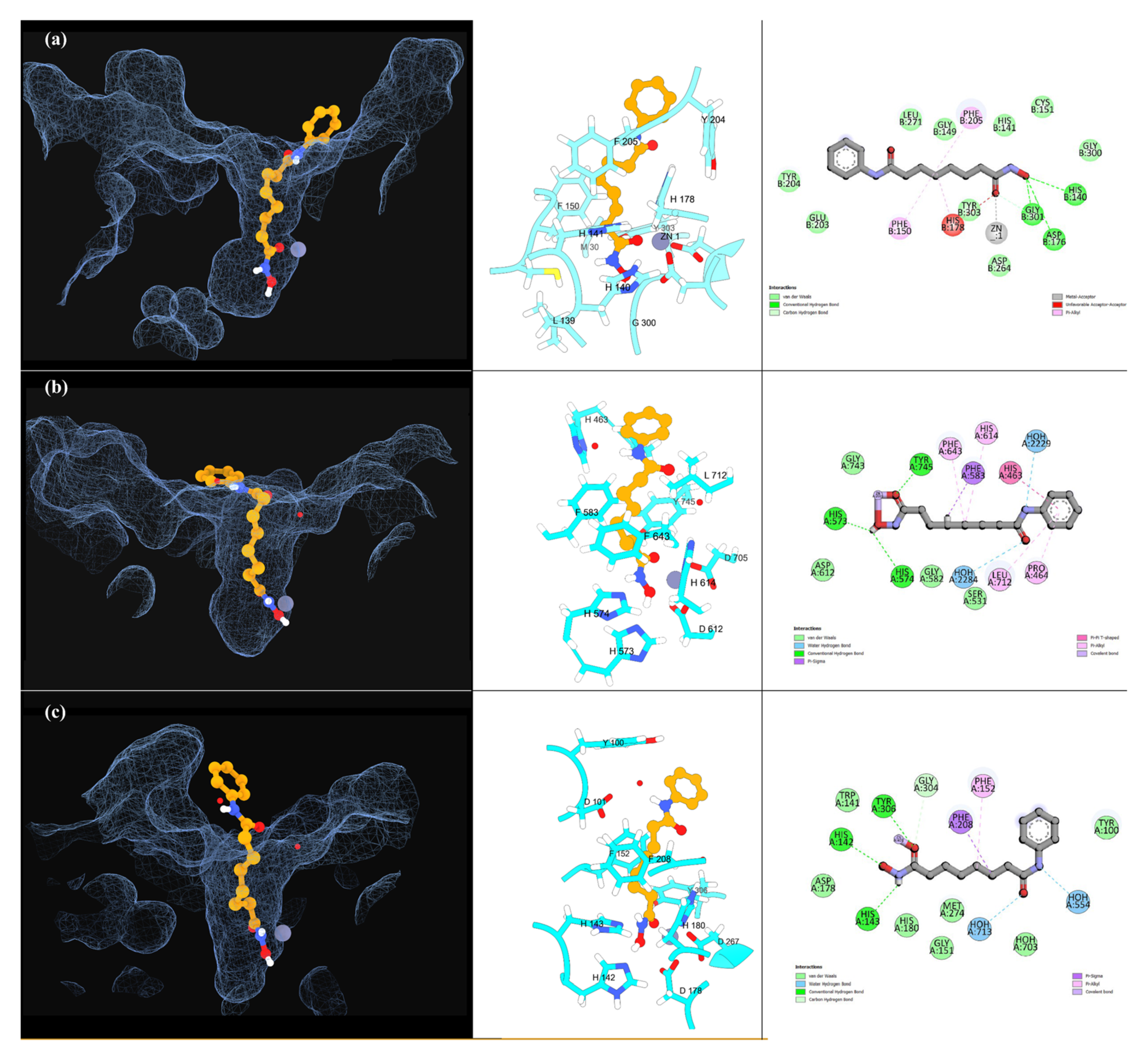
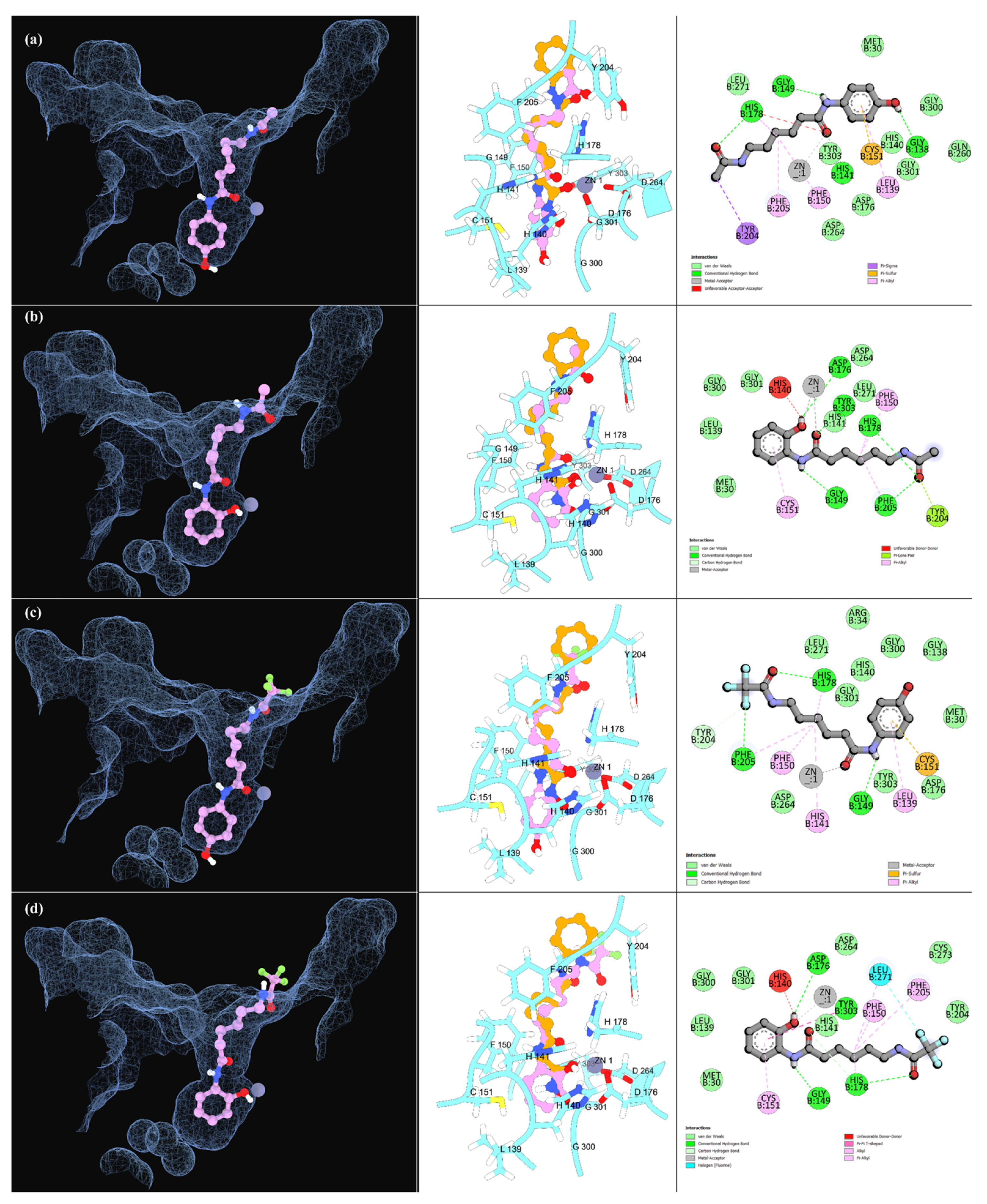
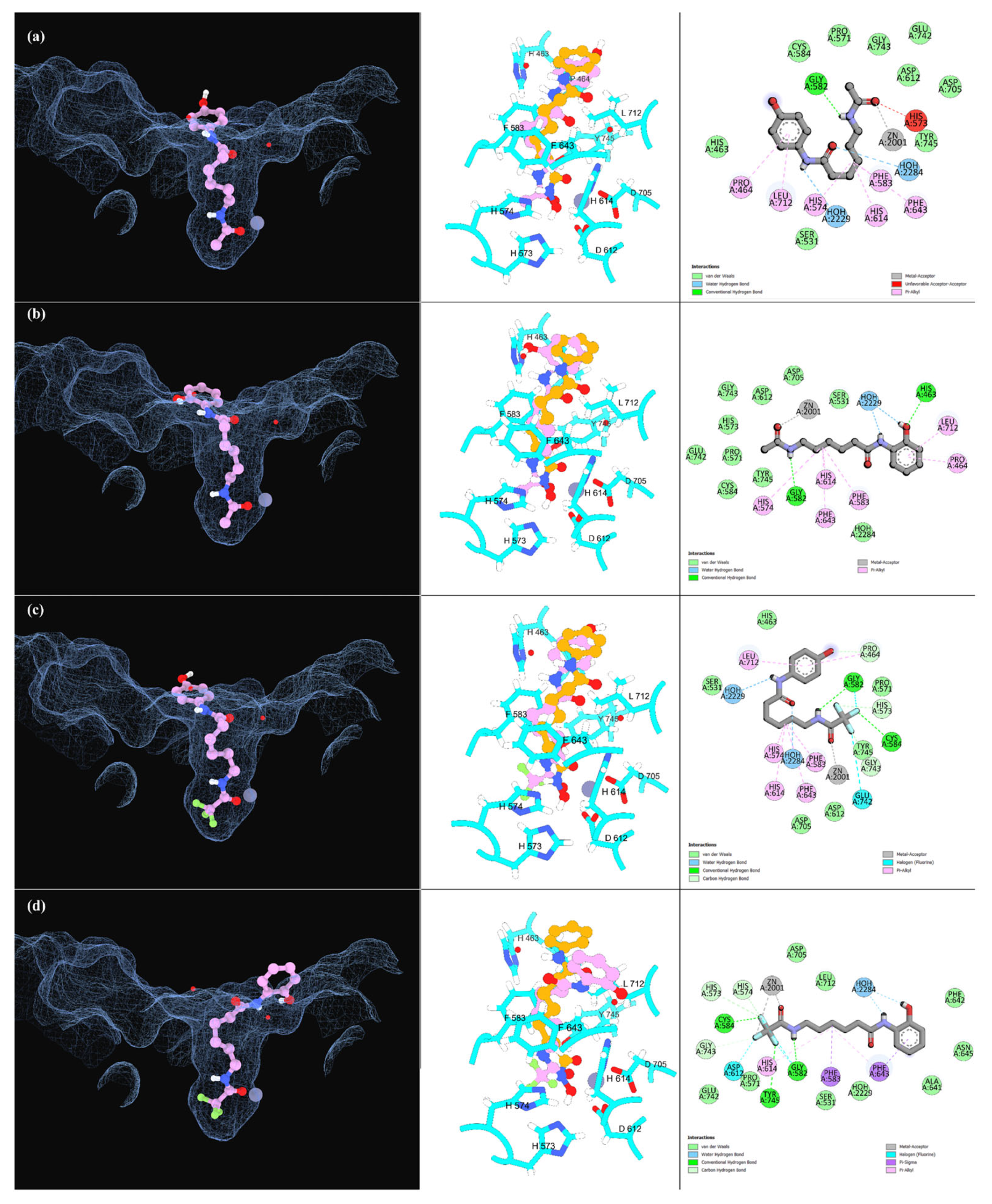
2.2.2. Molecular Docking Simulations
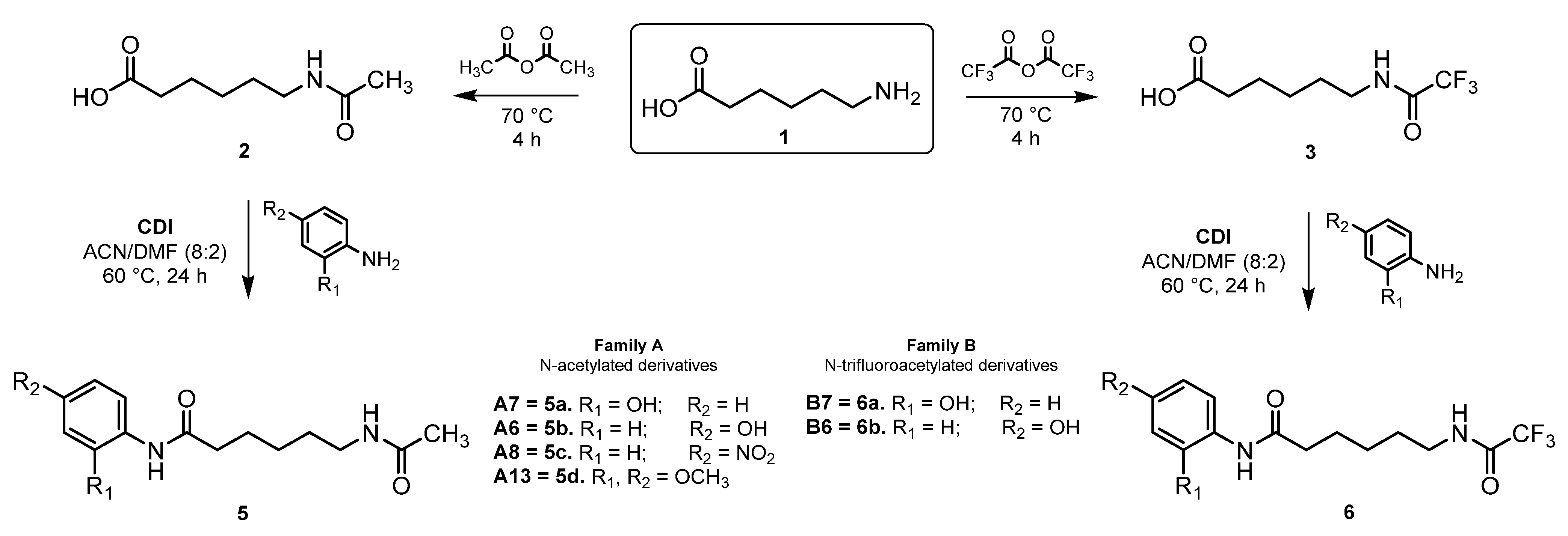
2.3. Chemistry
2.4. Biological Evaluation
2.4.1. Assessment of HDAC Inhibitory Activity
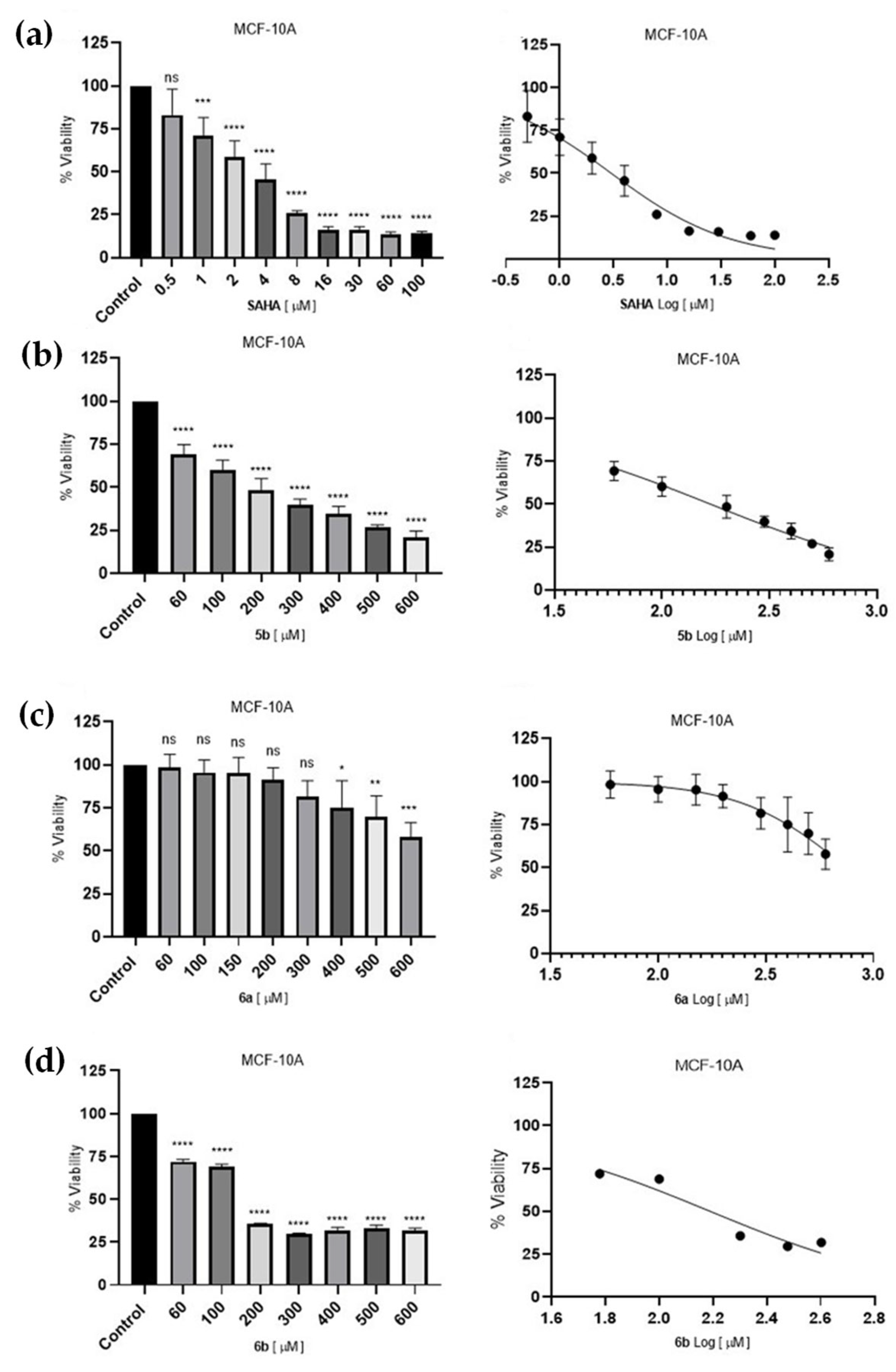
2.4.2. Cell Survival Assessment of Breast Cells Lines
3. Materials and Methods
3.1. Computational Design
3.1.1. Ligands Construction
3.1.2. ADMET Properties Prediction
3.1.3. Molecular Docking Simulations
3.1.4. Hit Identification
3.2. Chemical Synthesis
3.2.1. General Procedure for the Synthesis of N-Acetylated and N-Trifluoroacetylated Analogs
3.2.2. General Procedure for Amide Coupling Reaction Using CDI
6-Acetamidohexanoic Acid (2)
6-Acetamido-N-(2-hydroxyphenyl) Hexanamide (5a)
6-Acetamido-N-(4-hydroxyphenyl) Hexanamide (5b)
6-Acetamido-N-(4-nitrophenyl) Hexanamide (5c)
6-Acetamido-N-(2,4-dimethoxyphenyl) Hexanamide (5d)
6-(2,2,2-trifluoroacetamido) Hexanoic Acid (3)
N-(2-hydroxyphenyl)-6-(2,2,2-trifluoroacetamido) Hexanamide (6a)
N-(4-hydroxyphenyl)-6-(2,2,2-trifluoroacetamido) Hexanamide (6b)
3.3. Biological Evaluation
3.3.1. Assessment of HDAC Inhibitory Activity in HeLa Nuclear Extracts
Calculation of HDAC Residual Activity
3.3.2. Cell Survival Assessment of Breast Cancer Cells Lines
3.3.3. Cell Survival Assessment of Non-Tumorigenic Epithelial Breast Cell Line
3.3.4. MTT Assay
3.3.5. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACN | acetonitrile |
| AcOEt | ethyl acetate |
| ADMET | absorption, distribution, metabolism, excretion, and toxicity |
| CDI | carbonyldiimidazole |
| DNA | deoxyribonucleic acid |
| DMF | dimethylformamide |
| HDACs | histone deacetylases |
| HRMS | high-resolution mass spectrometry |
| HDACis | histone deacetylase inhibitors |
| NMR | nuclear magnetic resonance |
| PAINs | pan-assay interference compounds |
| PDB | Protein Data Bank |
| SAHA | suberoylanilide hydroxamic acid |
| SAR | structure–activity relationship |
| TI | Therapeutic Index |
| ZBG | zinc-binding group |
References
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Das, U.N. Introduction to Genes, Oncogenes, and Anti-Oncogenes. In Molecular Biochemical Aspects of Cancer; Das, U.N., Ed.; Springer: New York, NY, USA, 2020; pp. 1–40. [Google Scholar] [CrossRef]
- Porter, N.J.; Christianson, D.W. Structure, Mechanism, and Inhibition of the Zinc-Dependent Histone Deacetylases. Curr. Opin. Struct. Biol. 2019, 59, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Dent, S.Y.R. Histone Modifying Enzymes and Cancer: Going Beyond Histones. J. Cell. Biochem. 2005, 96, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A.; Richon, V.M.; Rifkind, R.A. Histone Deacetylase Inhibitors: Inducers of Differentiation or Apoptosis of Transformed Cells. JNCI J. Natl. Cancer Inst. 2000, 92, 1210–1216. [Google Scholar] [CrossRef]
