
Beyond the Basics: Exploring Pharmacokinetic Interactions and Safety in Tyrosine-Kinase Inhibitor Oral Therapy for Solid Tumors


Abstract
1. Introduction
2. Tyrosine-Kinase Receptors as Therapeutical Targets
3. Pharmacokinetic Drug–Drug Interactions
3.1. DDIs Affecting TKI Absorption
3.2. DDIs Affecting TKI Distribution
3.3. DDIs Affecting TKI Metabolization
3.3.1. DDIs Affecting ALK TKIs
3.3.2. DDIs Affecting EGFR TKIs
3.3.3. DDIs Affecting VEGF TKIs
3.3.4. DDIs Affecting Multiple Target TKIs
3.3.5. Potential Interactions with Complementary and Alternative Medicines
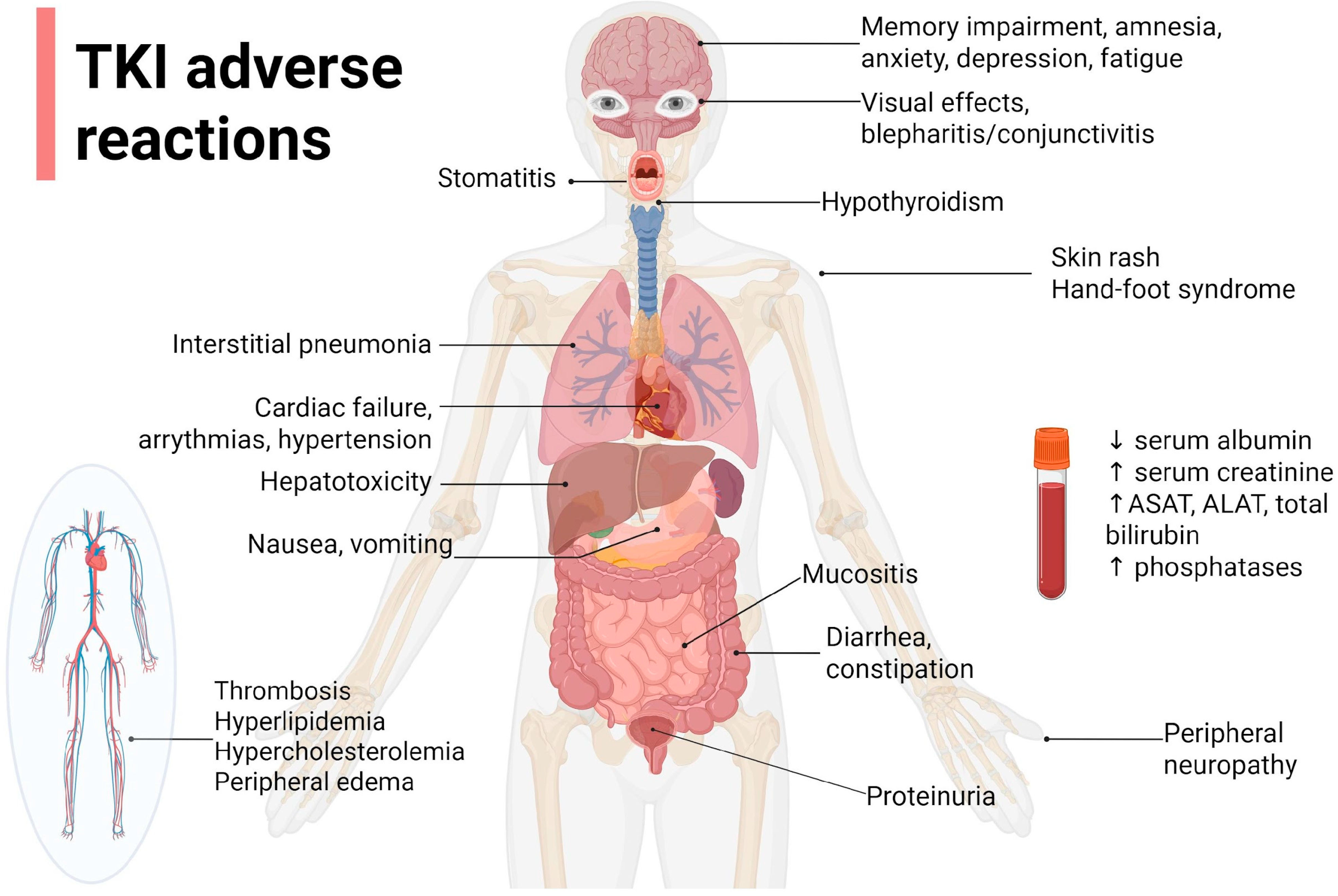
4. TKI Safety
4.1. Safety of ALK TKIs
4.2. Safety of EGFR TKIs
4.3. Safety of MET-TKIs
4.4. Safety of Multiple Target TKIs
4.5. Cardiac Failure (CF)
4.6. Hepatotoxicity

{kind=link}
{kind=link}
| Adverse Reaction | Monitoring Parameters | Management | Ref. |
|---|---|---|---|
| Nausea and/or vomiting | Ca2+, Mg2+, K+ levels Signs of dehydration | Take with food if drug PK allows Antiemetic treatment (5 HT3 antagonists, metoclopramide, ginger); pay attention to QT prolongation or 5HT3 antagonists | Rimassa et al. [95] |
| Stomatitis/mucositis | Good oral hygiene, non-alcoholic mouthwashes, consumption of non-irritating food; initiate antimicrobial/antifungal therapy if needed | Shyam Sunder et al. [96] | |
| Diarrhea | Number of stools Ca2+, Mg2+, K+ levels Stool test for C. difficile Evaluate patient’s baseline bowel patten to assess the severity | Loperamide, probiotics, diosmectite, rehydration salts if needed, low fiber, low-fat diet, 8–10 glasses of water/day | Yang, J. C. H. et al. [97] Zhou et al. [98] |
| Constipation | Evaluate patient’s baseline bowel patten to assess the severity | High-fiber meals, lactulose (pay attention to fluid intake to avoid obstruction), irritant laxative only when needed. Pay attention to opioid association | Zhou et al. [73] |
| Peripheral edemas | Serum albumin | Compression socks, limb elevation, diuretics (DDI potential, electrolyte perturbances) | Girard et al. [57] |
| Hepatotoxicity | ASAT, ALAT, total bilirubin | Lower doses or treatment interruption, depending on grade | Qian et al. [99] |
| Body weight increase/decrease | Body weight | Dietary regime, physical activity; evaluate cachexia! | Kodama et al. [100] |
| Rash | First week—presents as sensory disturbances, erythema, and edema, followed by papulopustular eruptions in the second week and crusting in the fourth week | Hydrating creams applied 2 times/day; topical corticoid applications; antibiotic treatment if needed (tetracycline/minocycline); do not use alcoholic-based solutions; wash with lukewarm water, not hot water | Vogel et al. [101] |
| Hyperlipidemia | Total lipid panel, triglycerides, cholesterol at baseline | Dietary measures (low-fat meals, fish, grains, fruits, vegetables), statins (rosuvastatin, pravastatin preferred because of CYP3A4 metabolism of most of the TKIs) | Blais et al. [102] |
| Hypertension | Blood pressure | Angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers or dihydropyridine calcium channel blockers; avoid non-dihydropyridine calcium channel blockers | Zhu et al. [103] |
| Ocular disorders: blepharitis, conjunctivitis, epiphora, periorbital rash | Ocular irritation, crusts on eyelids, excessive tear production | Warm compress, eyelid hygiene, corticosteroids, or anti-inflammatory medications | Agustoni et al. [104] |
| Hypothyroidism | TSH, FT4; assess baseline thyroid function | levothyroxine | Gabora et al. [105] |
| Cardiac failure | FEVS | Angiotensin-converting enzyme inhibitors, loop diuretics, potassium-sparing diuretics, beta blockers | AlShatnawi et al. [106] |
| QTc prolongation | EKG | Lower doses or interrupt/discontinue TKI treatment | Abu Rmilah et al. [107] |
| Fatigue | Brief Fatigue Inventory (BFI) questionnaire Functional Assessment of Cancer Therapy-Fatigue (FACT-F) Patient-Reported Outcomes Measurement Information System Cancer Fatigue Short Form (PROMISE-CF-SF) | Treat underlying causes: hypothyroidism, anemia, anxiety, depression Dexamethasone, methylphenidate, L-carnitine Drug holiday Nutrition and exercise therapy | Takahashi [108] |
5. Emerging Strategies and Further Steps
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| VEGF | vascular endothelial growth factor |
| TKI | tyrosine-kinase inhibitor |
| CML | chronic myeloid leukemia |
| DDIs | drug–drug interaction |
| CYP450 | cytochrome P450 |
| P-gp | P-glycoprotein |
| RTKs | tyrosine-kinase receptors |
| EGFRs | epidermal growth factor receptors |
| VEGFRs | vascular endothelial growth factor receptors |
| PDGFRs | platelet-derived growth factor receptors |
| ROR1, ROR2 | receptor tyrosine kinase-like orphan receptors 1 and 2 |
| NSCLC | non-small-cell lung cancer |
| RCC | renal cell carcinoma |
| HCC | hepatocellular carcinoma |
| GIST | gastrointestinal stromal tumor |
| PPIs | proton pump inhibitors |
| OS | overall survival |
| PFS | progression-free survival |
| mRCC | metastatic renal cell carcinoma |
| ABC | ATP-binding cassette |
| BCRP | breast cancer resistance protein |
| CNS | central nervous system |
| cmax | maximum plasma concentration |
| PXR | pregnane X receptor |
| OATP1B1 | organic anion transporting polypeptide 1B1 |
| ALK | anaplastic lymphoma kinase |
| INR | international normalized ratio |
| CPK | creatine-phosphokinase |
| CAM | complementary and alternative medicine |
| CTCAE | Common Terminology Criteria for Adverse Events |
| AE | adverse event |
| ALAT | alanine aminotransferase |
| ASAT | aspartate aminotransferase |
| EKG | electrocardiogram |
| MET | mesenchymal epithelial transition factor receptor |
| CF | cardiac failure |
| ICSR | individual case safety report |
| TDM | therapeutic drug monitoring |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- INSP. Romania—National Cancer Profile Report 2023; INSP: Bucharest, Romania, 2023. [Google Scholar]
- Cooper, G.M. The Cell: A Molecular Approach, 2nd ed.; Sinauer Associates: Sunderland, MA, USA, 2000. Available online: https://www.ncbi.nlm.nih.gov/books/NBK9840/ (accessed on 10 January 2025).
- Feitelson, M.A.; Arzumanyan, A.; Kulathinal, R.J.; Blain, S.W.; Holcombe, R.F.; Mahajna, J.; Marino, M.; Martinez-Chantar, M.L.; Nawroth, R.; Sanchez-Garcia, I.; et al. Sustained proliferation in cancer: Mechanisms and novel therapeutic targets. Semin. Cancer Biol. 2015, 35, S25–S54. [Google Scholar] [CrossRef]
- Fernald, K.; Kurokawa, M. Evading apoptosis in cancer. Trends Cell Biol. 2013, 23, 620–633. [Google Scholar] [CrossRef]
- Huang, L.; Jiang, S.; Shi, Y. Tyrosine kinase inhibitors for solid tumors in the past 20 years (2001–2020). J. Hematol. Oncol. 2020, 13, 143. [Google Scholar]
- Vener, C.; Banzi, R.; Ambrogi, F.; Ferrero, A.; Saglio, G.; Pravettoni, G.; Sant, M. First-line imatinib vs. second- And third-generation TKIs for chronic-phase CML: A systematic review and meta-analysis. Blood Adv. 2020, 4, 2723–2735. [Google Scholar] [CrossRef]
- Shao, J.; Markowitz, J.S.; Bei, D.; An, G. Enzyme- and transporter-mediated drug interactions with small molecule tyrosine kinase inhibitors. J. Pharm. Sci. 2014, 103, 3810–3833. [Google Scholar]
- Gay, C.; Toulet, D.; Le Corre, P. Pharmacokinetic drug-drug interactions of tyrosine kinase inhibitors: A focus on cytochrome P450, transporters, and acid suppression therapy. Hematol. Oncol. 2017, 35, 259–280. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; Liu, Y.; Zhang, J.; Fang, Z.; Fang, H.; Gong, Y.; Lv, X. Prevalence of potential drug–drug interactions in outpatients of a general hospital in China: A retrospective investigation. Int. J. Clin. Pharm. 2020, 42, 1190–1196. [Google Scholar] [CrossRef] [PubMed]
- Baye, A.M.; Fanta, T.G.; Siddiqui, M.K.; Dawed, A.Y. The Genetics of Adverse Drug Outcomes in Type 2 Diabetes: A Systematic Review. Front. Genet. 2021, 12, 675053. [Google Scholar] [CrossRef]
- Badiu, R.; Bucsa, C.; Mogosan, C.; Dumitrascu, D. Statin drug-drug interactions in a Romanian community pharmacy. Med. Pharm. Rep. 2016, 89, 273–278. [Google Scholar] [CrossRef]
- Saraon, P.; Pathmanathan, S.; Snider, J.; Lyakisheva, A.; Wong, V.; Stagljar, I. Receptor tyrosine kinases and cancer: Oncogenic mechanisms and therapeutic approaches. Oncogene 2021, 40, 4079–4093. [Google Scholar] [CrossRef]
- Tomuleasa, C.; Tigu, A.B.; Munteanu, R.; Moldovan, C.S.; Kegyes, D.; Onaciu, A.; Gulei, D.; Ghiaur, G.; Einsele, H.; Croce, C.M. Therapeutic advances of targeting receptor tyrosine kinases in cancer. Signal Transduct. Target. Ther. 2024, 9, 201. [Google Scholar] [PubMed]
- Ebrahimi, N.; Fardi, E.; Ghaderi, H.; Palizdar, S.; Khorram, R.; Vafadar, R.; Ghanaatian, M.; Rezaei-Tazangi, F.; Baziyar, P.; Ahmadi, A.; et al. Receptor tyrosine kinase inhibitors in cancer. Cell. Mol. Life Sci. 2023, 80, 104. [Google Scholar] [CrossRef]
- Nouiakh, L.; Oualla, K.; Ouafki, I.; Berrad, S.; Erraichi, H.; Amaadour, L.; Benbrahim, Z.; Arifi, S.; Mellas, N. Tyrosine Kinase Inhibitors against Cancer: Their Safety in 216 Moroccan Patients. J. Cancer Ther. 2021, 12, 116–126. [Google Scholar] [CrossRef]
- Archana, M.; Palathingal, M.; Damodharan, K.A.; Ashisha, P.; Marathakam, N.A. An updated Review on Therapeutic Potential of Entrectinib as a Promising TrK, ROS 1, and ALK Inhibitor in Solid Tumors and Lung Cancer. J. Pharm. Res. Int. 2021, 33, 413–421. [Google Scholar] [CrossRef]
- Bridoux, M.; Simon, N.; Turpin, A. Proton Pump Inhibitors and Cancer: Current State of Play. Front. Pharmacol. 2022, 13, 798272. [Google Scholar] [CrossRef]