- Shah, R.R. Safety and Tolerability of Histone Deacetylase (HDAC) Inhibitors in Oncology. Drug Saf. 2019, 42, 235–245. [Google Scholar] [CrossRef]
- Grayson, D.R.; Kundakovic, M.; Sharma, R.P. Is There a Future for Histone Deacetylase Inhibitors in the Pharmacotherapy of Psychiatric Disorders? Mol. Pharmacol. 2010, 77, 126–135. [Google Scholar] [CrossRef]
- Ho, T.C.S.; Chan, A.H.Y.; Ganesan, A. Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight. J. Med. Chem. 2020, 63, 12460–12484. [Google Scholar] [CrossRef]
- Yoshida, M.; Kudo, N.; Kosono, S.; Ito, A. Chemical and Structural Biology of Protein Lysine Deacetylases. Proc. Jpn. Acad. Ser. B 2017, 93, 297–321. [Google Scholar] [CrossRef]
- Łukasiewicz, S.; Czeczelewski, M.; Forma, A.; Baj, J.; Sitarz, R.; Stanisławek, A. Breast Cancer-Epidemiology, Risk Factors, Classification, Prognostic Markers, and Current Treatment Strategies-An Updated Review. Cancers 2021, 13, 4287. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Neiband, M.S.; Benvidi, A.; Mani-Varnosfaderani, A. Development of Classification Models for Identification of Important Structural Features of Isoform-Selective Histone Deacetylase Inhibitors (Class I). Mol. Divers. 2020, 24, 1077–1094. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Johnstone, R.W. New and Emerging HDAC Inhibitors for Cancer Treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Ceccacci, E.; Minucci, S. Inhibition of Histone Deacetylases in Cancer Therapy: Lessons from Leukaemia. Br. J. Cancer 2016, 114, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Rassool, F.V. HDAC Inhibitors: Roles of DNA Damage and Repair. In Advances in Cancer Research; Grant, S., Ed.; Academic Press: Cambridge, MA, USA, 2012; Volume 116, pp. 87–129. [Google Scholar] [CrossRef]
- Gryder, B.E.; Sodji, Q.H.; Oyelere, A.K. Targeted Cancer Therapy: Giving Histone Deacetylase Inhibitors All They Need To Succeed. Future Med. Chem. 2012, 4, 505–524. [Google Scholar] [CrossRef]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef]
- Cappellacci, L.; Perinelli, D.R.; Maggi, F.; Grifantini, M.; Petrelli, R. Recent Progress in Histone Deacetylase Inhibitors as Anticancer Agents. Curr. Med. Chem. 2020, 27, 2449–2493. [Google Scholar] [CrossRef]
- Miller, T.A.; Witter, D.J.; Belvedere, S. Histone Deacetylase Inhibitors. J. Med. Chem. 2003, 46, 5097–5116. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, J.; Jiang, Q.; Zhang, L.; Song, W. Zinc Binding Groups for Histone Deacetylase Inhibitors. J. Enzym. Inhib. Med. Chem. 2018, 33, 714–721. [Google Scholar] [CrossRef]
- Suzuki, T.; Miyata, N. Non-hydroxamate Histone Deacetylase Inhibitors. Curr. Med. Chem. 2005, 12, 2867–2880. [Google Scholar] [CrossRef]