- Uchiyama, A.A.T.; Silva, P.A.I.A.; Lopes, M.S.M.; Yen, C.T.; Ricardo, E.D.; Mutão, T.; Pimenta, J.R.; Machado, L.M.; Shimba, D.S.; Peixoto, R.D. Proton pump inhibitors and oncologic treatment efficacy: A practical review of the literature for oncologists. Curr. Oncol. 2021, 28, 783–799. [Google Scholar] [CrossRef]
- Keyvanjah, K.; DiPrimeo, D.; Li, A.; Obaidi, M.; Swearingen, D.; Wong, A. Pharmacokinetics of neratinib during coadministration with lansoprazole in healthy subjects. Br. J. Clin. Pharmacol. 2017, 83, 554–561. [Google Scholar] [CrossRef]
- Moreau-Bachelard, C.; Letailleur, V.; Bompas, E.; Soulié, P.; Paul, J.; Raoul, J.L. Effect of Concomitant Proton Pump Inhibitors with Pazopanib on Cancer Patients: A Retrospective Analysis. Cancers 2022, 14, 4721. [Google Scholar] [CrossRef]
- Vishwanathan, K.; Dickinson, P.A.; Bui, K.; Cassier, P.A.; Greystoke, A.; Lisbon, E.; Moreno, V.; So, K.; Thomas, K.; Weilert, D.; et al. The Effect of Food or Omeprazole on the Pharmacokinetics of Osimertinib in Patients With Non-Small-Cell Lung Cancer and in Healthy Volunteers. J. Clin. Pharmacol. 2018, 58, 474–484. [Google Scholar] [CrossRef]
- Rassy, E.; Cerbone, L.; Auclin, E.; Benchimoll-Zouari, A.; Flippot, R.; Alves Costa Silva, C.; Colomba, E.; Geraud, A.; Guida, A.; Mir, O.; et al. The Effect of Concomitant Proton Pump Inhibitor and Cabozantinib on the Outcomes of Patients with Metastatic Renal Cell Carcinoma. Oncologist 2021, 26, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Morita, T.; Onozuka, M.; Saito, K.; Sano, K.; Hanada, K.; Kondo, M.; Nakamura, Y.; Kishino, T.; Nakagawa, H.; et al. Impact of Q141K on the Transport of Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors by ABCG2. Cells 2019, 8, 763. [Google Scholar] [CrossRef]
- Shukla, S.; Chen, Z.S.; Ambudkar, S.V. Tyrosine kinase inhibitors as modulators of ABC transporter-mediated drug resistance. Drug Resist. Updates 2012, 15, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Alhaffar, D.; Han, Y.; Darling, J.; Skaar, T.C.; Fausel, C.A.; Hanna, N.H. Prevalence of the concurrent administration of contraindicated medications in patients with cancer treated with tyrosine kinase inhibitors (TKIs): A pilot study from the IU Simon Comprehensive Cancer Center. J. Clin. Oncol. 2021, 39 (Suppl. S15), e18714. [Google Scholar] [CrossRef]
- Wind, S.; Giessmann, T.; Jungnik, A.; Brand, T.; Marzin, K.; Bertulis, J.; Hocke, J.; Gansser, D.; Stopfer, P. Pharmacokinetic Drug Interactions of Afatinib with Rifampicin and Ritonavir. Clin. Drug Investig. 2014, 34, 173–182. [Google Scholar] [CrossRef]
- Calvo, E.; Lee, J.S.; Kim, S.W.; Moreno, V.; deCastro Carpeno, J.; Weilert, D.; Laus, G.; Mann, H.; Vishwanathan, K. Modulation of Fexofenadine Pharmacokinetics by Osimertinib in Patients With Advanced EGFR-Mutated Non–Small Cell Lung Cancer. J. Clin. Pharmacol. 2019, 59, 1099–1109. [Google Scholar] [CrossRef]
- Taguchi, T.; Masuo, Y.; Sakai, Y.; Kato, Y. Short-lasting inhibition of hepatic uptake transporter OATP1B1 by tyrosine kinase inhibitor pazopanib. Drug Metab. Pharmacokinet. 2019, 34, 372–379. [Google Scholar] [CrossRef]
- Timm, A.; Kolesar, J.M. Crizotinib for the treatment of non-small-cell lung cancer. Am. J. Health-Syst. Pharm. 2013, 70, 943–947. [Google Scholar] [CrossRef]
- Fogli, S.; Porta, C.; Del Re, M.; Crucitta, S.; Gianfilippo, G.; Danesi, R.; Rini, B.I.; Schmidinger, M. Optimizing treatment of renal cell carcinoma with VEGFR-TKIs: A comparison of clinical pharmacology and drug-drug interactions of anti-angiogenic drugs. Cancer Treat. Rev. 2020, 84, 101966. [Google Scholar] [CrossRef]
- Ferri, N.; Colombo, E.; Tenconi, M.; Baldessin, L.; Corsini, A. Drug-Drug Interactions of Direct Oral Anticoagulants (DOACs): From Pharmacological to Clinical Practice. Pharmaceutics 2022, 14, 1120. [Google Scholar] [CrossRef]
- Hulin, A.; Gelé, T.; Fenioux, C.; Kempf, E.; Sahali, D.; Tournigand, C.; Ollero, M. Pharmacology of Tyrosine Kinase Inhibitors. Clin. J. Am. Soc. Nephrol. 2024, 19, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Chen, J.; Long, X.; Wang, J. Dose adjustment for tyrosine kinase inhibitors in non-small cell lung cancer patients with hepatic or renal function impairment (Review). Oncol. Rep. 2020, 45, 413–426. [Google Scholar] [CrossRef] [PubMed]
- van Leeuwen, R.W.F.; Jansman, F.G.A.; Hunfeld, N.G.; Peric, R.; Reyners, A.K.L.; Imholz, A.L.T.; Brouwers, J.R.B.J.; Aerts, J.G.; van Gelder, T.; Mathijssen, R.H.J. Tyrosine Kinase Inhibitors and Proton Pump Inhibitors: An Evaluation of Treatment Options. Clin. Pharmacokinet. 2017, 56, 683–688. [Google Scholar] [CrossRef]
- Fermier, N.; Bourguignon, L.; Goutelle, S.; Bleyzac, N.; Tod, M. Identification of Cytochrome P450-Mediated Drug–Drug Interactions at Risk in Cases of Gene Polymorphisms by Using a Quantitative Prediction Model. Clin. Pharmacokinet. 2018, 57, 1581–1591. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, X.; Ning, C.; Wang, Z.; Wang, Y.; Zheng, M.; Zhang, S.; Lu, Y.; Zhang, Y.; Li, N.; et al. Influences of ABC transporter and CYP3A4/5 genetic polymorphisms on the pharmacokinetics of lenvatinib in Chinese healthy subjects. Eur. J. Clin. Pharmacol. 2020, 76, 1125–1133. [Google Scholar] [CrossRef]
- Ozeki, T.; Nagahama, M.; Fujita, K.; Suzuki, A.; Sugino, K.; Ito, K.; Miura, M. Influence of CYP3A4/5 and ABC transporter polymorphisms on lenvatinib plasma trough concentrations in Japanese patients with thyroid cancer. Sci. Rep. 2019, 9, 5404. [Google Scholar] [CrossRef]
- Takimoto, T.; Kijima, T.; Otani, Y.; Nonen, S.; Namba, Y.; Mori, M.; Yokota, S.; Minami, S.; Komuta, K.; Uchida, J.; et al. Polymorphisms of CYP2D6 Gene and Gefitinib-Induced Hepatotoxicity. Clin. Lung Cancer 2013, 14, 502–507. [Google Scholar] [CrossRef]
- Sarah, A.; Dondi, E.; De Francia, S. Tyrosine kinase inhibitors: The role of pharmacokinetics and pharmacogenetics. Expert Opin. Drug Metab. Toxicol. 2023, 19, 733–739. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Chen, Q.; Zhan, Y.; Wang, Q.; Hu, C.; Yu, C.; Guo, Z.; Chen, X.; Zhong, D. Pharmacokinetic Drug Interactions of Apatinib With Rifampin and Itraconazole. J. Clin. Pharmacol. 2018, 58, 347–356. [Google Scholar] [CrossRef]
- O’Bryant, C.L.; Wenger, S.D.; Kim, M.; Thompson, L.A. crizotinib: Una nueva opción de tratamiento para cáncer pulmonar de célula no pequeña ALK positivo. Ann. Pharmacother. 2013, 47, 189–197. [Google Scholar]
- Hurtado, F.K.; de Braud, F.; De Castro Carpeño, J.; de Miguel Luken, M.J.; Wang, D.; Scott, J.; Lau, Y.Y.; McCulloch, T.; Mau-Sorensen, M. Effect of ceritinib on the pharmacokinetics of coadministered CYP3A and 2C9 substrates: A phase I, multicenter, drug–drug interaction study in patients with ALK + advanced tumors. Cancer Chemother. Pharmacol. 2021, 87, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, N.; Nagao, S.; Takanashi, K.; Kato, M.; Kaneko, A.; Morita, K.; Shindoh, H.; Ishigai, M. Preclinical evaluation of the potential for cytochrome P450 inhibition and induction of the selective ALK inhibitor, alectinib. Xenobiotica 2017, 47, 1042–1051. [Google Scholar] [CrossRef] [PubMed]
- Morcos, P.N.; Cleary, Y.; Guerini, E.; Dall, G.; Bogman, K.; De Petris, L.; Viteri, S.; Bordogna, W.; Yu, L.; Martin-Facklam, M.; et al. Clinical Drug–Drug Interactions Through Cytochrome P450 3A (CYP3A) for the Selective ALK Inhibitor Alectinib. Clin. Pharmacol. Drug Dev. 2017, 6, 280–291. [Google Scholar] [CrossRef]
- Nakagawa, T.; Fowler, S.; Takanashi, K.; Youdim, K.; Yamauchi, T.; Kawashima, K.; Sato-Nakai, M.; Yu, L.; Ishigai, M. In vitro metabolism of alectinib, a novel potent ALK inhibitor, in human: Contribution of CYP3A enzymes. Xenobiotica 2018, 48, 546–554. [Google Scholar] [CrossRef]
- Vishwanathan, K.; Dickinson, P.A.; So, K.; Thomas, K.; Chen, Y.; De Castro Carpeño, J.; Dingemans, A.C.; Kim, H.R.; Kim, J.; Krebs, M.G.; et al. The effect of itraconazole and rifampicin on the pharmacokinetics of osimertinib. Br. J. Clin. Pharmacol. 2018, 84, 1156–1169. [Google Scholar] [CrossRef]
- Liu, L.; Wang, Q.; Xie, C.; Xi, N.; Guo, Z.; Li, M.; Hou, X.; Xie, N.; Sun, M.; Li, J.; et al. Drug interaction of ningetinib and gefitinib involving CYP1A1 and efflux transporters in non-small cell lung cancer patients. Br. J. Clin. Pharmacol. 2021, 87, 2098–2110. [Google Scholar] [CrossRef]
- Santarpia, M.; De Pas, T.M.; Altavilla, G.; Spaggiari, L.; Rosell, R. Moving towards molecular-guided treatments: Erlotinib and clinical outcomes in non-small-cell lung cancer patients. Future Oncol. 2013, 9, 327–345. [Google Scholar] [CrossRef] [PubMed]
- Draper, A. A concise review of the changing landscape of hepatocellular carcinoma. Am. J. Manag. Care 2020, 26 (Suppl. S10), S211–S219. [Google Scholar] [PubMed]
- Foxx-Lupo, W.T.; Sing, S.; Alwan, L.; Tykodi, S.S. A Drug Interaction Between Cabozantinib and Warfarin in a Patient With Renal Cell Carcinoma. Clin. Genitourin. Cancer 2016, 14, e119–e121. [Google Scholar] [CrossRef]
- Arora, B.; Gota, V.; Menon, H.; Sengar, M.; Nair, R.; Patial, P.; Banavali, S.D. Therapeutic drug monitoring for imatinib: Current status and Indian experience. Indian J. Med. Paediatr. Oncol. 2013, 34, 224–228. [Google Scholar] [CrossRef]
- Boudou-Rouquette, P.; Tlemsani, C.; Blanchet, B.; Huillard, O.; Jouinot, A.; Arrondeau, J.; Thomas-Schoemann, A.; Vidal, M.; Alexandre, J.; Goldwasser, F. Clinical pharmacology, drug-drug interactions and safety of pazopanib: A review. Expert Opin. Drug Metab. Toxicol. 2016, 12, 1433–1444. [Google Scholar] [CrossRef] [PubMed]