- Li, Y.; Woster, P.M. Discovery of a New Class of Histone Deacetylase Inhibitors with a Novel Zinc Binding Group. MedChemComm 2015, 6, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Stopper, D.; Biermann, L.; Watson, P. Exploring Alternative Zinc-Binding Groups in Histone Deacetylase (HDAC) Inhibitors Uncovers DS-103 as a Potent Ethylhydrazide-Based HDAC Inhibitor with Chemosensitizing Properties. ChemRxiv 2024. [Google Scholar] [CrossRef] [PubMed]
- He, X. Medicinal Chemistry Updates of Novel HDACs Inhibitors (2020 to Present). Eur. J. Med. Chem. 2022, 227, 113946. [Google Scholar] [CrossRef] [PubMed]
- Geurs, S.; Clarisse, D.; De Bosscher, K.; D’hooghe, M. The Zinc-Binding Group Effect: Lessons from Non-Hydroxamic Acid Vorinostat Analogs. J. Med. Chem. 2023, 66, 7698–7729. [Google Scholar] [CrossRef]
- Micheletti, G.; Boga, C.; Drius, G.; Bordoni, S.; Calonghi, N. Suberoylanilide Hydroxamic Acid Analogs with Heteroaryl Amide Group and Different Chain Length: Synthesis and Effect on Histone Deacetylase. Mol. Basel Switz. 2024, 29, 238. [Google Scholar] [CrossRef]
- Pietruś, W.; Kafel, R.; Bojarski, A.J.; Kurczab, R. Hydrogen Bonds with Fluorine in Ligand-Protein Complexes—The PDB Analysis and Energy Calculations. Mol. Basel Switz. 2022, 27, 1005. [Google Scholar] [CrossRef]
- Nielsen, T.K.; Hildmann, C.; Riester, D.; Wegener, D.; Schwienhorst, A.; Ficner, R. Complex Structure of a Bacterial Class 2 Histone Deacetylase Homologue with a Trifluoromethylketone Inhibitor. Acta Cryst. Sect. F Struct. Biol. Cryst. Commun. 2007, 63 Pt 4, 270–273. [Google Scholar] [CrossRef]
- Madsen, A.S.; Olsen, C.A. A Potent Trifluoromethyl Ketone Histone Deacetylase Inhibitor Exhibits Class-Dependent Mechanism of Action. MedChemComm 2016, 7, 464–470. [Google Scholar] [CrossRef]
- Leuci, R.; Brunetti, L.; Laghezza, A.; Loiodice, F.; Tortorella, P.; Piemontese, L. Importance of Biometals as Targets in Medicinal Chemistry: An Overview About the Role of Zinc (II) Chelating Agents. Appl. Sci. 2020, 10, 4118. [Google Scholar] [CrossRef]
- Shen, S.; Kozikowski, A.P. Why Hydroxamates May Not Be the Best Histone Deacetylase Inhibitors—What Some May Have Forgotten or Would Rather Forget? ChemMedChem 2016, 11, 15–21. [Google Scholar] [CrossRef]
- Bonomi, R.; Mukhopadhyay, U. Novel Histone Deacetylase Class IIa Selective Substrate Radiotracers for PET Imaging of Epigenetic Regulation in the Brain. PLoS ONE 2015, 10, e0133512. [Google Scholar] [CrossRef]