- Studentova, H.; Volakova, J.; Spisarova, M.; Zemankova, A.; Aiglova, K.; Szotkowski, T.; Melichar, B. Severe tyrosine-kinase inhibitor induced liver injury in metastatic renal cell carcinoma patients: Two case reports assessed for causality using the updated RUCAM and review of the literature. BMC Gastroenterol. 2022, 22, 49. [Google Scholar] [CrossRef]
- Spencer, S.A.; Riley, A.C.; Matthew, A.; Di Pasqua, A.J. Brigatinib: Novel ALK Inhibitor for Non–Small-Cell Lung Cancer. Ann. Pharmacother. 2019, 53, 621–626. [Google Scholar] [CrossRef]
- Zhao, T.; Li, X.; Chen, Y.; Du, J.; Chen, X.; Wang, D.; Wang, L.; Zhao, S.; Wang, C.; Meng, Q.; et al. Risk assessment and molecular mechanism study of drug-drug interactions between rivaroxaban and tyrosine kinase inhibitors mediated by CYP2J2/3A4 and BCRP/P-gp. Front. Pharmacol. 2022, 13, 914842. [Google Scholar] [CrossRef]
- Paranjpe, R.; Basatneh, D.; Tao, G.; De Angelis, C.; Noormohammed, S.; Ekinci, E.; Abughosh, S.; Ghose, R.; Trivedi, M.V. Neratinib in HER2-Positive Breast Cancer Patients. Ann. Pharmacother. 2019, 53, 612–620. [Google Scholar] [CrossRef]
- El-Goly, A.M.M. Lines of Treatment of COVID-19 Infection. In COVID-19 Infections and Pregnancy; Elsevier: Amsterdam, The Netherlands, 2021; pp. 91–144. [Google Scholar]
- Ali, M.J.; Hanif, M.; Haider, M.A.; Ahmed, M.U.; Sundas, F.; Hirani, A.; Khan, I.A.; Anis, K.; Karim, A.H. Treatment Options for COVID-19: A Review. Front. Med. 2020, 7, 480. [Google Scholar] [CrossRef]
- Anwar, K.; Nguyen, L.; Nagasaka, M.; Ou, S.H.I.; Chan, A. Overview of Drug-Drug Interactions Between Ritonavir-Boosted Nirmatrelvir (Paxlovid) and Targeted Therapy and Supportive Care for Lung Cancer. JTO Clin. Res. Rep. 2023, 4, 100452. [Google Scholar] [CrossRef]
- Keene, M.R.; Heslop, I.M.; Sabesan, S.S.; Glass, B.D. Complementary and alternative medicine use in cancer: A systematic review. Complement. Ther. Clin. Pract. 2019, 35, 33–47. [Google Scholar] [CrossRef]
- Loquai, C.; Schmidtmann, I.; Garzarolli, M.; Kaatz, M.; Kähler, K.C.; Kurschat, P.; Meiss, F.; Micke, O.; Muecke, R.; Muenstedt, K.; et al. Interactions from complementary and alternative medicine in patients with melanoma. Melanoma Res. 2017, 27, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Rodseeda, C.; Yamanont, P.; Pinthong, D.; Korprasertthaworn, P. Inhibitory effects of Thai herbal extracts on the cytochrome P450 3A-mediated the metabolism of gefitinib, lapatinib and sorafenib. Toxicol. Rep. 2022, 9, 1846–1852. [Google Scholar] [CrossRef] [PubMed]
- Collado-Borrell, R.; Escudero-Vilaplana, V.; Romero-Jiménez, R.; Iglesias-Peinado, I.; Herranz-Alonso, A.; Sanjurjo-Sáez, M. Oral antineoplastic agent interactions with medicinal plants and food: An issue to take into account. J. Cancer Res. Clin. Oncol. 2016, 142, 2319–2330. [Google Scholar] [CrossRef] [PubMed]
- Prely, H.; Herledan, C.; Caffin, A.G.; Baudouin, A.; Larbre, V.; Maire, M.; Schwiertz, V.; Vantard, N.; Ranchon, F.; Rioufol, C. Real-life drug–drug and herb–drug interactions in outpatients taking oral anticancer drugs: Comparison with databases. J. Cancer Res. Clin. Oncol. 2022, 148, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; He, W.; Gao, X.; Li, B.; Mei, C.; Xu, R.; Chen, H. Resveratrol overcomes gefitinib resistance by increasing the intracellular gefitinib concentration and triggering apoptosis, autophagy and senescence in PC9/G NSCLC cells. Sci. Rep. 2015, 5, 17730. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, R.; Chang, Z.; Wang, X. Resveratrol activates CD8+ T cells through IL-18 bystander activation in lung adenocarcinoma. Front. Pharmacol. 2022, 13, 1031438. [Google Scholar] [CrossRef]
- Huang, K.-Y.; Wang, T.-H.; Chen, C.-C.; Leu, Y.-L.; Li, H.-J.; Jhong, C.-L.; Chen, C.-Y. Growth Suppression in Lung Cancer Cells Harboring EGFR-C797S Mutation by Quercetin. Biomolecules 2021, 11, 1271. [Google Scholar] [CrossRef]
- Corina, G.; Christine, H.; Klein, G. Oncologists’ experiences of discussing complementary and alternative treatment options with their cancer patients. A qualitative analysis. Support. Care Cancer 2016, 24, 3857–3862. [Google Scholar] [CrossRef]
- Ergun, Y.; Numune, A.; Yildirim Ozdemir, N.; Toptas, S.; Kurtipek, A.; Eren, T.; Yazici, O.; Ali Nahit Sendur, M.; Akinci, B.; Ucar, G.; et al. Drug-drug interactions in patients using tyrosine kinase inhibitors: A multicenter retrospective study. JBUON 2019, 24, 1719–1726. [Google Scholar] [PubMed]
- U.S. Department of Health and Human Services. Common Terminology Criteria for Adverse Events (CTCAE) Version 5.0; National Cancer Institute: Bethesda, MD, USA, 2017.
- Minasian, L.M.; O’Mara, A.; Mitchell, S.A. Clinician and Patient Reporting of Symptomatic Adverse Events in Cancer Clinical Trials: Using CTCAE and PRO-CTCAE® to Provide Two Distinct and Complementary Perspectives. Patient Relat. Outcome Meas. 2022, 13, 249–258. [Google Scholar] [CrossRef]
- Dikopf, A.; Wood, K.; Salgia, R. A safety assessment of crizotinib in the treatment of ALK-positive NSCLC patients. Expert Opin. Drug Saf. 2015, 14, 485–493. [Google Scholar] [CrossRef]
- Girard, N.; Audigier-Valette, C.; Cortot, A.B.; Mennecier, B.; Debieuvre, D.; Planchard, D.; Zalcman, G.; Moro-Sibilot, D.; Cadranel, J.; Barlési, F. ALK-rearranged non-small cell lung cancers: How best to optimize the safety of crizotinib in clinical practice? Expert Rev. Anticancer Ther. 2015, 15, 225–233. [Google Scholar] [CrossRef]
- Bauer, T.M.; Felip, E.; Solomon, B.J.; Thurm, H.; Peltz, G.; Chioda, M.D.; Shaw, A.T. Clinical Management of Adverse Events Associated with Lorlatinib. Oncologist 2019, 24, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Reed, M.; Rosales, A.L.S.; Chioda, M.D.; Parker, L.; Devgan, G.; Kettle, J. Consensus Recommendations for Management and Counseling of Adverse Events Associated With Lorlatinib: A Guide for Healthcare Practitioners. Adv. Ther. 2020, 37, 3019–3030. [Google Scholar] [CrossRef]
- Cheng, X.; Lv, X.; Qu, H.; Li, D.; Hu, M.; Guo, W.; Ge, G.; Dong, R. Comparison of the inhibition potentials of icotinib and erlotinib against human UDP-glucuronosyltransferase 1A1. Acta Pharm. Sin. B 2017, 7, 657–664. [Google Scholar] [CrossRef]
- Solassol, I.; Pinguet, F.; Quantin, X. FDA- and EMA-Approved Tyrosine Kinase Inhibitors in Advanced EGFR-Mutated Non-Small Cell Lung Cancer: Safety, Tolerability, Plasma Concentration Monitoring, and Management. Biomolecules 2019, 9, 668. [Google Scholar] [CrossRef]
- Kucharczuk, C.R.; Ganetsky, A.; Vozniak, J.M. Drug-Drug Interactions, Safety, and Pharmacokinetics of EGFR Tyrosine Kinase Inhibitors for the Treatment of Non–Small Cell Lung Cancer. J. Adv. Pr. Oncol. 2018, 9, 189–200. [Google Scholar]
- Gao, X.; Le, X.; Costa, D.B. The safety and efficacy of osimertinib for the treatment of EGFR T790M mutation positive non-small-cell lung cancer. Expert Rev. Anticancer Ther. 2016, 16, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.H.; Lee, T.Y.; Chang, G.C. Hepatitis B flare during osimertinib targeted therapy in a lung cancer patient with a resolved hepatitis B virus infection. Eur. J. Cancer 2020, 130, 272–274. [Google Scholar] [CrossRef]
- Kang, Y.; Meng, F. Acute fulminant hepatitis associated with osimertinib administration in a lung cancer patient with chronic hepatitis B: The first mortality case report. Thorac. Cancer 2022, 13, 1091–1094. [Google Scholar] [CrossRef]
- Cortot, A.; Le, X.; Smit, E.; Viteri, S.; Kato, T.; Sakai, H.; Park, K.; Camidge, D.R.; Berghoff, K.; Vlassak, S.; et al. Safety of MET Tyrosine Kinase Inhibitors in Patients With MET Exon 14 Skipping Non-small Cell Lung Cancer: A Clinical Review. Clin. Lung Cancer 2022, 23, 195–207. [Google Scholar] [CrossRef]
- Iaxx, R.; Lefort, F.; Domblides, C.; Ravaud, A.; Bernhard, J.C.; Gross-Goupil, M. An Evaluation of Cabozantinib for the Treatment of Renal Cell Carcinoma: Focus on Patient Selection and Perspectives. Ther. Clin. Risk Manag. 2022, 18, 619–632. [Google Scholar] [CrossRef]
- Huillard, O.; Boissier, E.; Blanchet, B.; Thomas-Schoemann, A.; Cessot, A.; Boudou-Rouquette, P.; Durand, J.P.; Coriat, R.; Giroux, J.; Alexandre, J.; et al. Drug safety evaluation of sorafenib for treatment of solid tumors: Consequences for the risk assessment and management of cancer patients. Expert Opin. Drug Saf. 2014, 13, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.H.; Kerkelä, R.; Force, T. Mechanisms of Cardiac Dysfunction Associated With Tyrosine Kinase Inhibitor Cancer Therapeutics. Circulation 2008, 118, 84–95. [Google Scholar] [CrossRef]
- Bouitbir, J.; Alshaikhali, A.; Panajatovic, M.; Abegg, V.; Paech, F.; Krähenbühl, S. Mechanisms of Cardiotoxicity Associated with Tyrosine Kinase Inhibitors in H9c2 Cells and Mice. Eur. Cardiol. Rev. 2020, 15, e33. [Google Scholar] [CrossRef]
- Cheng, M.; Yang, F.; Liu, J.; Yang, D.; Zhang, S.; Yu, Y.; Jiang, S.; Dong, M. Tyrosine Kinase Inhibitors-Induced Arrhythmias: From Molecular Mechanisms, Pharmacokinetics to Therapeutic Strategies. Front. Cardiovasc. Med. 2021, 8, 758010. [Google Scholar] [CrossRef]
- Patras de Campaigno, E.; Bondon-Guitton, E.; Laurent, G.; Montastruc, F.; Montastruc, J.L.; Lapeyre-Mestre, M.; Despas, F. Identification of cellular targets involved in cardiac failure caused by PKI in oncology: An approach combining pharmacovigilance and pharmacodynamics. Br. J. Clin. Pharmacol. 2017, 83, 1544–1555. [Google Scholar] [CrossRef] [PubMed]
- van Leeuwen, R.W.F.; van Gelder, T.; Mathijssen, R.H.J.; Jansman, F.G.A. Drug–drug interactions with tyrosine-kinase inhibitors: A clinical perspective. Lancet Oncol. 2014, 15, e315–e326. [Google Scholar] [CrossRef] [PubMed]
- Paech, F.; Bouitbir, J.; Krähenbühl, S. Hepatocellular Toxicity Associated with Tyrosine Kinase Inhibitors: Mitochondrial Damage and Inhibition of Glycolysis. Front. Pharmacol. 2017, 8, 367. [Google Scholar] [CrossRef]