- Decroos, C.; Bowman, C.M.; Moser, J.-A.S.; Christianson, K.E.; Deardorff, M.A.; Christianson, D.W. Compromised Structure and Function of HDAC8 Mutants Identified in Cornelia de Lange Syndrome Spectrum Disorders. ACS Chem. Biol. 2014, 9, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Hai, Y.; Christianson, D.W. Histone Deacetylase 6 Structure and Molecular Basis of Catalysis and Inhibition. Nat. Chem. Biol. 2016, 12, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Millard, C.J.; Watson, P.J.; Celardo, I.; Gordiyenko, Y.; Cowley, S.M.; Robinson, C.V.; Fairall, L.; Schwabe, J.W.R. Class I HDACs Share a Common Mechanism of Regulation by Inositol Phosphates. Mol. Cell 2013, 51, 57–67. [Google Scholar] [CrossRef]
- Porter, N.J.; Mahendran, A.; Breslow, R.; Christianson, D.W. Unusual Zinc-Binding Mode of HDAC6-Selective Hydroxamate Inhibitors. Proc. Natl. Acad. Sci. USA 2017, 114, 13459–13464. [Google Scholar] [CrossRef]
- Christianson, D.W. Chemical Versatility in Catalysis and Inhibition of the Class IIb Histone Deacetylases. Acc. Chem. Res. 2024, 57, 1135–1148. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond: In Structural Chemistry and Biology; Oxford University Press: New York, NY, USA, 2001. [Google Scholar]
- Erdélyi, M. Halogen Bonding in Solution. Chem. Soc. Rev. 2012, 41, 3547–3557. [Google Scholar] [CrossRef]
- Lu, A.; Lou, H.; Shi, M.; Yuan, Y.; Liu, J.; Tang, F. Design, Synthesis and Docking Studies on Benzamide Derivatives as Histone Deacetylase Inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 4924–4927. [Google Scholar] [CrossRef]
- Curcio, A.; Rocca, R.; Alcaro, S.; Artese, A. The Histone Deacetylase Family: Structural Features and Application of Combined Computational Methods. Pharmaceuticals 2024, 17, 620. [Google Scholar] [CrossRef]
- Raucci, A.; Castiello, C.; Mai, A.; Zwergel, C.; Valente, S. Heterocycles-Containing HDAC Inhibitors Active in Cancer: An Overview of the Last Fifteen Years. ChemMedChem 2024, 19, e202400194. [Google Scholar] [CrossRef]
- La, M.T.; Jeong, B.-H.; Kim, H.-K. Design and Synthesis of Novel N-(2-Aminophenyl)benzamide Derivatives as Histone Deacetylase Inhibitors and Their Antitumor Activity Study. Bull. Korean Chem. Soc. 2021, 42, 740–743. [Google Scholar] [CrossRef]
- Kim, J.H.; Ali, K.H.; Oh, Y.J.; Seo, Y.H. Design, Synthesis, and Biological Evaluation of Histone Deacetylase Inhibitor with Novel Salicylamide Zinc Binding Group. Medicine 2022, 101, e29049. [Google Scholar] [CrossRef] [PubMed]
- Olaoye, O.O.; Watson, P.R.; Nawar, N.; Geletu, M.; Sedigni, A.; Bukhari, S.; Raouf, Y.S. Unique Molecular Interaction with the Histone Deacetylase 6 Catalytic Tunnel: Crystallographic and Biological Characterization of a Model Chemotype. J. Med. Chem. 2021, 64, 2691–2704. [Google Scholar] [CrossRef]
- Kozikowski, A.P.; Shen, S.; Pardo, M.; Tavares, M.T.; Szarics, D.; Benoy, V. Brain Penetrable Histone Deacetylase 6 Inhibitor SW-100 Ameliorates Memory and Learning Impairments in a Mouse Model of Fragile X Syndrome. ACS Chem. Neurosci. 2019, 10, 1679–1695. [Google Scholar] [CrossRef]