- Saran, C.; Brouwer, K.L.R. Hepatic Bile Acid Transporters and Drug-induced Hepatotoxicity. Toxicol. Pathol. 2023, 51, 405–413. [Google Scholar] [CrossRef]
- Lee, S.K.; Choi, J.Y.; Jung, E.S.; Kwon, J.H.; Jang, J.W.; Bae, S.H.; Yoon, S.K. An Immunological Perspective on the Mechanism of Drug Induced Liver Injury: Focused on Drugs for Treatment of Hepatocellular Carcinoma and Liver Transplantation. Int. J. Mol. Sci. 2023, 24, 5002. [Google Scholar] [CrossRef]
- Shah, R.R.; Morganroth, J.; Shah, D.R. Hepatotoxicity of Tyrosine Kinase Inhibitors: Clinical and Regulatory Perspectives. Drug Saf. 2013, 36, 491–503. [Google Scholar] [CrossRef]
- Rimassa, L.; Danesi, R.; Pressiani, T.; Merle, P. Management of adverse events associated with tyrosine kinase inhibitors: Improving outcomes for patients with hepatocellular carcinoma. Cancer Treat. Rev. 2019, 77, 20–28. [Google Scholar] [CrossRef]
- Shyam Sunder, S.; Sharma, U.C.; Pokharel, S. Adverse effects of tyrosine kinase inhibitors in cancer therapy: Pathophysiology, mechanisms and clinical management. Signal Transduct. Target. Ther. 2023, 8, 262. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.-H.; Reguart, N.; Barinoff, J.; Köhler, J.; Uttenreuther-Fischer, M.; Stammberger, U.; O’Brien, D.; Wolf, J.; Cohen, E.E. Diarrhea associated with afatinib: An oral ErbB family blocker. Expert Rev. Anticancer Ther. 2013, 13, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yang, Y.; Zhang, L.; Cheng, Y.; Han, B.; Lu, Y.; Wang, C.; Wang, Z.; Yang, N.; Fan, Y.; et al. Expert consensus of management of adverse drug reactions with anaplastic lymphoma kinase tyrosine kinase inhibitors. ESMO Open 2023, 8, 101560. [Google Scholar] [CrossRef]
- Qian, J.; Zhang, X.; Zhang, B.; Yan, B.; Wang, L.; Gu, P.; Wang, W.; Wang, H.; Han, B. Tyrosine Kinase Inhibitor-Related Hepatotoxicity in Patients with Advanced Lung Adenocarcinoma: A Real-World Retrospective Study. Cancer Manag. Res. 2020, 12, 3293–3299. [Google Scholar] [CrossRef] [PubMed]
- Kodama, T.; Sato, S.; Miyazaki, K.; Okauchi, S.; Sasatani, Y.; Ohara, G.; Kagohashi, K.; Satoh, H. Body Weight Change in Non-small Cell Lung Cancer Patients Treated With EGFR-TKI. Cancer Diagn. Progn. 2022, 2, 373–379. [Google Scholar] [CrossRef]
- Vogel, W.H.; Jennifer, P. Management Strategies for Adverse Events Associated With EGFR TKIs in Non–Small Cell Lung Cancer. J. Adv. Pr. Oncol. 2016, 7, 723–735. [Google Scholar]
- Blais, N.; Adam, J.P.; Nguyen, J.; Grégoire, J.C. Evaluation and Management of Dyslipidemia in Patients Treated with Lorlatinib. Curr. Oncol. 2021, 28, 265–272. [Google Scholar] [CrossRef]
- Zhu, X.; Wu, S. Risks and management of hypertension in cancer patients undergoing targeted therapy: A review. Clin. Hypertens. 2022, 28, 14. [Google Scholar] [CrossRef]
- Agustoni, F.; Platania, M.; Vitali, M.; Zilembo, N.; Haspinger, E.; Sinno, V.; Gallucci, R.; de Braud, F.; Garassino, M.C. Emerging toxicities in the treatment of non-small cell lung cancer: Ocular disorders. Cancer Treat. Rev. 2014, 40, 197–203. [Google Scholar] [CrossRef]
- Gabora, K.; Piciu, A.; Bădulescu, I.C.; Larg, M.I.; Stoian, I.A.; Piciu, D. Current evidence on thyroid related adverse events in patients treated with protein tyrosine kinase inhibitors. Drug Metab. Rev. 2019, 51, 562–569. [Google Scholar] [CrossRef] [PubMed]
- AlShatnawi, M.N.; Shawashreh, R.A.; Sunoqrot, M.A.; Yaghi, A.R. A systematic review of epidermal growth factor receptor tyrosine kinase inhibitor-induced heart failure and its management. Egypt. J. Intern. Med. 2022, 34, 85. [Google Scholar] [CrossRef]
- Abu Rmilah, A.A.; Lin, G.; Begna, K.H.; Friedman, P.A.; Herrmann, J. Risk of QTc prolongation among cancer patients treated with tyrosine kinase inhibitors. Int. J. Cancer 2020, 147, 3160–3167. [Google Scholar] [CrossRef]
- Takahashi, S. Fatigue and its management in cancer patients undergoing VEGFR-TKI therapy. Expert Opin. Drug Saf. 2022, 21, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Alrowais, F.; Alotaibi, S.S.; Hilal, A.M.; Marzouk, R.; Mohsen, H.; Osman, A.E.; Alneil, A.A.; Eldesouki, M.I. Clinical Decision Support Systems to Predict Drug–Drug Interaction Using Multilabel Long Short-Term Memory with an Autoencoder. Int. J. Environ. Res. Public Health 2023, 20, 2696. [Google Scholar] [CrossRef] [PubMed]
- Chan, F.Y.; Ku, Y.E.; Lie, W.N.; Chen, H.Y. Web-Based Explainable Machine Learning-Based Drug Surveillance for Predicting Sunitinib- and Sorafenib-Associated Thyroid Dysfunction: Model Development and Validation Study. JMIR Form. Res. 2025, 9, e67767. [Google Scholar] [CrossRef]
- Church, D.; Kerr, R.; Domingo, E.; Rosmarin, D.; Palles, C.; Maskell, K.; Tomlinson, I.; Kerr, D. “Toxgnostics”: An unmet need in cancer medicine. Nat. Rev. Cancer 2014, 14, 440–445. [Google Scholar] [CrossRef]
- van der Kleij, M.B.A.; Guchelaar, N.A.D.; Mathijssen, R.H.J.; Versluis, J.; Huitema, A.D.R.; Koolen, S.L.W.; Steeghs, N. Therapeutic Drug Monitoring of Kinase Inhibitors in Oncology. Clin. Pharmacokinet. 2023, 62, 1333–1364. [Google Scholar] [CrossRef]
- Ferrer, F.; Tetu, P.; Dousset, L.; Lebbe, C.; Ciccolini, J.; Combarel, D.; Meyer, N.; Paci, A.; Bouchet, S. Tyrosine kinase inhibitors in cancers: Treatment optimization—Part II. Crit. Rev. Oncol. Hematol. 2024, 200, 104385. [Google Scholar] [CrossRef]
- Westerdijk, K.; Steeghs, N.; Tacke, C.S.J.; van der Graaf, W.T.A.; van Erp, N.P.; van Oortmerssen, G.; Hermens, R.P.M.G.; Desar, I.M.E. Therapeutic drug monitoring to personalize dosing of imatinib, sunitinib, and pazopanib: A mixed methods study on barriers and facilitators. Cancer Med. 2023, 12, 21041–21056. [Google Scholar] [CrossRef]

| Indication | TKI | Molecular Target |
|---|---|---|
| Non-small-cell lung cancer (NSCLC) | Gefitinib | EGFR |
| Erlotinib | EGFR | |
| Afatinib | EGFR, Her2, Her 4 | |
| Osimertinib | EGFR (including T790M mutation) | |
| Dacomitinib | EGFR | |
| Alectinib | ALK | |
| Brigatinib | ALK | |
| Ceritinib | ALK | |
| Lorlatinib | ALK, ROS-1 | |
| Crizotinib | ALK, ROS-1 | |
| Entrectinib | ALK, ROS-1, TRK | |
| Renal cell carcinoma (RCC) | Sunitinib | VEGFR, PDGFR, KIT |
| Pazopanib | VEGFR, PDGFR, KIT | |
| Cabozantinib | MET, VEGFR, RET | |
| Axitinib | VEGFR | |
| Hepatocellular carcinoma (HCC) | Sorafenib | VEGFR, PDGFR, RAF |
| Regorafenib | VEGFR, PDGFR, KIT | |
| Cabozantinib | MET, VEGFR, RET | |
| Breast cancer | Lapatinib | EGFR, Her 2 |
| Neratinib | EGFR, Her 2 | |
| Abemaciclib | CDK4/6 | |
| Palbociclib | CDK4/6 | |
| Ribociclib | CDK4/6 | |
| Gastrointestinal stromal tumors (GISTs) | Imatinib | KIT, PDGFR |
| Sunitinib | KIT, PDGFR, VEGFR | |
| Regorafenib | KIT, PDGFR, VEGFR | |
| Ripretinib | KIT, PDGFR | |
| Thyroid cancer | Vandetanib | RET, VEGFR, EGFR |
| Cabozantinib | MET, VEGFR, RET | |
| Lenvatinib | EGFT, VEGFR | |
| Colorectal cancer | Regorafenib | KIT, PDGFR, VEGFR |
| Melanoma | Vemurafenib | BRAF V600E |
| Dabrafenib | BRAF V600E | |
| Trametinib | MEK |
| TKI | Interacting Drug | Impact of the DDI | Severity |
|---|---|---|---|
| Erlotinib | Omeprazole (and other proton pump inhibitors) | Increased gastric pH reduces drug solubility, leading to decreased absorption and lower plasma levels. | Major |
| Gefitinib | Antacids (e.g., aluminum hydroxide/magnesium) | Elevated gastric pH from antacids decreases solubility, resulting in reduced absorption. | Moderate |
| Pazopanib | Esomeprazole (and similar agents) | Concomitant PPI use significantly lowers pazopanib bioavailability due to pH-dependent solubility. | Major |
| Neratinib | Lansoprazole | Decreased absorption due to altered gastric pH, reducing bioavailability. | Major |
| TKI Target | TKI | DDI | DDI Management |
|---|---|---|---|
| ALK | lorlatinib | + strong CYP3A4 inhibitors: ↑ AUC and cmax | Avoid combination; if not possible, ↓ lorlatinib dose to 75 mg |
| crizotinib | + strong CYP3A4 inhibitors: ↑ AUC; + strong CYP3A4 inducers: ↓ AUC | Avoid combination; if not possible, ↓ dose by 50% when co-administration with strong inhibitors is mandatory | |
| ceritinib | strong CYP3A4 inhibitor and weak CYP2C9 inhibitor → affects CYP3A4 substrates | Caution at co-administration with narrow-therapeutic-index drugs; monitor INR if co-administered with warfarin | |
| EGFR | osimertinib | + strong CYP3A4 inducers: ↓ AUC | Avoid combination; if not possible, ↑ dose to 160 mg |
| erlotinib | + strong CYP3A4 inhibitors/inducers, P-gp inhibitors, coumarin-derived anticoagulants, cigarette smoke | Initial dose of 300 mg for current smokers instead of 150 mg for non-smokers; ↑ dose by 50 mg if association with strong inducers is mandatory; ↓ dose by 50 mg if association with strong inhibitors is mandatory | |
| VEGFR | sorafenib | + strong CYP3A4 inducers | Avoid combination; if not possible, dose adjustment |
| regorafenib | + strong CYP3A4 inducers, inhibitors | Avoid combination; if not possible, dose adjustment | |
| cabozantinib | + strong CYP3A4 inducers, inhibitors | Avoid combination; if not possible, ↑ dose by 20 mg if association with strong inducers is mandatory; ↓ dose by 20 mg if association with strong inhibitors is mandatory | |
| apatinib | + strong CYP3A4 inducers | Avoid combination; if not possible, dose adjustment | |
| Multiple targets | imatinib | + strong CYP3A4 inducers | Avoid combination; if not possible, ↑ dose by 50% if association with strong inducers is mandatory |
| pazopanib | + strong CYP3A4 inhibitors/inducers, P-gp or BCRP inhibitors | Avoid combination; if not possible, ↓ dose by 50% when co-administration with strong inhibitors is mandatory |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Budău, L.V.; Pop, C.; Mogoșan, C. Beyond the Basics: Exploring Pharmacokinetic Interactions and Safety in Tyrosine-Kinase Inhibitor Oral Therapy for Solid Tumors. Pharmaceuticals 2025, 18, 959. https://doi.org/10.3390/ph18070959
Budău LV, Pop C, Mogoșan C. Beyond the Basics: Exploring Pharmacokinetic Interactions and Safety in Tyrosine-Kinase Inhibitor Oral Therapy for Solid Tumors. Pharmaceuticals. 2025; 18(7):959. https://doi.org/10.3390/ph18070959
Chicago/Turabian StyleBudău, Laura Veronica, Cristina Pop, and Cristina Mogoșan. 2025. "Beyond the Basics: Exploring Pharmacokinetic Interactions and Safety in Tyrosine-Kinase Inhibitor Oral Therapy for Solid Tumors" Pharmaceuticals 18, no. 7: 959. https://doi.org/10.3390/ph18070959
APA StyleBudău, L. V., Pop, C., & Mogoșan, C. (2025). Beyond the Basics: Exploring Pharmacokinetic Interactions and Safety in Tyrosine-Kinase Inhibitor Oral Therapy for Solid Tumors. Pharmaceuticals, 18(7), 959. https://doi.org/10.3390/ph18070959