- Zhang, X.-H.; Ma, Q.; Wu, H.P.; Khamis, M.Y.; Li, Y.H.; Ma, L.Y. A Review of Progress in Histone Deacetylase 6 Inhibitors Research: Structural Specificity and Functional Diversity. J. Med. Chem. 2021, 64, 1362–1391. [Google Scholar] [CrossRef]
- Kim, J.Y.; Cho, H.; Yoo, J.; Kim, G.W.; Jeon, Y.H.; Lee, S.W.; Kwon, S.H. Pathological Role of HDAC8: Cancer and Beyond. Cells 2022, 11, 3161. [Google Scholar] [CrossRef]
- Bora-Tatar, G.; Dayangaç-Erden, D.; Demir, A.S.; Dalkara, S.; Yelekçi, K.; Erdem-Yurter, H. Molecular Modifications on Carboxylic Acid Derivatives as Potent Histone Deacetylase Inhibitors: Activity and Docking Studies. Bioorg. Med. Chem. 2009, 17, 5219–5228. [Google Scholar] [CrossRef]
- Fass, D.M. Effect of Inhibiting Histone Deacetylase with Short-Chain Carboxylic Acids and Their Hydroxamic Acid Analogs on Vertebrate Development and Neuronal Chromatin. ACS Med. Chem. Lett. 2010, 2, 39. [Google Scholar] [CrossRef]
- Ediriweera, M.K. Fatty Acids as Histone Deacetylase Inhibitors: Old Biochemistry Tales in a New Life Sciences Town. Drug Discov. Today 2023, 28, 103569. [Google Scholar] [CrossRef]
- Meyners, C.; Baud, M.G.J.; Fuchter, M.J.; Meyer-Almes, F.-J. Kinetic Method for the Large-Scale Analysis of the Binding Mechanism of Histone Deacetylase Inhibitors. Anal. Biochem. 2014, 460, 39–46. [Google Scholar] [CrossRef]
- Schweipert, M.; Amurthavasan, A.; Meyer-Almes, F.-J. Continuous Enzyme Activity Assay for High-Throughput Classification of Histone Deacetylase 8 Inhibitors. Explor. Target. Anti-Tumor Ther. 2023, 4, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Kopranovic, A.; Meyer-Almes, F.-J. Rapid Determination of Kinetic Constants for Slow-Binding Inhibitors and Inactivators of Human Histone Deacetylase 8. Int. J. Mol. Sci. 2024, 25, 5593. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.M.; Fang, J.; Silvers, T.; Delosh, R. Exposure Time Versus Cytotoxicity for Anticancer Agents. Cancer Chemother. Pharmacol. 2019, 84, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Bieszczad, B.; Garbicz, D.; Switalska, M. Improved HDAC Inhibition, Stronger Cytotoxic Effect and Higher Selectivity against Leukemias and Lymphomas of Novel, Tricyclic Vorinostat Analogues. Pharmaceuticals 2021, 14, 851. [Google Scholar] [CrossRef]
- Marijon, H.; Lee, D.H.; Ding, L.; Sun, H. Co-targeting Poly(ADP-Ribose) Polymerase (PARP) and Histone Deacetylase (HDAC) in Triple-Negative Breast Cancer: Higher Synergism in BRCA Mutated Cells. Biomed. Pharmacother. 2018, 99, 543–551. [Google Scholar] [CrossRef]
- Wawruszak, A.; Borkiewicz, L.; Okon, E.; Kukula-Koch, W.; Afshan, S.; Halasa, M. Vorinostat (SAHA) and Breast Cancer: An Overview. Cancers 2021, 13, 4700. [Google Scholar] [CrossRef]
- Cheshmazar, N.; Mivehroud, M.H.; Hemmati, S.; Abolhasani, H. Key Structural Requirements of Benzamide Derivatives for Histone Deacetylase Inhibition: Design, Synthesis and Biological Evaluation. Future Med. Chem. 2024, 16, 859–872. [Google Scholar] [CrossRef]
- Kraft, F.B.; Biermann, L.; Hubner, L.S.; Hanl, M. Hydrazide-Based Class I Selective HDAC Inhibitors Completely Reverse Chemoresistance Synergistically in Platinum-Resistant Solid Cancer Cells. J. Med. Chem. 2024, 67, 17796–17819. [Google Scholar] [CrossRef]
- He, J.; Wang, S.; Liu, X.; Lin, R. Synthesis and Biological Evaluation of HDAC Inhibitors with a Novel Zinc Binding Group. Front. Chem. 2020, 8, 256. [Google Scholar] [CrossRef]
- Riddhidev, B.; Endri, K.; Sabitri, L.; Borkiewicz, L. Rational Design of Metabolically Stable HDAC Inhibitors: An Overhaul of Trifluoromethyl Ketones. Eur. J. Med. Chem. 2022, 244, 114807. [Google Scholar] [CrossRef]
- Galindo-Alvarez, N.L.; Mendoza-Figueroa, H.L.; Rosales-Hernández, M.C.; Bakalara, N.; Correa-Basurto, J. Decrease in Cell Viability of Breast Cancer Cells by a Di-hydroxylated Derivative of N-(2-hydroxyphenyl)-2-propylpentanamide. Anticancer. Agents Med. Chem. 2022, 22, 1802–1812. [Google Scholar] [CrossRef] [PubMed]
- Yue, K.; Qin, M.; Huang, C.; Chou, C.J.; Jiang, Y.; Li, X. Comparison of Three Zinc Binding Groups for HDAC Inhibitors—A Potency, Selectivity and Enzymatic Kinetics Study. Bioorg. Med. Chem. Lett. 2022, 70, 128797. [Google Scholar] [CrossRef] [PubMed]
- Indrayanto, G.; Putra, G.S.; Suhud, F. Validation of In-Vitro Bioassay Methods: Application in Herbal Drug Research. Profiles Drug Subst. Excip. Relat. Methodol. 2021, 46, 273–307. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, A. In Vitro Assessment of the Genotoxic Hazard of Novel Hydroxamic Acid- and Benzamide-Type Histone Deacetylase Inhibitors (HDACi). Int. J. Mol. Sci. 2020, 21, 4747. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Conrad, J.; Paras, N.A.; Vaz, R.J. Model of P-Glycoprotein Ligand Binding and Validation with Efflux Substrate Matched Pairs. J. Med. Chem. 2024, 67, 5854–5865. [Google Scholar] [CrossRef]
- Morales-Herrejón, G.; Mendoza-Figueroa, H.L.; Martínez-Archundía, M.; Correa-Basurto, J. The Importance of Structured Water in HDAC8 for Correct Binding Pose Applied for Drug Design of Anticancer Molecules. Anticancer. Agents Med. Chem. 2024, 24, 1109–1125. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Santos-Martins, D.; Forli, S.; Ramos, M.J.; Olson, A.J. AutoDock4Zn: An Improved AutoDock Force Field for Small-Molecule Docking to Zinc Metalloproteins. J. Chem. Inf. Model. 2014, 54, 2371–2379. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp | ADMET Properties Predictions | Score | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| SAHA | MW | miLogP | HBA | HBD | TPSA | nRotB | Sol | GI abs | Gp-P | 80 |
| 264.32 | 2.47 | 5 | 3 | 78.42 | 8 | Good | High | No | ||
| * Lipinski | * Ghose | * Veber | * Egan | * Muegge | ** PAINs | ** Brenk | Mutagenic | Tumorogenic | ||
| 0 | 0 | 0 | 0 | 0 | 0 | 2 | Yes | Yes | ||
| Cytotoxic | Irritant | R. effects | ImmunoTox | HepatoTox | OTP | Inhibition of metabolism CYPs? | ||||
| Yes | No | No | No | No | Yes | No | ||||
| A6 | MW | miLogP | HBA | HBD | TPSA | nRotB | Sol | GI abs | Gp-P | 92 |
| 264.32 | 1.33 | 5 | 2 | 78.42 | 7 | Good | High | No | ||
| * Lipinski | * Ghose | * Veber | * Egan | * Muegge | ** PAINs | ** Brenk | Mutagenic | Tumorogenic | ||
| 0 | 0 | 0 | 0 | 0 | 0 | 0 | No | No | ||
| Cytotoxic | Irritant | R. effects | ImmunoTox | HepatoTox | OTP | Inhibition of metabolism CYPs? | ||||
| No | No | No | No | No | No | No | ||||
| A7 | MW | miLogP | HBA | HBD | TPSA | nRotB | Sol | GI abs | Gp-P | 89 |
| 264.32 | 1.55 | 5 | 2 | 78.42 | 7 | Good | High | No | ||
| * Lipinski | * Ghose | * Veber | * Egan | * Muegge | ** PAINs | ** Brenk | Mutagenic | Tumorogenic | ||
| 0 | 0 | 0 | 0 | 0 | 0 | 1 | No | No | ||
| Cytotoxic | Irritant | R. effects | ImmunoTox | HepatoTox | OTP | Inhibition of metabolism CYPs? | ||||
| No | No | No | No | No | No | No | ||||
| B6 | MW | miLogP | HBA | HBD | TPSA | nRotB | Sol | GI abs | Gp-P | 90 |
| 318.30 | 2.22 | 5 | 2 | 78.42 | 8 | Good | High | No | ||
| * Lipinski | * Ghose | * Veber | * Egan | * Muegge | ** PAINs | ** Brenk | Mutagenic | Tumorogenic | ||
| 0 | 0 | 0 | 0 | 0 | 0 | 0 | No | No | ||
| Cytotoxic | Irritant | R. effects | ImmunoTox | HepatoTox | OTP | Inhibition of metabolism CYPs? | ||||
| No | No | No | No | No | No | No | ||||
| B7 | MW | miLogP | HBA | HBD | TPSA | nRotB | Sol | GI abs | Gp-P | 88 |
| 318.30 | 2.43 | 5 | 3 | 78.42 | 8 | Good | High | No | ||
| * Lipinski | * Ghose | * Veber | * Egan | * Muegge | ** PAINs | ** Brenk | Mutagenic | Tumorogenic | ||
| 0 | 0 | 0 | 0 | 0 | 0 | 1 | No | No | ||
| Cytotoxic | Irritant | R. effects | ImmunoTox | HepatoTox | OTP | Inhibition of metabolism CYPs? | ||||
| No | No | No | No | No | No | No | ||||
| HDAC1 a | HDAC6-DC2 b | HDAC8 c | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ligands | ADMET Score | ΔG (Kcal/mol) | Interactions Score | % Binding | ΔG (Kcal/mol) | Interactions Score | % Binding | ΔG (Kcal/mol) | Interactions Score | % Binding |
| SAHA | 80 | −7.20 | 25.0 | 100 | −7.51 | 38.4 | 92.9 | −7.30 | 27.6 | 84.6 |
| A2 | 91 | −6.98 | 20.0 | 100 | −7.57 | 30.2 | 100 | −6.85 | 30.8 | 100 |
| A3 | 91 | −7.64 | 24.6 | 100 | −7.86 | 31.6 | 100 | −6.92 | 28.8 | 92.3 |
| A6 | 92 | −7.37 | 33.4 | 100 | −7.35 | 30.2 | 92.9 | −7.18 | 32.2 | 100 |
| A7 | 89 | −6.80 | 23.8 | 100 | −7.67 | 35.2 | 100 | −7.11 | 35.2 | 92.3 |
| A8 | 93 | −8.69 | 22.6 | 100 | −12.18 | 18.0 | 78.6 | −10.26 | 27.0 | 92.3 |
| A10 | 93 | −8.04 | 25.4 | 100 | −7.42 | 26.5 | 85.1 | −8.06 | 33.2 | 92.3 |
| A11 | 90 | −7.57 | 32.0 | 100 | −7.50 | 34.4 | 100 | −7.13 | 33.2 | 92.3 |
| A12 | 88 | −7.16 | 20.4 | 100 | −11.24 | 33.4 | 78.6 | −9.39 | 26.3 | 92.3 |
| A13 | 85 | −7.30 | 22.2 | 100 | −7.52 | 28.8 | 100 | −7.12 | 33.0 | 92.3 |
| A23 | 83 | −7.95 | 22.3 | 100 | −8.28 | 31.0 | 100 | −7.30 | 31.8 | 92.3 |
| B2 | 92 | −7.44 | 27.2 | 100 | −6.66 | 36.8 | 100 | −6.21 | 34.6 | 100 |
| B6 | 90 | −7.45 | 33.4 | 100 | −6.63 | 35.8 | 100 | −6.25 | 39.0 | 100 |
| B7 | 88 | −7.26 | 34.6 | 100 | −7.17 | 43.1 | 85.7 | −5.90 | 34.6 | 100 |
| B14 | 85 | −6.01 | 35.0 | 100 | −6.55 | 39.2 | 100 | −6.35 | 37.0 | 100 |
| B17 | 91 | −7.31 | 31.0 | 100 | −6.59 | 35.4 | 100 | −6.05 | 37.4 | 100 |
| B20 | 88 | −7.64 | 29.0 | 100 | −7.19 | 38.6 | 100 | −6.31 | 40.0| | 92.3 |
| Cytotoxic Activity IC50 (µM) and Selectivity Index | |||||
|---|---|---|---|---|---|
| Compound | MDA-MB-231 | MCF-7 | MCF-10A | MCF-10A/ MDA-MB-231 | MCF-10A/ MCF-7 |
| SAHA | 3.50 ± 0.56 | 4.23 ± 1.59 | 3.02 ± 0.32 | 0.86 | 0.71 |
| 5b | 121.3 ± 26.65 | 227.7 ± 52.85 | 168.6 ± 20.55 | 1.39 | 0.74 |
| 6a | 225.1 ± 17.60 | 253.8 ± 34.4 | >600 | >2.66 | >2.36 |
| 6b | 76.74 ± 8.52 | 45.70 ± 6.91 | 154.6 ± 21.6 | 2.01 | 3.38 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morales-Herrejón, G.; García-Vázquez, J.B.; Fernández-Pomares, C.; Bakalara, N.; Correa-Basurto, J.; Mendoza-Figueroa, H.L. Computationally Guided Design, Synthesis, and Evaluation of Novel Non-Hydroxamic Histone Deacetylase Inhibitors, Based on N-Trifluoroacetamide as a Zinc-Binding Group, Against Breast Cancer. Pharmaceuticals 2025, 18, 351. https://doi.org/10.3390/ph18030351
Morales-Herrejón G, García-Vázquez JB, Fernández-Pomares C, Bakalara N, Correa-Basurto J, Mendoza-Figueroa HL. Computationally Guided Design, Synthesis, and Evaluation of Novel Non-Hydroxamic Histone Deacetylase Inhibitors, Based on N-Trifluoroacetamide as a Zinc-Binding Group, Against Breast Cancer. Pharmaceuticals. 2025; 18(3):351. https://doi.org/10.3390/ph18030351
Chicago/Turabian StyleMorales-Herrejón, Gerardo, Juan Benjamín García-Vázquez, Cynthia Fernández-Pomares, Norbert Bakalara, José Correa-Basurto, and Humberto L. Mendoza-Figueroa. 2025. "Computationally Guided Design, Synthesis, and Evaluation of Novel Non-Hydroxamic Histone Deacetylase Inhibitors, Based on N-Trifluoroacetamide as a Zinc-Binding Group, Against Breast Cancer" Pharmaceuticals 18, no. 3: 351. https://doi.org/10.3390/ph18030351
APA StyleMorales-Herrejón, G., García-Vázquez, J. B., Fernández-Pomares, C., Bakalara, N., Correa-Basurto, J., & Mendoza-Figueroa, H. L. (2025). Computationally Guided Design, Synthesis, and Evaluation of Novel Non-Hydroxamic Histone Deacetylase Inhibitors, Based on N-Trifluoroacetamide as a Zinc-Binding Group, Against Breast Cancer. Pharmaceuticals, 18(3), 351. https://doi.org/10.3390/ph18030351